
Introduction
Urologic cancers account for approximately 10% of all cancer deaths in the USA and include bladder, kidney, prostate and testicular cancers [1].
The establishment and progression of malignancy involves broad changes in gene expression that are determined by both genetic and epigenetic events. Genetic events include chromosome rearrangements and duplications as well as translocations, deletions, and single base pair mutations. Epigenetic modifications are somatically heritable changes that modify gene expression without altering the DNA sequence. Among these are histone modifications, DNA methylation, and miRNA expression [2].
Histone Modifications
Post-translational modification of the histone protein N terminal tails can alter the structure of the nucleosome and change the compaction state of chromatin. Common modifications include methylation, acetylation, phosphorylation, ubiquitylation and sumoylation [2]. Among these, histone acetylation and methylation are best described in cancer epigenetic dysregulation [2].
Histone acetylation neutralizes the positive charge of lysine residues, weakening their electrostatic interactions with DNA [3]. This leads to a more relaxed state of the chromatin and is associated with transcriptional activation. Addition of the acetyl group is carried out by histone acetyltransferases (HATs), and its removal is catalyzed by histone deacetylases (HDACs) [4]. HDACs are classified into four distinct groups based on their homology to yeast histone deacetylases. Class I HDACs encompass HDAC1, 2, 3 and 8 and, with the exception of HDAC8 that can be located in the nucleus or cytoplasm, are exclusively located in the nucleus. Class II HDACs include HDAC4, 5, 6, 7, 9 and 10 and can be present in the nucleus or the cytoplasm [5, 6]. Class III HDACs are the sirtuins, proteins which require the cofactor NAD+ to be active. Unlike Class I and Class II HDACs, sirtuins are not inhibited by known pharmacologic HDAC inhibitors (HDACi) such as Vorinostat and Trichostatin A (TSA) [5, 6].
Histone methylation occurs at lysine and arginine residues and, depending on the target, can lead to activation or repression of gene expression [7]. Methylation is catalyzed by histone methyltransferases (HTMs) while demethylation is performed by histone demethylases (HDMs). There are currently two histone demethylase families, the lysine specific demethylases (LSD) and the JmjC-domain-containing histone demethylases (JHDMs) [7]. The LSDs comprise LSD1 and LSD2, which are dependent on FAD to be catalytically active [8]. The JHDMs in turn, catalyze the hydroxylation of the lysine methylgroup and require two factors to be catalytically active: Fe(II) and 2-oxoglutarate [7].
In cancer, histone modifications have been associated with both activation and repression of gene expression. Modifications such as histone 3 methylation at lysine 4 (H3K4me), histone 3 di-methylation at lysine 4 (H3K4me2), histone 3 tri-methylation at lysine 4 (H3K4me3), histone 3 acetylation at lysine 9 (H3K9ac), histone 3 methylation at lysine 9 (H3K9me) and histone 3 acetylation at lysine 27 (H3K27ac) are associated with active chromatin whereas histone 3 tri-methylation at lysine 36 (H3K36me3), histone 3 tri-methylation at lysine 9 (H3K9me3) and histone 3 methylation at lysine 27 (H3K27me) are associated with repressive chromatin [9].
DNA methylation
DNA methylation results from addition of a methyl group to the 5-carbon of a cytosine residue by the enzyme DNA methyltransferase (DNMT) [10]. DNMT forms a complex with CpG dinucleotides that allows the transfer of a methyl group to the cytosine residue [10]. Many CpG sites are located in the promoter regions of genes. Collectively they are known as CpG islands. In general, DNA methylation of CpG islands located in gene promoters leads to transcriptional repression. However, there are exceptions to this classical view, in which promoter hypermethylation is associated with increased gene expression [11-14]. This occurs in instances where DNA methylation drives the use of an alternative transcription start site or inhibits the binding of a repressive protein [15]. Increased gene expression in the context of promoter hypermethylation is associated with an increase in H3K4me3, a histone mark characteristic of gene activation [15].
In eukaryotes, DNA methylation is mediated by three DNMTs: DNMT1 is responsible for the maintenance of methylation patterns after DNA replication whereas DNMT3A and DNMT3B carry out de novo methylation [4]. Any alteration that affects the activity of these enzymes can lead to an imbalance in methylation that provides the basis, or contributes, to the initiation of carcinogenesis.
miRNAs
miRNAs are small endogenous non-coding RNAs (ncRNAs), 21-25 nucleotides in length, that regulate gene expression by targeting specific messenger RNAs (mRNAs) for translational repression or degradation. Expression patterns of miRNAs differ between normal and tumor tissues [16, 17]. Depending on their target, miRNAs can act either as tumor suppressors or oncogenes; downregulation of an miRNA that targets an oncogene, or an overexpression of an miRNA that targets a tumor suppressor gene, can promote carcinogenesis [16, 17].
Epigenetic drugs
Two strategies for epigenetic therapy are currently in use: small molecules that inhibit epigenetic-modifying enzymes and manipulation of miRNA expression.
Amongst the small molecule inhibitors are HDAC inhibitors and DNMT inhibitors. HDAC inhibitors (HDACi) are classified into 4 groups according to their chemical structures: hydroxamates (SB393, Vorinostat, Panobinostat), cyclic peptides (Romidepsin), benzamides (Entinostat and Mocetinostat) and aliphatic fatty acids (Valproic Acid) [18].
The majority of HDACi inhibit zinc-dependent HDACs by interacting with the zinc ion. In cancer cells, the inhibition of histone deacetylation restores expression of tumor suppressor genes that were previously silenced by epigenetic mechanisms [18, 19].
DNMT inhibitors are divided into nucleoside analogues and non-nucleoside analogs [4]. Nucleoside analogues, such as Azacitidine, Decitabine and FdCyd, are cytosine analogs modified at the C5 position. Inside the cell they are metabolized and incorporated into DNA molecules [4]. DNA methyltransferases can bind to these modified nucleotides but their modification at C5 prevents their methylation. It also prevents the dissociation of the enzyme thereby reducing DNMT activity at other sites [4]. Non-nucleoside analogues, such as Hydralazine, Procainamide and MG98, inhibit methylation by binding to the catalytic region of the enzyme [4].
Another focus of epigenetic therapy is the manipulation of miRNA expression and activity. Several strategies have been employed to silence miRNAs that are overexpressed in cancer. These include anti-miRNA oligonucleotides (AMOs), peptide nucleic acids (PNAS), miRNA-masking antisense oligonucleotides (miR-mask) and miRNA sponges [16]. Restoration of miRNA expression that has been downregulated in cancer is achieved by administration of synthetic miRNAs or by induced expression of miRNA coding genes using viral constructs, such as adenovirus-associated vectors [16].
Dysregulation of epigenetic marks leads to changes in gene expression that, in cancer cells, can result in activation of oncogenes or inactivation of tumor suppressor genes, both of which can contribute to cancer. Unlike genetic mutations, however, epigenetic changes are reversible. Therefore, the development of drugs capable of restoring the normal epigenetic patterns of cells has great therapeutic potential. In this review we discuss the efficacy of this novel therapeutic approach through the analysis of clinical trials of epigenetic therapies conducted in prostate, kidney and bladder cancers.
Methods
We performed a comprehensive literature review and searched for clinical trials from the United States (https://clinicaltrials.gov/) and European (https://www.clinicaltrialsregister.eu/) databases. Relevant articles on the subject were also retrieved from PubMed database using keywords encapsulating all types of epigenetic therapies and urologic cancers (examples: “epigenetic therapy” AND “urologic cancer”, “prostate cancer” AND “HDACi”, “kidney cancer” AND “DNMTi”). To guarantee that most of the data on the subject was included, the reference sections of the captured articles were also filtered for relevant articles.
Prostate cancer - epigenetics
Dysregulation of epigenetic-modifying enzymes disturbs normal epigenetic patterns and is associated with cancer development and progression. In prostate cancer, DNA methyltransferases are upregulated [20, 21]. Histone-modifying enzymes, such as HDACs are upregulated in prostate cancer [22]. HMTs and HDMs show variable changes in expression with a tendency for upregulation of HMTs and lower expression of HDMs [23, 24]. Prognostically, overexpression of HDAC2 is associated with a shortened time before prostate cancer recurrence as shown in a subgroup of patients with Gleason Score 7 carcinomas, [6].
Specific histone modifications have also been associated with prostate cancer [25, 26]. The levels of histone marks H3Ac and H3K9me2 are significantly lower in tumor tissue when compared to normal tissue [26]. Conversely, an increase in H3K27me3 is found in metastatic tissue relative to localized tumors and normal prostatic tissue [25]. Finally, higher levels of H3K4me1 are associated with a higher probability of recurrence [26].
Changes in DNA methylation are also evident in prostate cancer and are targets for epigenetic therapy. The CCDN2, GSTP1 and RARβ2 genes, involved in cell cycle control, DNA repair mechanisms and hormonal responses respectively, are hypermethylated in prostate cancer. Alteration of their normal methylation status is correlated with poor clinical prognosis [26]. As with many epigenetic alterations, these biomarkers are useful in diagnosis and prognosis of disease [25, 26].
Finally, miRNA levels are also altered in prostate cancer, affecting the expression of genes involved in cell cycle control, apoptosis, migration, and invasion [27]. Levels of miRNAs also have the potential to be used as biomarkers for diagnosis and prognosis [25]. As an example, miR-141 is upregulated in prostate cancer [25, 28]. Serum levels of this miRNA can distinguish between tumor and healthy tissue and higher levels of miR-141 are associated with worse prognosis [28]. miR-449a is another miRNA that is downregulated in prostate cancer. It targets HDAC1, so its downregulation contributes to overexpression of this enzyme, showing that epigenetic-modifying enzymes are often regulated epigenetically [27, 29].
Prostate cancer - current treatment
Prostate cancer treatment is disease stage-specific. Epigenetic therapies have thus far been limited to the advanced form of castrate resistant prostate cancer (CRPC). Currently there are several chemotherapeutic agents approved for the treatment of advanced CRPC: Sipuleucel T, Docetaxel, Cabazitaxel, Abiraterone, Alpharadin and Enzalutamide [30-35]. Although all of these agents have shown efficacy, strategies for the sequence of administration and their combination are still being optimized [36]. Treatment resistance is a major concern with some of these agents, including Abiraterone and Enzalutamide, reflecting the need for ongoing development of novel therapeutic strategies [36]. Since epigenetic dysregulation contributes to the development of treatment resistance, epigenetic therapy is an intriguing addition to the CRPC therapy arsenal [37].
Prostate cancer - pre-clinical data
Pre-clinical studies in prostate cancer cell lines demonstrate that treatment with HDACi can restore susceptibility to chemotherapeutic agents such as taxanes, antiandrogens, and mTOR inhibitors [38-40]. Combined therapy using HDACi and taxanes prevents tumor growth and increases cell death rate when compared to a monotherapeutic approach [38]. Liu et al showed that low doses of the HDACi Panobinostat can restore the susceptibility of prostate cancer cells to hormonal therapy with the nonsteroidal antiandrogen Bicalutamide [39]. The HDACi Belinostat (PXD101) can also downregulate the androgen receptor, preventing the onset of castration resistant prostate cancer in vivo in the context of hormonal therapy [41].
An in vitro study of the HDACi Panobinostat in combination with the mTOR inhibitor Rapamycin in prostate cancer cell lines resulted in a decrease in HIF1- α expression leading to inhibition of angiogenesis [40]. Combined therapy with these agents was more efficient than either one administrated alone [40].
Cancer stem cells are thought to be responsible for treatment resistance and tumor recurrence [42] and present epigenetic alterations that contribute to their ability to resist therapy [42]. Epigenetic therapeutics may therefore have the potential to target not only the bulk tumor but also this key subset of cells [42]. A study carried out by Frame et al revealed that prostate stem-like cells are more resistant to radiotherapy [43]. However, combined therapy with HDACi restored sensibility to radiotherapy [43]. Additionally, prostate stem-like cells treated jointly with the HDACi Trichostatin A and radiotherapy showed a significant reduction in the number of cell colonies formed when compared to treatment with radiation alone [43].
Cancer is a heterogeneous disease and the identification of biomarkers that predict whether a specific therapy (including epigenetic therapies) will be beneficial, is essential to improving cancer treatment. Recently, it has been reported that prostate cells positive for the presence of androgen receptor and cellular prostatic acid phosphatase show greater response to treatment with HDACi than cells without this pattern of expression [44].
At the level of DNA methylation, reversion of methylation can restore expression of genes silenced by this epigenetic mechanism. Treatment of human prostate cancer cells with Procainamide, a non-nucleoside DNMT inhibitor, results in a decrease in GSTP1 methylation levels and a consequent increase in gene expression [45]. In vivo, treatment of immunodeficient mice carrying xenograft tumors with Procainamide resulted in a significant reduction in tumor size, suggesting clinical efficacy [45].
Resistance to hormonal therapy in prostate cancer is mediated by several mechanisms. Alterations at the DNA level include androgen receptor gene amplifications and point mutations [46]. However, these modifications account for only a minority of cases. Downstream activation of the androgen receptor pathway and activation of an alternative signaling pathway can also contribute to hormonal therapy resistance [46]. Hypermethylation of the androgen receptor promoter region correlates with decreased androgen receptor expression and is also associated with the development of hormonal therapy resistance [47]. In vivo studies reveal that long-term treatment of prostate cancer cells with the DNMTi Azacitidine led to a significant reduction in cell proliferation due to increased androgen receptor expression. Moreover, androgen receptor induction restored sensitivity to the antiandrogen agent Bicalutamide [47].
The DNA demethylating agent, Disulfiram, has also been tested in prostate cancer cells. Treatment with Disulfiram resulted in the reestablishment of APC and RARβ gene expression, both of which are known to be hypermethylated and inactive in prostate cancer [48]. Cell growth inhibition was observed in vitro, and in vivo using prostate cancer xenograft models [48].
Finally, microRNAs modulators have been tested in preclinical studies as potential therapeutic options for prostate cancer. miR-16 regulates the expression of genes involved in cell-cycle control and apoptosis such as CDK1, CDK2 and BCL2 [49, 50]. Transfection of a synthetic miR-16 reduced the proliferative capacity of several prostate cancer cell lines [49]. In vivo, Takeshita et al used the atelocollagen method to deliver miR-16 to bone metastases via the mouse tail vein. They subsequently observed a suppression in metastasis growth, indicating not only efficacy of the treatment but also of the delivery method [49].
Like HDACi and DNMTi, antisense oligonucleotides can restore the sensitivity of cancer cells to chemotherapeutic agents. Upregulation of the Bcl2 and CLU genes in prostate cancer is linked to chemoresistance and cancer progression [51, 52]. Knockdown of these genes by antisense oligonucleotides decreases gene expression and reestablishes tumor sensitivity to taxane-based chemotherapy [51, 52]. Furthermore, transfection of miR-449a into prostate cancer cells lines caused cell cycle arrest and a decrease in HDAC1 levels, an effect also observed after knockdown of HDAC1 using an siRNA [29]. The inhibitory effect of miR-449a on cell cycle progression was associated with increased expression of the protein p27 [29].
These results demonstrate the potential for epigenetic therapies to advance prostate cancer treatment.
Prostate cancer - clinical data
Clinical trials using HDAC inhibitors for the treatment of prostate cancer showed a PSA response in five studies, three of which resulted in a decrease in PSA levels of ≥50% (Table 1: NCT01075308, NCT00667862, NCT00330161, NCT00106418, [53-57]). The best results were obtain with the administration of the HDACi Panobinostat (Table1: NCT00667862, [54, 55]. Stable disease was reported in two clinical trials, but in one of them conversion from an unfavorable circulating tumor cell profile to a favorable one was observed in 64% of the patients (Table 1: NCT01075308, NCT00667862, NCT00330161, [53, 54, 56]).
Most common side effects were grade 2 fatigue and nausea. In addition, HDACi SB939 caused five patients to experience one or more grade 3 complications (Table 1: NCT01075308, [53]). More severe side effects were noted with HDACi Panobinostat, resulting in 71, 4% of the patients experiencing one or more grade 3 adverse effects and four subjects reporting grade 4 adverse effects (Table1: NCT00667862, [54]). Another trial using Panobinostat reported no grade 4 toxicities when administered as monotherapy [55], however, when administered in combination with Docetaxel, seven patients experienced grade 4 toxicities [55] (Table 1). HDACi Vorinostat also showed a complex side effect profile. When administered alone in patients pre-treated with chemotherapeutic agents it led to the development of grade 3/4 toxicities in 48% of the patients, with 41% of the patients forced to discontinue therapy due to their severity (Table 1: NCT00330161, [56]). A second trial of Vorinostat in combination with docetaxel was terminated early due to excessive toxicity as five patients experienced dose-limiting toxicities, including two patients experiencing neutropenic fever and sepsis. The other three patients reported an anaphylactic reaction, a myocardial infarction and a gastrointestinal bleed, respectively (Table 1: NCT00565227, [58]). Finally, a trial of the HDACi Romidepsin in metastatic prostate cancer resulted in no grade 4 toxicities, and grade 3 events represented only 4.7% of all reported adverse effects (Table 1: NCT00106418, [57]).
Curcumin, a compound found in the spice turmeric, is another HDACi [59]. A trial of Curcumin in prostate cancer showed no PSA response when used in combination with radiotherapy (Table 1: NCT01917890, [60]). However, there was a significant reduction in urinary symptoms, one of the most common side effects of radiotherapy (Table 1: NCT01917890, [60]). Two additional trials testing Curcumin in the treatment of prostate cancer are ongoing (Table 1: NCT02095717, NCT02064673).
DNMT inhibitors have also showed promising results in clinical trials of prostate cancer. When treated with the DNMTi disulfiram, five patients achieved a transient demethylation response. No grade 4 adverse effects were observed in this trial but 6 patients were forced to quit due to treatment toxicity (Table 1: NCT01118741, [61]). The DNMTi Azacitidine was trialed in chemonaive patients with CRPC in combination with combined androgen blockade (CAB). PSA doubling time increased relative to patients receiving only CAB and no grade 4 toxicities were reported, although 4 patients had to stop treatment due to grade 3 toxicities [62].
Azacitidine has also been tested in combination with Docetaxel and Prednisone in CRPC (Table 1: NCT00503984). Therapeutic response was assessed by magnetic resonance imaging with a complete response considered the disappearance of target lesions, and a partial response considered a ≥30% decrease in the sum of the longest diameter of targeted lesions. Complete and partial responses were achieved by only one and two patients, respectively. A PSA response was observed in 10 patients. Despite some positive results, the study was terminated due to withdrawal of funding (Table 1: NCT00503984). More studies are needed to assess the clinical potential of this agent.
Another category of epigenetic drugs with clinical potential in cancer treatment are HDM inhibitors. Phenelzine is a monoamine oxidase A (MAOA) inhibitor used in the treatment of psychiatric disease. MAOA is an enzyme responsible for the deamination of neurotransmitters, such as dopamine, serotonin and norepinephrine, that are important in a variety of neurological and psychiatric illnesses [63]. MAOA has close homology to LSD1, a histone demethylase, which catalyzes removal of the methyl group from H3K4me1 and H3K4me2. As a result of this homology, Phenelzine is able to bind and inhibit LSD1. [63]. Lower levels of H3K4me2 are correlated with higher risk of recurrence in prostate cancer [64]. Phenelzine is currently being trialed as a monotherapy for the treatment of recurrent prostate cancer and in combination with docetaxel for the treatment of progressive prostate cancer. Given that Phenelzine is already an approved medication, positive responses in these clinical trials will open the door to using this class of epigenetic drugs in clinical practice in the near future.
In the area of miRNA modulation, four clinical trials of antisense oligonucleotides have reported a positive PSA response, with three trials describing a PSA response >=50% (Table 1: NCT01188187, NCT00471432, NCT00085228, [65-68]). One of these trials, evaluating the efficacy of the antisense oligonucleotide OGX-011 in combination with docetaxel or mitoxantrone, reported a PSA decrease ≥50% in 6 of 14 patients with CRPC (Table 1: NCT00471432, [66]). Most of the adverse effects were grade 1 and 2 only, however adverse effects of grade 3 or higher affected 60% of the patients receiving the antisense oligonucleotide in combination with docetaxel/prednisone and 73% of the patients receiving the OGX-011 in combination with mitoxantrone/prednisone (Table 1: NCT00471432, [66]). The most common grade 3 or higher adverse effects in both groups were fatigue and lymphopenia (Table 1: NCT00471432, [66]). OGX-011 was also tested in combination with docetaxel and prednisone in a phase III clinical trial but, despite some positive results observed in phase II of the study where 58% of the patients had a PSA response >=50%, no significant results were observed in phase III (Table 1: NCT00471432, NCT01188187, [66, 69]). In the group receiving the combined therapy OGX-011/docetaxel/prednisone, 41% of the patients had to discontinue the treatment program due to adverse effects of the therapy (≥3 grade) (Table 1: NCT01188187, [69]).
Administration of the antisense oligonucleotide LY2181308 to decrease the expression of survivin, an anti-apoptotic gene involved in therapy resistance was tested in a randomized phase 2 clinical trial performed by Wiecho et al for the treatment of CRPC [70]. The patients allocated in the group treated with LY2181308 reported higher incidence of grade 3 and 4 adverse effects without any improvement in progression free survival or overall survival of the patients [70].
Disappointing results were obtained with another antisense oligonucleotide, Oblimersen, that was trailed by Sternberg et al both alone and in combination with docetaxel (Table 1: NCT00085228, [67]). The authors observed a PSA response in 46% of the patients treated with docetaxel alone, versus 37% of the patients treated with the antisense oligonucleotide and docetaxel (Table 1: NCT00085228, [67]). In the group of patients receiving the combined therapy, major toxic events were observed in 40.7% of the patients, compared to 22.8% of the patients receiving the antisense oligonucleotide alone, indicating that docetaxel increased oblimersen-related toxicity. This suggests that combined therapy with antisense oligonucleotides and taxanes might not the best therapeutic approach (Table 1: NCT00085228, [67]). In another clinical trial Oblimersen was administrated in combination with mitoxantrone, with 2 patients out of 25 showing a PSA decrease equal or superior to 50%. One patient had a PSA response inferior to 50% while stable disease was observed in a further five patients [68].
Although no clinical benefits were observed in a study testing the antisense oligonucleotides ISIS 3512 and ISIS 5132, two patients who received the oligonucleotide ISIS 3512 and one patient who received ISIS 5132 did not show disease progression for at least five months [71]. Finally, results are currently unavailable from eight completed clinical trials and two other trials are ongoing (Table 1).
Kidney cancer - epigenetics
Genetic and epigenetic dysregulation of genes involved in pathways such as the hypoxia-inducible pathway, the mTOR pathway, and the cMET-RAF-MEK-ERK pathway contribute to the progression of kidney cancer [72]. Changes in the levels of epigenetic-modifying enzymes are an important factor in altering expression of genes involved in cancer-related pathways. In renal cell carcinoma (RCC), an increase in the levels of histone demethylases such as UTX, JMJD2 and EZH2, results in a reduction in H3K27me and promotes progression of the disease [73, 74]. Also in RCC, almost 60% of patients overexpress HDAC1 and HDAC2 [75]. No prognostic value has been associated with these alterations [75]. HDAC3 is also highly expressed, but only in the papillary carcinoma subset [75].
At the level of individual epigenetic changes, low levels of H3K4me2, H3K18Ac, and H3K9me2 are associated with poor prognosis and lower survival probability in RCC. , Mechanistically, H3K4me2 and H3K18Ac are associated with active transcription while H3K9me2 is associated with transcriptional repression [73, 74]. H3K27me is another histone modification that correlates with poor clinical outcome, result of overexpression of histone demethylases in RCC [73, 74].
Clear cell RCC is the most common form of renal cell carcinoma, and is associated with inactivation of the tumor suppressor gene von-Hippel Lindau (VHL) by either genetic and epigenetic factors [76, 77]. VHL inactivation in both sporadic and familial forms can occur due to point mutations or deletions at the genetic level, or due to DNA hypermethylation at the gene promoter [76, 77].
DNA methylation also affects the regulation of several genes in RCC and has the potential to be used as biomarker and as a therapeutic target [78]. For example, hypermethylation of RASSF1A and HIC in patients with RCC is associated with a poor prognosis [79, 80].
Several miRNAs showed altered expression in RCC, resulting in changes to important cellular functions such as apoptosis, angiogenesis and the epithelial mesenchymal transition [27, 81]. Examples of miRNAs with altered expression in RCC include miR-210, miR-34a, miR-30c, miR-29b and miR-23b [27, 81].
Kidney cancer - current treatment
Radical nephrectomy is the standard of care for localized renal cell carcinoma. However, high rates of recurrence after surgery demand the development of new adjuvant therapies. Both radiotherapy and hormone therapy have proven ineffective in advanced stages of disease and chemotherapy has a response rate inferior to 10% [82, 83].
Kidney cancer - pre-clinical data
Preclinical studies using epigenetic drugs for kidney cancer treatment show some promise. In renal cancer cell lines, the HDACi Panobinostat induced cell cycle arrest and apoptosis and also resulted in a reduction in tumor size in xenograft mice models [84]. Pre-clinical studies of DNMT inhibitors in kidney cancer have also shown promise with evidence of reactivation of silenced genes and growth inhibition of cancer cells [85, 86].
One ongoing issue in RCC treatment is resistance to immunomodulatory therapy with interferons. This can occur via promoter hypermethylation and silencing of interferon response genes [87]. Treatment of renal cancer cell lines with 5-Aza-2’-deoxycitidine (5-Aza-dC), increased expression of interferon response genes and restored interferon induced apoptosis [87]. In addition, treatment of RCC cells with the antisense DNMT1 oligonucleotide MG98 also restored susceptibility to interferon therapy [88].
Downregulation of miR-30c in RCC is associated with promotion of the epithelial mesenchymal transition [89]. Reduced expression of this miRNA is associated with hypoxia and VHL cell status with lower levels of miR-30c being observed in VHL-deficient RCC cell lines [89]. Transfection of a RNA mimic to restore miR-30c levels caused an increase in E-cadherin expression and reduced cell migration capacity [89].
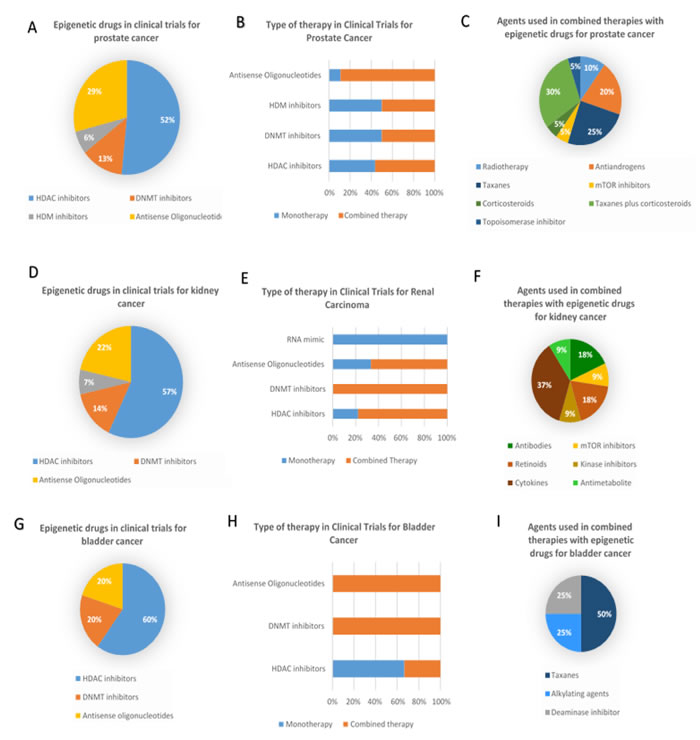
Figure 1: Epigenetic therapies in clinical trials for prostate, bladder and kidney cancers. A. Percentage of clinical trials employing each types of epigenetic therapeutic agents in prostate cancer; B. Percentage of clinical trials using mono or combined therapy as therapeutic strategy with the different classes of epigenetic drugs in prostate cancer; C. Percentage of clinical trials where different agents are used in combined therapies for prostate cancer; D. Percentage of clinical trials employing each types of epigenetic therapeutic agents in kidney cancer; E. Percentage of clinical trials using mono or combined therapy as therapeutic strategy with the different classes of epigenetic drugs in kidney cancer; F. Percentage of clinical trials where different agents are used in combined therapies for kidney cancer G. Percentage of clinical trials employing each types of epigenetic therapeutic agents in bladder cancer; H. Percentage of clinical trials using mono or combined therapy as therapeutic strategy with the different classes of epigenetic drugs in bladder cancer; I. Percentage of clinical trials where different agents are used in combined therapies for bladder cancer
Kidney cancer - clinical trials
Trials of HDACi in RCC have shown mixed responses. HDACi Vorinostat used as a monotherapy showed an objective response in 36% of patients, however 63% of the patients presented disease progression at 6 months (Table 2: s). When used in combination with Bevacizumab, 48, 6 % of the patients showed stable disease at 6 months (Table 2: NCT00324870). By contrast, treatment with Panobinostat alone resulted in no objective responses, and a median of progression-free survival of 1.7 months (Table 2: NCT00550277, [90]). Treatment was generally well tolerated, but 7 patients reported thrombocytopenia grade 3 or higher (Table 2: NCT00550277, [90]). In a separate small phase I trial, the HDACi Entinostat was administered in combination with Isotretinoin. One patient, who had presented with disease progression after treatment with cytokines and anti-angiogenic therapy, subsequently showed stable disease [91]. The patient had a reduction in tumor size after 4 months of therapy and did not show signs of disease progression at 12 months [91]. However, the number of patients enrolled in the study (2) was insufficient to draw any conclusions [91].
The only DNMT inhibitor tested in RCC is Decitabine. When used in combination with the cytokine IL-2 in a phase II study of advanced RCC, three out of five patients showed stable disease [92]. Another study combining Decitabine with interferon-α was terminated early due to low accrual (Table 1, NCT00561912).
Treatment with antisense oligonucleotides has also resulted in stabilization of disease in some RCC patients. The antisense oligonucleotide GTI-2040, targeting the R2 subunit of ribonucleotide reductase, was tested in patients with metastatic disease in a phase II trial and generated a partial response for approximately eight months [93]. One patient experienced a dose limiting toxicity (grade 3 diarrhea) and adverse effects of all grades were reported in this trial including grade 4 pancytopenia, pulmonary embolism and bone pain [93]. In a trial reported by Winquist et al, in which the antisense oligonucleotide MG98 was administered to 15 patients, no objective responses were observed with nine patients presenting progression of the disease. MG98 targets DNMT1 but no decrease in enzyme activity was observed [94]. Also, grade 3 and 4 adverse effects forced 8 patients to discontinue treatment, primarily due to elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels [94].
miRNA mimics have also been trialed in RCC. MRX34, an miRNA mimic of the tumor suppressor miRNA34, was tested in a phase I clinical trial for advanced or metastatic cancers, including RCC, but the trial was terminated early due to serious immunologic adverse events (Table 2: NCT01829971).
Table 1: Clinical trials of epigenetic drugs in prostate cancer
Drug |
Combined Therapy |
Enzimatic Class |
Approval Stage |
Status |
Indication |
Results |
Reference/Clinical trial identification |
SB939 |
- |
HDAC inhibitor |
Phase 2 |
Completed |
Castration Resistance Prostate Cancer (CRPC) |
6% of the patients had a PSA response |
Eigl et al. 2015 (NCT01075308) |
Panobinostat |
- |
HDAC inhibitor |
Phase 2 |
Completed |
CRPC |
14,3% of the patients had a PSA decrease <50% but no objective responses were seen 11,4% of the patients had stable disease for at least 24 weeks |
Rathkopf et al. 2013 (NCT00667862) |
Panobinostat |
Docetaxel |
HDAC inhibitor |
Phase 1 |
Completed |
CRPC |
63% had a PSA decrease >= 50% |
Rathkopf et al. 2010 |
Panobinostat |
Radiotherapy |
HDAC inhibitor |
Phase 1 |
Completed |
Prostate Cancer, esophageal cancer and neck cancer |
No study results or publications provided |
NCT00670553 |
Panobinostat |
Docetaxel/prednisone |
HDAC inhibitor |
Phase 2 |
Completed |
CRPC |
No study results or publications provided |
NCT00878436 |
Panobinostat |
Bicalutamide |
HDAC inhibitor |
Phase 1 |
Completed |
CRPC |
No study results or publications provided |
NCT00663832 |
Vorinostat |
- |
HDAC inhibitor |
Phase 2 |
Completed |
Progressive metastatic prostate cancer |
No PSA declines >=50% were observed Median of progression free survival=2,8 months with a median overall survival of 11,7 months |
Bradley et al. 2010 (NCT00330161) |
Vorinostat
|
Docetaxel |
HDAC inhibitor |
Phase 1 |
Terminated
|
Advanced solid tumor including prostate cancer, urothelial carcinoma and kidney cancer |
This study was terminated due to excessive toxicity as five patients experienced dose-limiting toxicities (DLT)
No responses were observed |
Schneider et al. 2012 (NCT00565227) |
Vorinostat |
Temsirolimus |
HDAC inhibitor |
Phase 1 |
Terminated |
Metastatic prostate cancer |
This study was terminated due to lack of efficacy |
NCT01174199 |
Vorinostat |
Androgen deprivation therapy |
HDAC inhibitor |
Phase 2 |
Completed |
Localized prostate cancer |
No study results or publications provided |
NCT00589472 |
Vorinostat |
- |
HDAC inhibitor |
Phase 1 |
Completed |
Advanced solid tumors including prostate cancer |
No study results or publications provided |
NCT00005634 |
Romidepsin |
- |
HDAC inhibitor |
Phase 2 |
Completed |
Metastatic prostate cancer |
No study results or publications provided |
NCT00106418 |
Romidepsin |
- |
HDAC inhibitor |
Phase 2 |
Completed |
metastatic castration-resistant prostate cancer (MCRPC) |
63% of the patients had progressive disease with a median time to progression of 49,5 days |
Molife et al. 2009
|
Curcumin |
Docetaxel |
HDAC inhibitor |
Phase 2 |
Ongoing |
MCRPC |
Final data collection date for primary outcome measure: January 2017 |
NCT02095717 |
Curcumin |
- |
HDAC inhibitor |
Phase 2 |
Ongoing |
Prostate cancer |
Estimated primary completion date: June 2020 |
NCT02064673 |
Curcumin |
Radiotherapy |
HDAC inhibitor |
- |
Completed |
Prostate cancer |
No PSA response was observed but the severity of radiotherapy related urinary symptoms was reduced, |
Hejazi J. et al. 2013 |
Dissulfiram |
- |
DNMT inhibitor |
Phase 1 |
Completed |
Non-metastatic recurrent prostate cancer |
Five patients achieve a transient demethylation response Six patients discontinue therapy due to adverse effects |
Schweizer et al. 2013 |
Azacitidine |
Combined Androgen Blockade (CAB) |
DNMT inhibitor |
Phase 2 |
Completed |
CRPC |
Overall median PSA doubling time increased significantly (2.8 vs 1.5 months of the baseline). |
Sonpavde et al. 2011 |
Azacitidine |
- |
DNMT inhibitor |
Phase 2 |
Completed |
Prostate cancer |
No study results or publications provided |
NCT00384839 |
Azacitidine |
Docetaxel/prednisone |
DNMT inhibitor |
Phase1/2 |
Terminated |
CRPC |
This study was terminated due to withdrawal of funding Complete and partial response were achieved by one and two patients, respectively |
NCT00503984 |
Phenelzine sulfate |
- |
HDM inhibitor/monoamine oxidase A inhibitor |
Phase 2 |
Ongoing |
Non-metastatic recurrent prostate cancer |
Study completion date: August 2018 |
NCT02217709 |
Phenelzine sulfate |
Docetaxel |
HDM inhibitor/monoamine oxidase A inhibitor |
Phase 2 |
Ongoing |
Progressive prostate cancer |
Final data collection date for primary outcome measure: January 2016 |
NCT01253642 |
OGX-011
|
Docetaxel/prednisone |
Antisense oligonucleotide that targets clusterin |
Phase 1 |
Completed
|
advanced cancer including prostate, bladder and kidney cancer |
Six patients with hormone-refractory prostate cancer had a PSA decline >=50%
|
Saad et al. 2011 (NCT00471432) |
OGX-011 |
Docetaxel/prednisone and docetaxel/mitoxantrone |
Antisense oligonucleotide that targets clusterin |
Phase 3 |
Completed |
MCRPC |
No objective responses were seen 41% of the patients discontinued treatment due to serious adverse events |
Chi et al. 2008 (NCT01188187) |
OGX-011 |
Cabazitaxel/prednisone |
Antisense oligonucleotide that targets clusterin |
Phase 3 |
Ongoing |
CRPC |
Study completion date: December 2016 |
NCT01578655 |
Oblimersen |
Docetaxel |
Antisense oligonucleotide that targets Bcl-2 |
Phase 2 |
Completed |
CRPC |
PSA response was observed in 46% and 37% of the patients treated with docetaxel alone and docetaxel+oblimersen, respectively |
Sternberq et al. 2009 (NCT00085228) |
Oblimersen sodium (Genasense) |
Mitoxantrone |
Antisense oligonucleotide that targets Bcl-2 |
Phase 1 |
Completed |
CRPC |
Two patients had a PSA reduction >=50%, 1 patient had a PSA resuction <50%, and 5 patients had stable disease |
Chi et al. 2001 |
OGX-427 |
Prednisone |
Antisense oligonucleotide that targets heat shock protein27 |
Phase 2 |
Completed |
CRPC |
No study results or publications provided |
NCT01120470 |
OGX-427 |
Abiraterone |
Antisense oligonucleotide that targets heat shock protein27 |
Phase 2 |
Ongoing |
MCRPC |
Study completion date: December 2017 |
NCT01681433 |
ISIS 1837 |
Docetaxel/prednisone |
Antisense oligonucleotide that targets eIF4E |
Phase 2 |
Completed |
Metastatic resistant castrate prostate cancer |
No study results or publications provided |
EudraCT Number: 2010-022239-12 |
ISIS 3521/ISIS 5132 |
- |
Antisense oligonucleotides that targets PKC-α and Raf-1, respectively |
Phase 2 |
Completed |
CRPC |
No objective responses were observed but three patients had stable disease for 5 or more months PSA values of five patients did not rise more than 25% for >=120 days |
Tolcher et al. 2002 |
LY2181308 |
Docetaxel/prednisone |
Antisense oligonucleotide that targets survivin |
Phase 2 |
Completed |
CRPC |
No differences in efficacy were observed between the control and the experimental group. Higher incidence of adverse effects in the LY2181308 treated group. |
Wiechno et al. 2014 |
Bladder cancer - epigenetics
Epigenetic modifications are important in bladder cancer development [95]. An increase in global histone methylation was reported in bladder cancer samples, particularly in the subset of patients with non-muscular invasive bladder cancer. In these patients, a global increase in H3K9 and H3K27 methylation was associated with high-grade tumors. However, the authors did not find any correlation between histone methylation and tumor recurrence or survival [96]. Interestingly, another study by the same group revealed that a decrease in methylation levels of other histone proteins, namely H3K4 and H3K20, could be a prognostic biomarker for muscle invasive bladder cancer [97]. The presence of different histone methylation patterns in muscle invasive and non-invasive bladder cancer, suggests that patients of these subgroups will respond differentially to epigenetic therapies affecting histone methylation. These data reinforce the need for biomarker discovery to predict responses to epigenetic therapy.
With respect to DNA methylation in bladder cancer, Friedrich et al reported hypermethylation of the genes DAPK, BCL2 and TERT in urine samples from patients with bladder cancer [98]. Detection of methylation patterns in urine samples has proven to be a good diagnostic strategy in bladder cancer. [99, 100]. Methylation of some gene promoters can also be indicative of prognosis, for example, methylation of the RUNX3 promoter is associated with a higher risk of progression and lower survival [101].
Differential miRNA expression is another epigenetic feature of bladder cancer and can distinguish between cancer patients and healthy subjects [27, 102]. The miRNAs implicated in bladder cancer target genes involved in cell cycle control, cell proliferation, cell differentiation and signal transduction pathways [27, 102]. Both upregulation and downregulation of miRNA expression can potentiate cancer development. In bladder cancer, loss of miR-200 is associated with epithelial mesenchymal transition while upregulation of miR-21 and miR-129 is associated with high grade tumors and poor prognosis, respectively [27, 102, 103].
Bladder cancer - current treatment
Muscle invasion is a critical factor in the selection of the right therapeutic option for bladder cancer. In the case of non-muscle invasive bladder cancer, the standard clinical approach is transurethelial resection followed by administration of chemotherapeutic or immunotherapeutic agents [104]. For muscle-invasive bladder cancer, a more aggressive form, the standard of care is radical cystectomy [105]. In cases of disease relapse, occurring in approximately 30% of patients, combinatory chemotherapy regimens containing cisplatin are used. These include MVAC (Methrotrexate, Vinblastine, Achiamycin and Cisplatin) and GC (Gemcitabine and Cisplatin). Despite positive early responses to these therapeutic modalities, the median survival rate after treatment is only 12 months [104-106]. For advanced and metastatic bladder cancer, where surgery is not a valid approach, the only treatment option is palliative chemotherapy. This reflects the need for the development and implementation of new therapeutic agents [104].
Bladder cancer - pre-clinical data
Proteomic studies after exposure of bladder cancer cells to HDAC inhibitors reveals that HDAC activity influences many cellular pathways involved in carcinogenesis [107]. The treatment of bladder cancer cell lines with these agents resulted in cell growth suppression and induction of cell death [107]. Wang et al showed that the HDACi Vorinostat was able to induced cell growth inhibition in bladder cancer cells in part due to downregulation of survivin, an apoptosis inhibitory protein [108]. Importantly, Vorinostat had a synergistic effect with chemotherapeutic agents including Cisplatin, Mitomycin c, and Adriamycin. Combined therapy of using Vorinostat and Cisplatin prevented cancer progression in an animal model [108].
Positive responses were also obtained in bladder cancer cells after administration of the DNMTi Belinostat. A significant decrease in cell proliferation was observed in vitro, and in vivo with the use of a transgenic mouse model [109].
Epigenetic drugs can be used in combination with other agents to enhance their efficacy. Shang et al evaluated the DNMT inhibitor 5-Aza-2-deoxycytidine in combination with chemotherapeutic agents in bladder transitional cell carcinoma cell lines. The authors demonstrated that DAC enhances susceptibility to Cisplatin, a common agent used as neoadjuvant therapy for bladder cancer, in a synergistic way. Both agents induced cell cycle arrest at G2/M phase, with Cisplatin also inducing tumor cell apoptosis [110].
Rieger et al compared the efficacy and efficiency of siRNAS and antisense oligonucleotides against Bcl-xL in bladder cancer cell lines. Their effect in combined therapy with chemotherapeutic agents was also tested. Both agents enhanced tumor cell apoptosis when administrated with Cisplatin, however Bcl-xL knockout was more efficient with siRNAs than with antisense oligonucleotides [111]. Another study showed that simultaneous knockout of Bcl-xL and survivin with siRNAs in bladder cancer cells led to greater sensitization of the cells to chemotherapeutic agents [112].
Table 2: Clinical trials of epigenetic drugs in kidney cancer
Drug |
Combined Therapy |
Enzimatic Class |
Approval Stage |
Status |
Indication |
Results |
Reference/Clinical trial identification |
Vorinostat |
Bevacizumab |
HDAC inhibitor |
Phase 1/2 |
completed |
Unresectable or metastatic kidney cancer |
48,6% of the patients had absence of disease progression at 6 months |
NCT00324870 |
Vorinostat |
Isotretinoin |
HDAC inhibitor |
Phase 1/2 |
Completed |
Advanced renal cell carcinoma |
MTD of Vorinostat in combination with isotretinoin=0,5 mg/kg |
NCT00324740 |
Vorinostat |
- |
HDAC inhibitor |
Phase 2 |
Completed |
Advanced renal cell carcinoma |
An objective response was observed in 36% of the patients |
NCT00278395 |
Vorinostat |
Pembrodizumab |
HDAC inhibitor |
Phase 1 |
Ongoing |
Advanced renal or urothelial cell carcinoma |
Final data collection date for primary outcome measure: May 2018 |
NCT02619253 |
Panobinostat |
- |
HDAC inhibitor |
Phase 2 |
Completed |
Refractory clear cell renal carcinoma |
Median of progression free survival=1,7 months 30% of the patients experienced serious adverse events |
Hainsworth et al. 2011 (NCT00550277)
|
Panobinostat |
Everolimus |
HDAC inhibitor |
Phase ½ |
Terminated |
Metastatic or unresectable renal cell cancer |
The study has been terminated (patients off study, principal investigator left institute) |
NCT01582009 |
Panobinostat |
Sorafenib |
HDAC inhibitor |
Phase 1 |
Ongoing |
Advanced renal cell carcinoma |
Study completion date: November 2016 |
NCT01005797 |
Entinostat |
Isotreitinoin |
HDAC inhibitor |
Phase 1 |
Completed |
solid tumor including kidney cancer, urothelial carcinoma and prostate cancer |
No objective responses were observed but stable disease was noticed in patients with kidney, prostate and pancreatic cancer |
Pili et al. 2012 |
Entinostat |
IL-2 |
HDAC inhibitor |
Phase 1/2 |
Ongoing |
Metastatic kidney cancer |
No date given for study completion |
NCT01038778 |
Decitabine |
Interferon alpha2B |
DNMT inhibitor |
Phase 2 |
Terminated |
Advanced renal cell carcinoma |
The study was terminated due to low accrual and unavailable treatment agent. |
NCT00561912 |
Decitabine |
IL-2 |
DNMT inhibitor |
Phase 1 |
Completed |
Melanoma or renal cell cancer |
Three patients with renal cell cancer had stable disease |
Gollob et al. 2006 |
GTI-2040 |
Capecitabine |
Antisense oligonucleotide that targets R2 subunit of ribonucleotide reductase |
Phase 2 |
Completed |
Advanced/metatastic renal cell carcinoma |
52% of the patients had stable disease with median duration of 4 months |
Desai et al. 2004 |
MG98 |
- |
Antisense oligonucleotide that targets DNMT1 |
Phase 2 |
Completed |
Metastatic renal carcinoma |
Six patients had stable disease but no objective responses were seen |
Whinquist E. et al 2006 |
Oblimersen |
Interferon alpha |
Antisense oligonucleotide that targets bcl2 |
Phase 2 |
Completed |
Metastatic renal cell cancer |
No study results or publications provided |
NCT00059813 |
MRX34 |
|
RNA mimic |
Phase 1 |
Terminated |
Renal cell carcinoma |
This study was terminated due to serious adverse events |
NCT01829971 |
Bladder cancer - clinical data
Relative few studies of epigenetic therapies have been undertaken in bladder cancer. Only one of three clinical trials employing HDAC inhibitors showed a positive response; in this trial, four of fifteen patients treated with the HDACi Belinostat in combination with Carboplatin or Paclitaxel showed complete or partial response to treatment, while five patients presented disease progression with a median time to progression of 136 days (Table 3, NCT00421889). Further studies employing this HDACi in combination with other agents are essential to confirm its therapeutic potential [109]. The only study using the HDACi Vorinostat terminated due to lack of efficacy (Table 3, NCT00363883). Three other phase 2 clinical trials employing epigenetic drugs for bladder cancer treatment are currently ongoing (Table 3: NCT02236195, NCT00978250, NCT01780545).
Table 3: Clinical trials of epigenetic drugs in bladder cancer
Drug |
Combined Therapy |
Enzimatic Class |
Approval Stage |
Status |
Indication |
Results |
Reference/Clinical trial identification |
Belinostat |
Carboplatin or paclitaxel |
HDAC inhibitor |
Phase 1/2 |
Phase 1 concluded, Phase 2 ongoing |
Bladder cancer |
Four out of fifteen patients had complete or partial response patients had progressive disease with median time to progression of 136 days |
NCT00421889 |
Vorinostat |
- |
HDAC inhibitor |
Phase 2 |
Terminated for futility |
Locally Recurrent or Metastatic Cancer of the Urothelium |
No objective response was observed |
NCT00363883 |
Mocetinostat |
- |
HDAC inhibitor |
Phase 2 |
Ongoing |
Patients with advanced urothelial Carcinoma and inactivating alterations of acetyltransferase genes |
Study completion date: December 2017 |
NCT02236195 |
FdCyd |
Tetrahydrouridine |
DNMT inhibitor |
Phase 2 |
Ongoing |
Advanced cancer including bladder Cancer |
Study completion date: May 2017 |
NCT00978250 |
OGX-427 |
Docetaxel |
Antisense oligonucleotide that targets heat shock protein 27 |
Phase 2 |
Ongoing |
Advanced urothelial Carcinoma |
Study completion date: February 2017 |
NCT01780545 |
Conclusions
Despite the obvious importance of epigenetics in the development of cancer, few epigenetic therapies have thus far reached advanced clinical testing. As the data above demonstrates, pre-clinical data has not translated into the hoped-for clinical responses. This is likely secondary to the nonspecific actions of epigenetic drugs and the consequent toxicities associated with their administration.
Many of the epigenetic therapies being tested have global epigenetic effects on both cancerous and non-cancerous tissues. Moreover, some of them have additional non-epigenetic effects that limit their efficacy. HDAC enzymes, for instance, target non-histone proteins involved in oncologic pathways unrelated to epigenetic regulation [113]. It is important to consider that the observed therapeutic responses to HDACi treatment may thus be the result of the altered activity of these proteins and not to the reversal of specific epigenetic marks [113]. Similarly, demethylating agents are not specific to genes involved in carcinogenesis but result in global demethylation of the genome, an epigenetic signature associated with genomic instability that can lead to severe side effects [113].
Of all the clinical trials analyzed in this study, 16 included evaluation of gene expression and/or DNA methylation as a secondary objective of the trial. We feel that analysis of gene expression and epigenetic patterns should be included in all clinical trials using epigenetic agents in order to assess the causal link between drug induced alterations and therapeutic responses. A better knowledge of the specific mechanism of action of these agents is essential to overcoming their clinical limitations and improving therapeutic success.
Antisense oligonucleotides (ASOs) avoid some of the issues described above as as they are designed to be more target specific. Thus far, two separate studies of ASOs as monotherapy have shown no objective responses [71, 94], however when used in combination with other agents the results have been more promising [66, 93]. However, ASOs also have their limitations secondary to toxicity and delivery efficiency [67, 69].
Currently, cancer treatment is determined largely according to cancer stage, even though patients with similar stage cancers may respond differently to the same type of therapy. The promise of “personalized medicine” is the idea that tailoring treatment to an individual patient will optimize efficacy while minimizing toxicity. Personalization should be an ongoing goal for all cancer therapy development, including epigenetic therapies. Biomarker development should thus be a central goal in the development of epigenetic therapies, both so that the correct patient receives the correct therapy, and to ensure that therapies that have value in a subset of patients are not passed over because of lack of efficacy in other patients. Epigenetic therapies are still in their infancy as a therapeutic class and pre-clinical promise has not yet translated into clinical efficacy. However, the development of target-specific agents, and the careful combination of epigenetic therapies with traditional modalities should enable them to achieve clinical success in the near future.
Conflicts of Interest
RAM has received honoraria from the Pfizer Advisory Board, Zodiac Advisory Board, Astrazeneca, the National Science Centre, Krakow, Poland, and an educational grant from Pierre Fabre. RAM is ad hoc consultant at the Ministry of Health, Brasília, Brazil. The other authors have no conflicts of interest related to this manuscript.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30.
2. Dawson MA, Kouzarides T. Cancer epigenetics: From mechanism to therapy. Cell. 2012;150:12-27.
3. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381-95.
4. Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37-50.
5. Gray SG, Ekström TJ. The human histone deacetylase family. Exp Cell Res. 2001;262:75-83.
6. Kaushik D, Vashistha V, Isharwal S, Sediqe SA. Histone deacetylase inhibitors in castration- resistant prostate cancer: molecular mechanism of action and recent clinical trials. Ther Adv Urol. 2015;7:388-95.
7. Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297-311.
8. Højfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12:917-30.
9. Hon GC, Hawkins RD, Ren B. Predictive chromatin signatures in the mammalian genome. Hum Mol Genet. 2009;18:R195-201.
10. Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pr Oncol. 2005;2:S4-11.
11. Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, Zhukova N, Walker EJ, Martin D, Merino D, Wasserman JD, Elizabeth C, Alon N, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: An integrative genomic and molecular study. Lancet Oncol. 2013;14:534-42.
12. Chao W-R, Lin W-L, Chen C-K, Han L-M, Lin J-C, Han C-P. Unusual c-KIT (+) squamous cell carcinoma of the uterine cervix showing paradoxical hypermethylation of the c-KIT proto-oncogene. Eur J Obstet Gynecol Reprod Biol. 2015;184:130-1.
13. Loeb DM, Evron E, Patel CB, Mohini Sharma P, Niranjan B, Buluwela L, Weitzman SA, Korz D, Sukumar S. Wilms’ tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res. 2001;61:921-5.
14. Castelo-Branco P, Leão R, Lipman T, Campbell B, Lee D, Price A, Zhang C, Heidari A, Stephens D, Boerno S, Coelho H, Gomes A, Domingos C, et al. A cancer specific hypermethylation signature of the TERT promoter predicts biochemical relapse in prostate cancer: A retrospective cohort study. Oncotarget. 2016;7:57726-57736. doi: 10.18632/oncotarget.10639.
15. Bert SA, Robinson MD, Strbenac D, Statham AL, Song JZ, Hulf T, Sutherland RL, Coolen MW, Stirzaker C, Clark SJ. Regional Activation of the Cancer Genome by Long-Range Epigenetic Remodeling. Cancer Cell. 2013;23(1):9-22.
16. Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775-89.
17. Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321-33.
18. Bose P, Dai Y, Grant S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol Ther. 2014;143:323-36.
19. Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541-52.
20. Patra SK, Patra A, Zhao H, Dahiya R. DNA Methyltransferase and Demethylase in Human Prostate Cancer. Mol Carcinog. 2002;33:163-71.
21. Subramaniam D, Thombre R, Dhar A, Anant S. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol. 2014;4:1-13.
22. Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M, Kristiansen G. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98:604-10.
23. Vieira F, Costa-Pinheiro P, Ramalho-Carvalho J, Pereira A, Menezes F, Antunes L, Carneiro I, Jero C, Oliveira J, Henrique R, Jeronimo C. Deregulated expression of selected histone methylases and demethylases in prostate carcinoma. Endocr Relat Cancer. 2014;21:51-61.
24. Islam ABMMK, Richter WF, Jacobs LA, Lopez-Bigas N, Benevolenskaya E V. Co-regulation of histone-modifying enzymes in cancer. PLoS One. 2011;6:e24023.
25. Valdés-Mora F, Clark SJ. Prostate cancer epigenetic biomarkers: next-generation technologies. Oncogene. 2014;34:1609-18.
26. Jeronimo C, Bastian PJ, Bjartell A, Carbone GM, Catto JWF, Clark SJ, Henrique R, Nelson WG, Shariat SF. Epigenetics in prostate cancer: Biologic and clinical relevance. Eur Urol. 2011;60:753-66.
27. Catto JWF, Alcaraz A, Bjartell AS, De Vere White R, Evans CP, Fussel S, Hamdy FC, Kallioniemi O, Mengual L, Schlomm T, Visakorpi T. MicroRNA in prostate, bladder, and kidney cancer: A systematic review. Eur Urol. 2011;59:671-81.
28. Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-agadjanyan EL, Peterson A, Noteboom J, Briant KCO, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008;105:10513-8.
29. Noonan EJ, Place RF, Pookot D, Basak S, Whitson JM, Hirata H, Giardina C, Dahiya R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene. 2009;28:1714-24.
30. Bono JS De, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Saad F, Staffurth JN, Mainwaring P, Harland S, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995-2005.
31. Miller K, Wit R De, Mulders P, Ph D, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Fléchon A, Ph D, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, Bono JS De, Ch B, Ph D, Investigators A. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N Engl J Med. 2012;367:1187-97.
32. Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Ph D, Frohlich MW, Schellhammer PF. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N Engl J Med. 2012;363:411-22.
33. Bono JS De, Oudard S, Ozguroglu M, Hansen S, Machiels J, Kocak I, Gravis G, Pompidou EG. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment : a randomised open-label trial. Lancet. 2010;376:1147-54.
34. Parker C, Widmark A, Johannessen DC, Hoskin P, Bottomley D, James ND, Solberg A, Syndikus I, Kliment J, Wedel S, Boehmer S, Oglio MD, Franzén L, Coleman R, Vogelzang NJ, Staudacher K, Shan M, Bruland ØS, Sartor O, Investigators A. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N Engl J Med. 2013;369:213-23.
35. Tannock I, Horti J, Pluzanska A, Chi KN, Oudard S, Théodore C, Rosenthal MA, Ph D, Eisenberger MA. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer. N Engl J Med. 2004;351:1502-12.
36. Silberstein JL, Pal SK, Lewis B, Sartor O. Current clinical challenges in prostate cancer. Transl Androl Urol. 2013;2:122-36.
37. Ellis L, Ku S-Y, Lasorsa E, Pili R. Epigenetics in Castration Resistant Prostate Cancer. In: Saad F, Eisenberger MA, editors. Management of Castration Resistant Prostate Cancer. New York, NY: Springer New York; 2014. p. 277-95.
38. Sharma NL, Groselj B, Hamdy FC, Kiltie AE. The emerging role of histone deacetylase (HDAC) inhibitors in urological cancers. BJU Int. 2013;111:537-42.
39. Liu X, Gomez-Pinillos A, Liu X, Johnson EM, Ferrari AC. Induction of bicalutamide sensitivity in prostate cancer cells by an epigenetic Puralpha-mediated decrease in androgen receptor levels. Prostate. 2010;70:179-89.
40. Verheul HMW, Salumbides B, Van Erp K, Hammers H, Qian DZ, Sanni T, Atadja P, Pili R. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin Cancer Res. 2008;14:3589-97.
41. Gravina GL, Marampon F, Muzi P, Mancini A, Piccolella M, Negri-Cesi P, Motta M, Lenzi A, Cesare E Di, Tombolini V, Jannini EA, Festuccia C. PXD101 potentiates hormonal therapy and prevents the onset of castration-resistant phenotype modulating androgen receptor, HSP90, and CRM1 in preclinical models of prostate cancer. Endocr Relat Cancer. 2013;20:321-37.
42. Shukla S, Meeran SM. Epigenetics of cancer stem cells: Pathways and therapeutics. Biochim Biophys Acta. 2014;1840:3494-502.
43. Frame FM, Pellacani D, Collins AT, Simms MS, Mann VM, Jones GDD, Meuth M, Bristow RG, Maitland NJ. HDAC inhibitor confers radiosensitivity to prostate stem-like cells. Br J Cancer. 2013;109:3023-33.
44. Chou Y-W, Lin F-F, Muniyan S, Lin FC, Chen C-S, Wang J, Huang C-C, Lin M-F. Cellular prostatic acid phosphatase (cPAcP) serves as a useful biomarker of histone deacetylase (HDAC) inhibitors in prostate cancer cell growth suppression. Cell Biosci. 2015;5:38.
45. Lin X, Asgari K, Putzi MJ, Gage WR, Yu X, Cornblatt BS, Kumar A, Piantadosi S, Deweese TL, Marzo AM De, Nelson WG. Reversal of GSTP1 CpG Island Hypermethylation and Reactivation of pi -Class Glutathione S-Transferase ( GSTP1 ) Expression in Human Prostate Cancer Cells by Treatment with Procainamide. Cancer Res. 2001;61:8611-6.
46. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33-9.
47. Gravina GL, Festuccia C, Millimaggi D, Dolo V, Tombolini V, De Vito M, Vicentini C, Bologna M. Chronic azacitidine treatment results in differentiating effects, sensitizes against bicalutamide in androgen-independent prostate cancer cells. Prostate. 2008;68:793-801.
48. Lin J, Haffner MC, Zhang Y, Lee BH, Nathaniel W, Britton J, Kachhap SK, Shim JS, Liu JO, Nelson G, Yegnasubramanian S, Carducci MA. Disulfiram Is a DNA Demethylating Agent and Inhibits Prostate Cancer Cell Growth. Prostate. 2011;71:333-43.
49. Takeshita F, Patrawala L, Osaki M, Takahashi R, Yamamoto Y, Kosaka N, Kawamata M, Kelnar K, Bader AG, Brown D, Ochiya T. Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol Ther. 2010;18:181-7.
50. Cimmino A, Calin GA, Fabbri M, Iorio M V, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Pnas. 2005;102:13944-9.
51. Miayake H1, Tolcher A GM. Chemosensitization and delayed androgen-independent recurrence of prostate cancer with the use of antisense Bcl-2 oligodeoxynucleotides. J Natl Cancer Inst. 2000;92:34-41.
52. Sowery RD, Hadaschik BA, So AI, Zoubeidi A, Fazli L, Hurtado-Coll A, Gleave ME. Clusterin knockdown using the antisense oligonucleotide OGX-011 re-sensitizes docetaxel-refractory prostate cancer PC-3 cells to chemotherapy. BJU Int. 2008;102:389-97.
53. Eigl BJ, North S, Winquist E, Finch D, Wood L, Sridhar SS, Powers J, Good J, Sharma M, Squire JA, Bazov J, Jamaspishvili T, Cox ME, Bradbury PA, Eisenhauer EA, Chi KN. A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Invest New Drugs. 2015;33:969-76.
54. Rathkopf DE, Picus J, Hussain A, Ellard S, Chi KN, Nydam T, Allen-Freda E, Mishra KK, Porro MG, Scher HI, Wilding G. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2013;72:537-44.
55. Rathkopf D, Wong BY, Ross RW, Anand A, Tanaka E, Woo MM, Hu J, Dzik-Jurasz A, Yang W, Scher HI. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2010;66:181-9.
56. Bradley D, Rathkopf D, Dunn R, Stadler WM, Liu G, Smith C, Pili R, Zwiebel J, Scher H, Hussain M, Arbor A. Vorinostat in Advanced Prostate Cancer Patients Progressing on Prior Chemotherapy (NCI Trial # 6862): Trial results and IL-6 analysis. A study by the DOD Prostate Cancer Clinical Trial Consortium and University of Chicago Phase II Consortium. Cancer. 2010;115:5541-9.
57. Molife LR, Attard G, Fong PC, Karavasilis V, Reid AHM, Patterson S, Riggs CE, Higano C, Stadler WM, McCulloch W, Dearnaley D, Parker C, de Bono JS. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann Oncol. 2009;21:109-13.
58. Schneider BJ, Kalemkerian GP, Bradley D, Smith DC, Egorin MJ, Daignault S, Dunn R, Hussain M. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Invest New Drugs. 2012;30:249-57.
59. Reuter S, Gupta SC, Park B, Goel A, Aggarwal BB. Epigenetic changes induced by curcumin and other natural compounds. Genes Nutr. 2011;6(2):93-108.
60. Hejazi J, Rastmanesh R, Taleban F-A, Molana S-H, Ehtejab G. A Pilot Clinical Trial of Radioprotective Effects of Curcumin Supplementation in Patients with Prostate Cancer. J Cancer Sci Ther. 2013;5:320-4.
61. Schweizer MT, Lin J, Blackford A, Bardia A, King S, Armstrong AJ, Rudek MA, Yegnasubramanian S, Carducci MA. Pharmacodynamic study of disulfiram in men with non-metastatic recurrent prostate cancer. Prostate Cancer Prostatic Dis. 2013;16:357-61.
62. Sonpavde G, Aparicio AM, Zhan F, North B, DeLaune R, Garbo LE, Rousey SR, Weinstein RE, Xiao L, Boehm KA, Asmar L, Fleming MT, Galsky MD, Berry WR, Von Hoff DD. Azacitidine favorably modulates PSA kinetics correlating with plasma DNA LINE-1 hypomethylation in men with chemonaïve castration-resistant prostate cancer. Urol Oncol Semin Orig Investig. 2011;29:682-9.
63. Culhane JC, Wang D, Yen PM, Cole PA. Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. J Am Chem Soc. 2010;132:3164-76.
64. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343-57.
65. Chi KN, Siu LL, Hirte H, Hotte SJ, Knox J, Kollmansberger C, Gleave M, Guns E, Powers J, Walsh W, Tu D, Eisenhauer E. A phase I study of OGX-011, a 2’-methoxyethyl phosphorothioate antisense to clusterin, in combination with docetaxel in patients with advanced cancer. Clin Cancer Res. 2008;14:833-9.
66. Saad F, Hotte S, North S, Eigl B, Chi K, Czaykowski P, Wood L, Pollak M, Berry S, Lattouf JB, Mukherjee SD, Gleave M, Winquist E. Randomized phase II trial of custirsen (OGX-011) in combination with docetaxel or mitoxantrone as second-line therapy in patients with metastatic castrate-resistant prostate cancer progressing after first-line docetaxel: CUOG trial P-06c. Clin Cancer Res. 2011;17:5765-73.
67. Sternberg CN, Dumez H, Van Poppel H, Skoneczna I, Sella A, Daugaard G, Gil T, Graham J, Carpentier P, Calabro F, Collette L, Lacombe D, de Balincourt C, et al. Docetaxel plus oblimersen sodium (Bcl-2 antisense oligonucleotide): An EORTC multicenter, randomized phase II study in patients with castration-resistant prostate cancer. Ann Oncol. 2009;20:1264-9.
68. Chi K, Gleave M, Klasa R, Murray N, Bryce C, Lopes de Menezes DE, D’Alessio A, Tolcher AW. A Phase I dose-finding study of combined treatment with an antisense BCL-2 oligonucleotide (Genesense) and mitoxantrone in patients with metastatic hormone-refractory prostate cancer. Clin Cancer Res. 2001;7:3920-7.
69. Chi K, Higano C, Reeves J, Feyerabend S, G G, Ferrero J, Jacobs C, Barnett-Griness O, Pande A, de Bono J. A Randomized Phase 3 Study Comparing First-line Docetaxel/Prednisone (DP) to DP Plus Custirsen in Men with Metsatic Castration-Resistant Prostate Cancer (MRCP). Ann Oncol. 2014;25:iv255-iv279.
70. Wiechno P, Somer BG, Mellado B, Chłosta PL, Cervera Grau JM, Castellano D, Reuter C, Stöckle M, Kamradt J, Pikiel J, Durán I, Wedel S, Callies S, André V, Hurt K, Brown J, Lahn M, Heinrich B. A randomised phase 2 study combining LY2181308 sodium (survivin antisense oligonucleotide) with first-line docetaxel/prednisone in patients with castration-resistant prostate cancer. Eur Urol. 2014;65:516-20.
71. Tolcher AW, Reyno L, Venner PM, Ernst SD, Moore M, Geary RS, Chi K, Hall S, Walsh W, Dorr A. A Randomized Phase II and Pharmacokinetic Study of the Antisense Oligonucleotides ISIS 3521 and ISIS 5132 in Patients with Hormone-refractory Prostate Cancer. Clin Cancer Res. 2002;8:2530-5.
72. Banumathy G, Cairns P. Signaling pathways in renal cell carcinoma. Cancer Biol Ther. 2010;10:658-64.
73. Ramakrishnan S, Ellis L, Pili R. Histone modifications: implications in renal cell carcinoma. Epigenomics. 2014;5:1-16.
74. Ellinger J, Kahl P, Mertens C, Rogenhofer S, Hauser S, Hartmann W, Bastian PJ, Büttner R, Müller SC, Von Ruecker A. Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. Int J Cancer. 2010;127:2360-6.
75. Fritzsche FR, Weichert W, Roske A, Gekeler V, Beckers T, Stephan C, Jung K, Scholman K, Denkert C, Dietel M, Kristiansen G. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer. 2008;8:381.
76. Hoffman A, Cairns P. The Epigenetics of Kidney Cancer and Bladder Cancer. Epigenomics. 2011;3:19-34.
77. Creighton C, Morgan M, Gunaratne P, Wheeler D, Gibbs R, Ozenberger B, Robertson A, Chu A, Beroukhim R, Cibulskis K, Signoretti S, Vandin F, Wu H, et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;449:43-9.
78. Shenoy N, Vallumsetla N, Zou Y, Galeas JN, Shrivastava M, Hu C, Susztak K, Verma A. Role of DNA methylation in renal cell carcinoma. J Hematol Oncol. 2015;8:1-13.
79. Ellinger J, Holl D, Nuhn P, Kahl P, Haseke N, Staehler M, Siegert S, Hauser S, Stief CG, Müller SC, Bastian PJ. DNA hypermethylation in papillary renal cell carcinoma. BJU Int. 2010;107:664-9.
80. Eggers H, Steffens S, Grosshennig A, Becker JU, Hennenlotter J. Prognostic and diagnostic relevance of hypermethylated in cancer 1 (HIC1) CpG island methylation in renal cell carcinoma. Int J Oncol. 2012;40:1650-8.
81. la Rosa AH-D, Acker M, Swain S, Manoharan M. The role of epigenetics in kidney malignancies. Cent Eur J Urol. 2015;68:157-64.
82. Martel C. Renal cell carcinoma: current status and future directions. Crit Rev Oncol Hematol. 2003;45:177-90.
83. Janowitz T, Welsh SJ, Zaki K, Mulders P, Eisen T. Adjuvant therapy in renal cell carcinoma-past, present, and future. Semin Oncol. 2013;40:482-91.
84. Cha TL, Chuang MJ, Wu ST, Sun GH, Chang SY, Yu DS, Huang SM, Huan SKH, Cheng TC, Chen TT, Fan PL, Hsiao PW. Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer Res. 2009;15:840-50.
85. Hu CY, Mohtat D, Yu Y, Ko Y, Shenoy N, Izquierdo MC, Seo A, Park D, Giricz O, Gundabolu K, Ware K, Bhagat T, Suzuki M, Liu S, Greally J, Susztak K, Verma A. Kidney cancer is characterized by aberrant methylation of tissue specific enhancers that are prognostic for overall survival. Clin Cancer Res. 2015;20:4349-60.
86. To KKW, Zhan Z, Bates SE. Aberrant promoter methylation of the ABCG2 gene in renal carcinoma. Mol Cell Biol Cell Biol. 2006;26:8572-85.
87. Reu FJ, Bae SI, Cherkassky L, Leaman DW, Lindner D, Beaulieu N, MacLeod a R, Borden EC. Overcoming resistance to interferon-induced apoptosis of renal carcinoma and melanoma cells by DNA demethylation. J Clin Oncol. 2006;24:3771-9.
88. Amato RJ, Stephenson J, Hotte S, Nemunaitis J, Bélanger K, Reid G, Martell RE. MG98, a Second-Generation DNMT1 Inhibitor, in the Treatment of Advanced Renal Cell Carcinoma. Cancer Invest. 2012;30:415-21.
89. Huang J, Yao X, Zhang J, Dong B, Chen Q, Xue W, Liu D, Huang Y. Hypoxia-induced downregulation of miR-30c promotes epithelial-mesenchymal transition in human renal cell carcinoma. Cancer Sci. 2013;104:1609-17.
90. Hainsworth JD, Infante JR, Spigel DR, Arrowsmith ER, Boccia R V, Burris H a. A phase II trial of panobinostat, a histone deacetylase inhibitor, in the treatment of patients with refractory metastatic renal cell carcinoma. Cancer Invest. 2011;29:451-5.
91. Pili R, Salumbides B, Zhao M, Altiok S, Qian D, Zwiebel J, Carducci MA, Rudek MA. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br J Cancer. 2012;106:77-84.
92. Gollob JA, Sciambi CJ, Peterson BL, Richmond T, Thoreson M, Moran K, Dressman HK, Jelinek J, Issa JJ. Phase I Trial of Sequential Low-Dose 5-Aza-2’-Deoxycytidine Plus High-Dose Intravenous Bolus Interleukin-2 in Patients with Melanoma or Renal Cell Carcinoma. Clin Cancer Res. 2006;12:4619-28.
93. Desai AA, Bukowski R, Murray P, Poiesz B, Quinn D, Saltzman M, Wright J, Young A, Stadler WM, Torti FM. 457 Interim evaluation of a multi-institution phase I/II study of antisense oligonucleotide GTI-2040 (G) and capecitabine (C) in patients with metastatic renal cell carcinoma (mRCC). Eur J Cancer Suppl. 2004;2:136.
94. Winquist E, Knox J, Ayoub J-P, Wood L, Wainman N, Reid GK, Pearce L, Shah A, Eisenhauer E. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: a National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Invest New Drugs. 2006;24:159-67.
95. Han H, Wolff EM, Liang G. Epigenetic Alterations in Bladder Cancer and Their Potential Clinical Implications. Adv Urol. 2012;2012:546917.
96. Ellinger J, Bachmann A, Göke F, Behbahani T, Baumann C, Heukamp LC, Rogenhofer S, Muller S. Alterations of Global Histone H3K9 and H3K27 Methylation Levels in Bladder Cancer. Urol Int. 2014;93:113-8.
97. Schneider A, Heukamp LC, Rogenhofer S, Fechner G, Bastian PJ, Ruecker A Von, Müller SC, Ellinger J. Global histone H4K20 trimethylation predicts cancer-specific survival in patients with muscle-invasive bladder cancer. BJU Int. 2011;108:290-6.
98. Friedrich MG, Weisenberger DJ, Cheng JC, Chandrasoma S, Siegmund KD, Gonzalgo ML, Toma MI, Huland H, Yoo C, Tsai YC, Nichols PW, Bochner BH, Jones PA, Liang G. Detection of Methylated Apoptosis-Associated Genes in Urine Sediments of Bladder Cancer Patients. Clin Cancer Res. 2004;10:7457-65.
99. Kim W-J, Kim Y. Epigenetic biomarkers in urothelial bladder cancer. Expert Rev Mol Diagn. 2009;9:259-69.
100. Hoque MO, Begum S, Topaloglu O, Rosenbaum E, Criekinge W Van, Westra WH, Zahurak M, Goodman SN, Sidransky D. Quantitation of Promoter Methylation of Multiple Genes in Urine DNA and Bladder Cancer Detection. J Natl Cancer Inst. 2006;98:996-1004.
101. Kim E, Kim Y, Kim W, Kim E, Kim Y, Jeong P, Ha Y, Bae S, Kim W. Methylation of the RUNX3 Promoter as a Potential Prognostic Marker for Bladder Tumor. J Urol. 2008;180:1141-5.
102. Dudziec E, Goepel JR, Catto JWF. Global epigenetic profiling in bladder cancer. Epigenomics. 2011;3:35-45.
103. Dyrskjøt L, Ostenfeld MS, Bramsen JB, Silahtaroglu AN, Lamy P, Ramanathan R, Fristrup N, Jensen JL, Andersen CL, Zieger K, Kauppinen S, Ulhøi BP, Kjems J, Borre M, Ørntoft TF. Genomic Profiling of MicroRNAs in Bladder Cancer : miR-129 Is Associated with Poor Outcome and Promotes Cell Death In vitro. Cancer Res. 2009;69:4851-61.
104. Griffiths TRL. Current perspectives in bladder cancer management. Int J Clin Pract. 2013;67:435-48.
105. Volpe A, Racioppi M, D’Agostino D, D’Addessi A, Marangi F, Totaro A, Pinto F, Sacco E, Battaglia S, Chiloiro G, Bassi PF. Advanced bladder cancer: new agents and new approaches. A review. Urol Oncol. 2013;31:9-16.
106. Pliarchopoulou K, Laschos K, Pectasides D. Current chemotherapeutic options for the treatment of advanced bladder cancer: a review. Urol Oncol. 2013;3:294-302.
107. Li QQ, Hao JJ, Zhang Z, Hsu I, Liu Y, Tao Z, Lewi K, Metwalli AR, Agarwal PK. Histone deacetylase inhibitor-induced cell death in bladder cancer is associated with chromatin modification and modifying protein expression: A proteomic approach. Int J Oncol. 2016;48:2591-607.
108. Wang D, Ouyang S, Tian Y, Yang Y, Li B, Liu X, Song Y. Intravesical Treatment with Vorinostat Can Prevent Tumor Progression in MNU Induced Bladder Cancer. 2013;2013:1-6.
109. Buckley MT, Yoon J, Yee H, Chiriboga L, Liebes L, Ara G, Qian X, Bajorin DF, Sun T-T, Wu X-R, Osman I. The histone deacetylase inhibitor belinostat (PXD101) suppresses bladder cancer cell growth in vitro and in vivo. J Transl Med. 2007;5:49.
110. Shang D, Liu Y, Matsui Y, Ito N, Nishiyama H, Kamoto T, Ogawa O. Demethylating Agent 5-Aza-2′-Deoxycytidine Enhances Susceptibility of Bladder Transitional Cell Carcinoma to Cisplatin. Urology. 2008;71(6):1220-5.
111. Rieger C, Huebner D, Temme A, Wirth MP, Fuessel S. Antisense- and siRNA-mediated inhibition of the anti-apoptotic gene Bcl-xL for chemosensitization of bladder cancer cells. Int J Oncol. 2015;47:1121-30.
112. Kunze D, Erdmann K, Froehner M, Wirth MP, Fuessel S. Enhanced inhibition of bladder cancer cell growth by simultaneous knockdown of antiapoptotic Bcl-xL and survivin in combination with chemotherapy. Int J Mol Sci. 2013;14:12297-312.
113. Rius M, Lyko F. Epigenetic cancer therapy: rationales, targets and drugs. Oncogene. 2012;31:4257-65.