
INTRODUCTION
Interstitial deletion of the long arm of chromosome 5 (del(5q)), is the most common karyotypic abnormality in myeloid neoplasms, observed in 10–15% of patients with myelodysplastic syndromes (MDS) [1–5] or primary acute myeloid leukemia (pAML) [6, 7], and in up to 40% of secondary myeloid leukemias (sAML) [8]. While a smaller fraction of more homogenous patients with the isolated del(5q) and classical 5q- syndrome show more favorable prognosis [9, 10], the majority of myeloid neoplasms with del(5q) are morphologically heterogeneous, and have additional cytogenetic abnormalities [7, 11, 12].
The commonly deleted regions (CDR) in del(5q) have been extensively studied with a distal region (CDR1:5q32-33) often deleted in the 5q- syndrome [2] and a proximal region (CDR2:5q31) in higher-risk MDS and AML [13]. SNP-array-based karyotyping helped to further refine the boundaries of the CDR (CDR1;145,299,747-153,828,955 and CDR2;137,500,665-139,471,723), and commonly retained regions (CRR1;from the centromere to 5q14.2 and CRR2;from 5q34 to the telomere) [5]. Patients with small interstitial deletions were shown to have a better outcome as compared to those with larger deletions [5]. Haploinsufficiency of several genes in the CDRs likely contributes to specific phenotypic features in del(5q). For instance, heterozygous deletions resulting in haploinsufficient expression of RPS14 is a key determinant of ineffective erythropoiesis [14], while thrombocytosis and megakaryocytic dysplasia may be related to haploinsufficient miR-145/miR-146a [15]. However, experimental knockdown of these genes did not result in a growth advantage and haploinsufficiency is not uniformly present in all cases to explain clonal dominance. Recurrent hemizygous mutations of genes within the deleted locus have not been identified with the notable exception CSNK1A1 missense mutations found in only 3/40 del(5q) cases, but absent in heterozygous configuration [16]. Moreover, most of del(5q) cases involve large deletions encompassing both CDRs, and thus identification of genes contributing to individual clinical phenotypes has been challenging. It is likely that pathogenetic mechanisms in del(5q) may involve hemizygous mutations or haploinsufficiency and be modified by additional somatic lesions affecting genes on other chromosomes. Furthermore, the position of del(5q) within the clonal hierarchy might also affect the phenotype and clinical behavior.
To characterize the genetic and genomic complexity and clonal hierarchy in myeloid neoplasms with 5q abnormalities, we used next generation sequencing (NGS), including whole exome sequencing (WES) and targeted multiamplicon deep sequencing in a cohort of patients with del(5q) in a comparison to patients with diploid chr5. In addition, to explore ancestral events in del(5q), we compared clonal size of individual somatic mutations with that of del(5q) identified by WES.
RESULTS
Detection of mutations within CDRs and CRRs in del(5q)
Using a combination of cytogenetic methods, including SNP-array karyotyping, FISH analysis and/or metaphase cytogenetics, we analyzed 1472 patients with myeloid neoplasms and identified various types of del(5q) in 178 (12%, isolated del(5q) N = 43, del(5q) with additional chromosome abnormality N = 135) patients and 5q uniparental disomy (UPD) in 8 (0.5%) (Table 1). After we defined large proximal (CRR1 5q11.1 to 5q14.2, 48400001-81634579) and distal (CRR2 5q34 to 5q35.3, 164213764-180915260) commonly retained genomic segments, as well as a region including the well-known CDR (5q31.1-5q33.1) as an interstitial deleted region (IDR) (5q14.2 to 5q34, 81634580-164213763), we correlated clinical characteristics with the extent of the deleted area. Interstitial deletions were frequently seen in low-risk MDS, while deletions involving the CRR1 and/or CRR2 (extended deletions) were more prevalent in high-risk MDS (RAEB1,RAEB2) and in AML compared to low-risk MDS (RCUD, RCMD, 5q-, MDS-U, RARS) (P < .0001, Figure 1A). No del(5q) case was observed within the MPN cohort (N = 81).
Table 1: Analysis cohort and frequency of 5q deletions and loss of heterozygosity
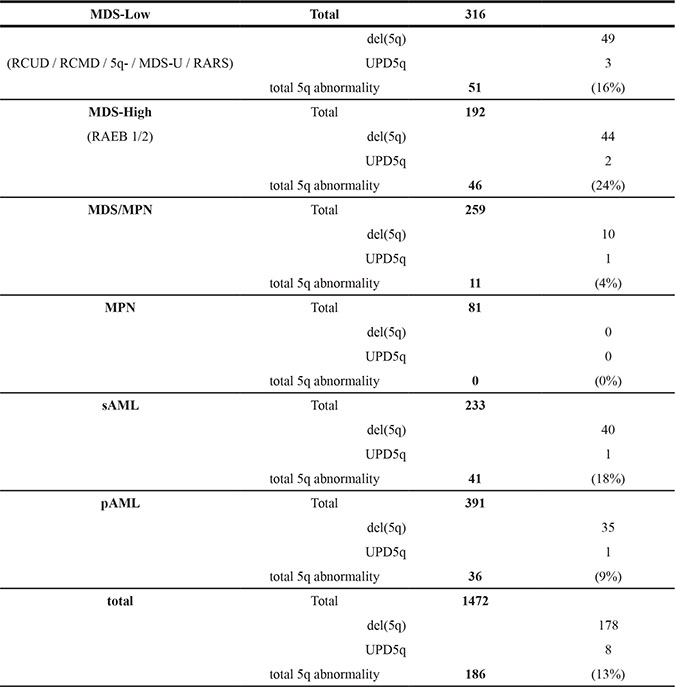
MDS, myelodysplastic syndromes; RCUD, refractory cytopenia with unilineage dysplasia; RCMD, refractory cytopenia with multilineage dysplasia; 5q-, MDS with isolated del(5q); MDS-U, MDS unclassifiable; RARS, refractory anemia with ring sideroblasts; RAEB, refractory anemia with excess blasts; MDS/MPN, myelodysplastic/myeloproliferative neoplasms, including CMML, JMML, MDS/MPN-U, RARS-T, and atypical CML; MPN, myeloproliferative neoplasms, including MPN, idiopathic MF, systemic mastocytosis, essential thrombocythemia, CML, and polycytemia vera ; pAML, primary acute myeloid leukemia; sAML, secondary acute myeloid leukemia, includes therapy related myeloid malignancies.
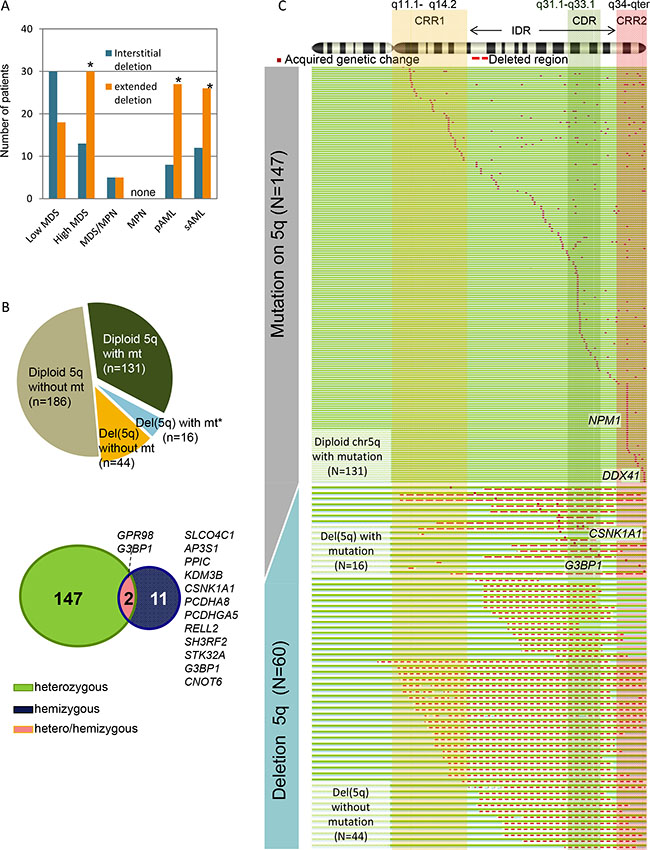
Figure 1: Whole spectrum of deletion 5q (del(5q)) and somatic mutations on chromosome 5q. (A) Number of various types of del(5q) lesions affecting interstitial and commonly retained regions (CRRs) in each disease phenotype. For detection of del(5q), SNP-array karyotyping and metaphase cytogenetics were applied (N = 1476). interstitial deletion: deletions within 5q14.2 to 5q34, extended deletion: deletions involving the CRR1 and/or CRR2, Low-risk MDS includes: refractory cytopenia with unilineage dysplasia, refractory cytopenia with multilineage dysplasia, MDS with isolated del(5q), MDS unclassifiable, and refractory anemia with ring sideroblasts. High-risk MDS includes refractory anemia with excess blasts. *P < .001. (B) Frequency of cases with mutations (mt) on chromosome 5 (chr5) in each copy-number status (diploid or deletion) as demonstrated by pie chart. *In a case with del(5q), a heterozygous mutation of a gene located on the non-deleted region of chr5 was identified. Venn diagram showing the number of mutated genes categorized by their zygosity (heterozygous, hemizygous, hemizygous and heterozygous). (C) Mutations on chr5 detected by whole-exome sequencing (N = 389) were shown in red dots (see Supplementary Table S3 for individual genes). Copy number status of chr5 was demonstrated as follows: diploid 5q, green and blue lines; and del(5q), a red dashed line. Two commonly retained regions (CRR1 and CRR2) and a commonly deleted region (CDR), defined by SNP-A karyotyping analyses, are represented by vertical rectangles.
Our investigations focused on mutational events in 5q genes in both diploid and 5q deletion cases; 329 non-del(5q) cases and 60 cases of del(5q), including 13 low-risk MDS, 8 high-risk MDS, 14 sAML, 4 MDS/MPN(3 CMML and 1 MDS/MPN-U) and 21 pAML. In addition, 8 selected genes on chr5 (e.g., DHX29, GPR98, APC, CSNK1A1, CSF1R, NPM1, SIMC1 and DDX41), found to be mutated in WES cohort studies were investigated in other cases (N = 627) by targeted sequencing (Supplementary Table S1). We identified 583 non-silent alterations on chr5 (6% of whole exome alterations) predominantly located on 5q (505 alterations/275 genes). After stringent filtering to avoid false positives [18], we narrowed the focus of our investigations to “tier 1” mutations. All candidate alterations were validated by Sanger sequencing and/or targeted deep sequencing of DNA from both neoplastic and germ line DNA. In the entire cohort we identified 257 somatic mutations in 159 genes through 5q; present in 26% (16/60) of del(5q) cases and in 40% (131/327) of 5q diploid cases (Figure 1B–1C), including well-known NPM1 mutations (N = 50 mutations), but also the newly identified recurrently mutated genes CSNK1A1, G3BP1 and DDX41 (Figure 2). No homozygous mutations were found within the long arm of chr5 in UPD cases.
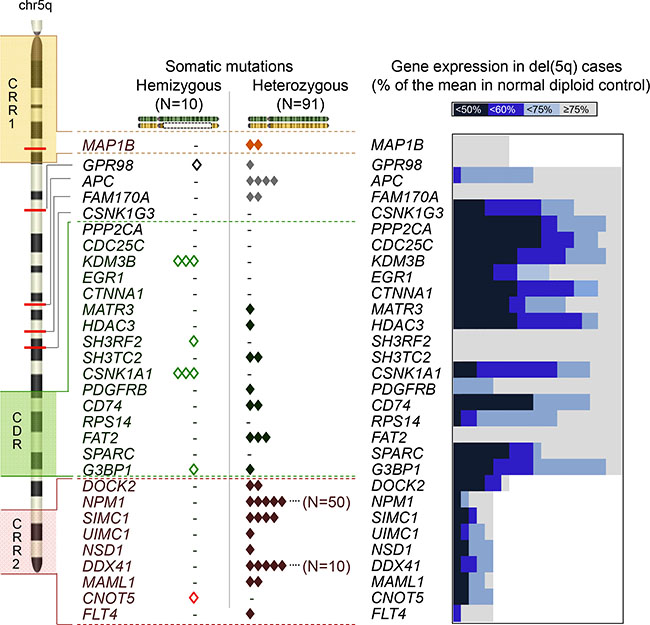
Figure 2: Zygosity of mutations and haploinsufficiency of the genes on chr5. An ideogram of chr5 in the left panel demonstrates 2 commonly retained regions (CRR1 and CRR2), a commonly deleted region (CDR) and the locations of the genes mutated in this study or previously reported to be pathologically important. Each somatic mutation was shown according to the configuration of zygosity; hemizygous (open diamonds) and heterozygous mutations (solid diamonds). In the right panel, the ratio (%) of the gene expression in each case with del(5q) (N = 5–21) was calculated as divided by the mean value in normal diploid chr5q samples (N = 17–162). The ratios in individual del(5q) cases were shown by gradient blue and gray plots as indicated in the figure. Decreased expressions less than 60% were considered haploinsufficient.
When we focused on mutations in the CDR, we found 70 alterations in 57 genes (27% of all alterations on 5q) including CSNK1A1 (5q32) and novel recurrently mutated genes G3BP1 (N = 2, 5q33.1). Hemizygous alterations in most of cases were found in the CDR (N = 8, Figure 1C, Figure 2). Mutations of CSNK1A1 (3/131 del(5q); 0/429 diploid cases) and KDM3B (3/60 del(5q); 0/331 diploid cases) were detected only in cases with del(5q). Mutations were found outside of the CDR, including APC (N = 4), FAM170A (N = 2) and GPR98 (N = 2), the latter previously found to be involved in germ line mutation in Usher syndrome. In serially studied specimens a GPR98 mutation appeared initially in a heterozygous configuration and was found later to be hemizygous upon evolution of del(5q). No mutations in CDC25C, CTNNA1, RPS14, PPP2CA and SPARC were identified. In genes corresponding to CRR1 we found 29 heterozygous alterations, including MAP1B. We also detected 96 alterations in CRR2, including NPM1 (N = 50), SIMC1 (N = 4), DDX41 (N = 10) and FLT4 (N = 1); these mutations occurred only in a heterozygous configuration.
Expression of frequently mutated genes on chr5
Haploinsufficiency caused by deletion of 5q involving multiple genes is likely the key pathogenetic mechanism in 5q- syndrome. We hypothesized that heterozygous hypomorphic mutations ultimately result in haploinsufficient expression, thereby phenocopying haploinsufficiency due to deletions. We investigated the expression levels of genes located on 5q in comparison between cases with/without del(5q) focusing on genes which have been previously reported or we have found affected by heterozygous mutations. For definition of haploinsufficiency we set the cut-off value < 60% of normal. In total, 12/27 genes showed haploinsufficient expression in del(5q), with the majority of haploinsufficient genes located in the CDR (7 genes) or CRR2 (3 genes) (Figure 2). Within the CDR, G3BP1, CD74 and CSNK1A1 exhibited both haploinsufficiency and somatic mutations, whereas PPP2CA, CTNNA1 and CDC25C showed haploinsufficient expression but no mutations. SH3RH2 and SH3TC2 genes did not display haploinsufficiency, while somatic events of these genes were noted. Other recurrently mutated genes located outside of the CDR: such as APC, DDX41 and MAML1 also showed haploinsufficient expression; however, mRNA levels of GPR98, FAM170A, a cluster of protocadherin family genes and NPM1 were not decreased in deletion cases.
Del(5q) and genetic events on other chromosomes
The associated mutational landscape outside of the del(5q) region may also affect the clinical and biological features of del(5q) cases. We thus analyzed the potential relationship of somatic mutations observed in other chromosomes in del(5q) cases. Globally, 5q- syndrome cases were associated with lower numbers of mutations (average 2.5 mutations/case) compared to IDR deletions (9.5 mutations) and patients with extreme deletions (involving CRR1 and CRR2; on average 18 mutations) by WES. TP53 mutations were associated with del(5q) as previously described by our group [19] and others [20–22]. In contrast, 10/15 of top mutated genes showed a significantly mutual exclusivity with del(5q) (e.g., TET2, NPM1, FLT3-ITD/TKD) (Figure 3A). Mutations of DNMT3A, SF3B1, ZRSR2, NRAS and BCOR were evenly distributed. The correlation of TP53 mutations with del(5q) was most prominent. TP53 mutations with del(5q) was mostly occurred with other chromosome abnormality, though, only 1 case was seen with isolated del(5q) (Supplementary Table S2). In low risk MDS, TP53 mutations were detected in 13% of del(5q) cases and in only 0.5% of diploid 5q cases (Figure 3B; P = .0001). Among high-risk MDS, TP53 was mutated in 42% of del(5q) vs. 4% of diploid 5q patients (Figure 3B; P < .0001). When we focused on the extent of the deletion, somatic TP53 mutations were particularly frequent (39%, 17/38) in cases whose deletion involved both CRR1 and CRR2, and in 32% (12/38) of interstitial deletion cases (Figure 3C, P = .03). Moreover, large deletions tended to be a part of complex karyotypes and 17p abnormality (Figure 3D).
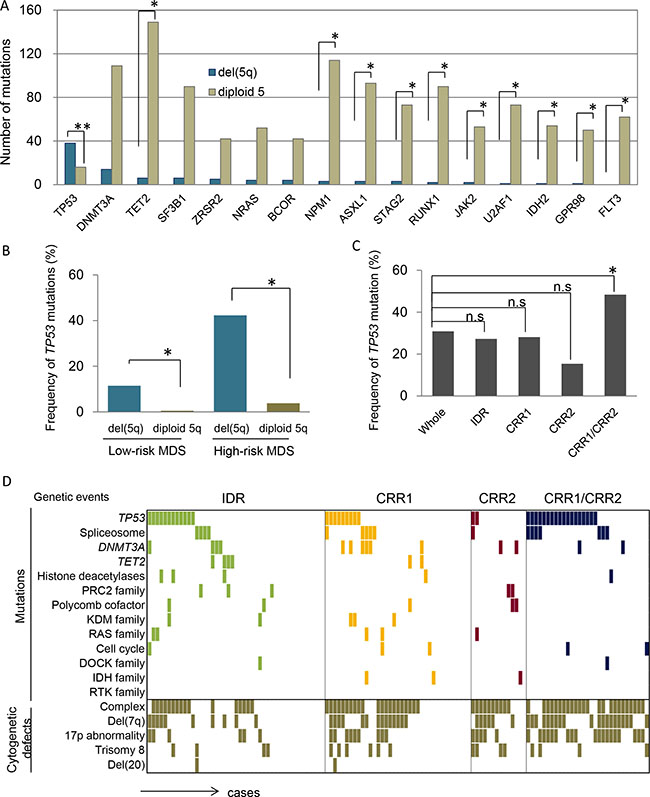
Figure 3: Presence of coexisting genetic events with del(5q). (A) In total, 1020 cases were analyzed for mutational landscape using NGS. Among them, 369 cases were MDS, 148 cases were MDS/MPN, 56 cases MPN, 146 cases were secondary-AML and 301 cases were primary-AML. Number of mutations were compared between cases with and without del(5q) (N = 894 and 126, respectively) *P < .005, **P < .0001. (B) Frequencies of TP53 mutations were compared between del(5q) and diploid chr5q cohorts (Low-risk MDS; RCUD, RCMD, 5q-synd, MDS-U and RARS, High-risk MDS; RAEB-I and RAEB-II), *P < .001. (C) Frequencies of TP53 mutations coexisting with subsets of del(5q) (as below) were individually compared to that with whole del(5q). IDR, interstitial deleted region (affecting q14.2-q34); CRR1, commonly retained region 1 (q11.1-q14.2); CRR2, commonly retained region 2 (q34-qter); CRR1/CRR2, deleted region spanning from CRR1 to CRR2, *P < .001. (D) Mutational spectrum associated with subsets of del(5q) were demonstrated according to the functionally relevant grouping of genes frequently mutated in myeloid neoplasms. Positive genes in each pathway were as follows: spliceosome, SF3B1, LUC7L2 and PRPF8; KDM family, KDM3B and KDM6B; Ras family, KRAS, NRAS and RIT1.
Prognostic impact
To investigate clinical implications, we initially assessed the impact of the deleted lesion on clinical outcomes, in which follow-up data were available. As expected, patients with isolated del(5q) showed better prognosis compared with del(5q) with other chromosomal abnormality (P < .001, Figure 4A). When we investigated the size of deletion, patients with both CRR1/CRR2 lesions (involving the 5q extremes) showed a worse prognosis compared with cases including CRR1 or CRR2, or with IDR lesions (P = .01, Figure 4A). We also investigated the impact of the presence of TP53 mutations (TP53MT) as the most common mutational event associated with del(5q). Predictably, survival among patients with del(5q) was inferior for TP53MT compared to wild type TP53 (TP53WT) cases (P < .001, Figure 4A). Furthermore, there were significant survival differences reflective of the previously described differences in the extent of deleted regions on 5q in TP53WT cases (P < .001, Figure 4B). TP53MT cases showed inferior outcome regardless of deleted region (Figure 4B).
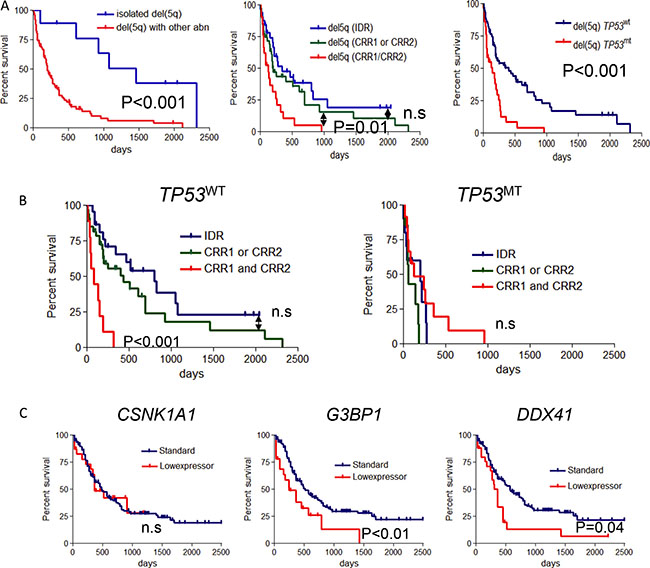
Figure 4: Survival analysis in the cases with del(5q). (A) (Left panel) Overall survival (OS) for patients with isolated del(5q) was compared among the del(5q) with other abnormality(del(5q) with abn). (Middle panel) Overall survival (OS) for patients with del(5q) was compared among the subgroups of 3 deleted regions as mentioned in Figure 1C. (Right panel) OS for patients with del(5q) were compared according to the TP53 mutational status. TP53MT, TP53 mutation; TP53WT, TP53 wild type. (B) OS was compared in 3 groups of each deleted region as above in the cohorts of TP53WT (left) and TP53MT (right). (C) OS was compared between low (under mean-1.5 S.D. in all cohort) and normal-range expression of 3 genes in each panel (CSNK1A1, G3BP1 and DDX41).
When we focused on the prognostic value of low-expressed genes in primary AML cohort, low expression of G3BP1 and DDX41 correlated with a shorter survival (P < .001 and P = .04, Figure 4C), an effect that was not seen in CSNK1A1 cases (Figure 4C). We also could not find association between low-expression and outcome in MAP1B, GPR98 and FAM170A which did not reach the haploinsufficiency cut off in our cohort (data not shown).
Del(5q) and hierarchical clonal architecture
It has been presumed that the del(5q) is the ancestral event in the myeloid neoplasms harboring this lesion [23]. Using deep sequencing and ’allelic imbalance’ we can determine the position of del(5q) in the hierarchical clonal architecture [24]. In our cohort, del(5q) was present in 17–98% of tumor cells and there was good correlation to the size of del(5q) clone by FISH (r = .94; Figure 5A). We identified three patterns of recurrent clonal architecture in del(5q) cases (Figure 5B) i) apparent pathogenic somatic mutations precede the deletion event (31%), ii) del(5q) appears to precede any other somatic mutation (19%) and iii) the succession cannot be determined because of expanded clones with similar size (“clonal saturation”) i.e., these cases were not informative. In our cohort, among the majority of cases in which del(5q) was a secondary lesion, in 64% of instances a TP53 mutation was the ancestral event and in 27% of cases the primordial lesion was DNMT3A mutation (Figure 5C). When we compared different time points in same case (MDS-phase and leukemic phase), the proportion of cells affected by TP53 mutation was more prominent than that of del(5q) (92% vs. 17%, Figure 5C right). In contrast, CSNK1A1 mutation occurred in the remaining allele after del(5q) (Figure 5D). We also detected 1 case in which del(5q) was asserted to be ancestral, and the TP53 mutation was detected as a secondary event.
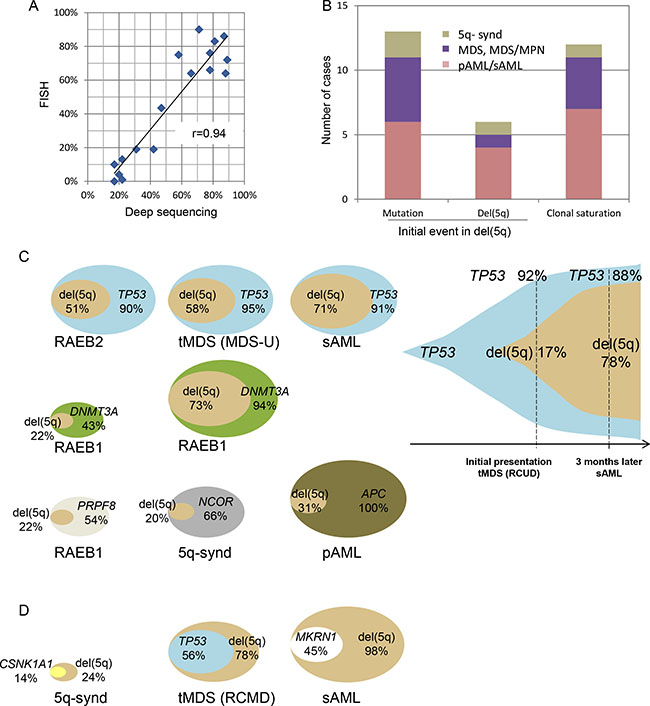
Figure 5: Ancestral mutations and subsequent del(5q) clones. (A) Correlation between FISH and deep sequencing on the detection of del(5q) clone. (B) Initial genetic events (mutations or del(5q)) were determined by the size of affected clonal cell populations based on the following assessments (bar graph). Variant allelic frequencies (VAF) of somatic mutations were adjusted by copy numbers and LOH was based on SNP-A results. Pathogenic genes mutated as initial events included TP53, DNMT3A, NCOR2 and PRPF8 as shown in Figure 5C. VAF of SNPs with a deleted-allele due to del(5q) were calculated for allelic imbalance as mentioned in the methods section. Inconclusive cases without discrimination were categorized as clonal saturation. Distribution of the disease phenotypes in each clonal pattern was demonstrated by colors as indicated. (C) Clonal architecture of the cases (N = 9) with initial driver mutations prior to del(5q) was demonstrated by overlaid double-oval figures (left) and a serial-assessment figure (right). Percentages indicate the fraction of the cells affected by mutations or del(5q). (D) In 3 other cases, del(5q) was shown to be a primary event.
DISCUSSION
Our study identified a cohort of 178 patients with various forms of del(5q) to answer several fundamental questions related to the pathogenesis of myeloid malignancies associated with these deletions: i) is del(5q) associated with recurrent hemizygous mutations; ii) may heterozygous mutations corresponding to haploinsufficient genes mimic the phenotype of the deletion; iii) are gene mutations on other chromosomes recurrent in del(5q); iv) what is the architecture and clonal evolution pattern in del(5q) myeloid neoplasms? Does 5q still stand as the primordial lesion in the light of data generated from the use of the new genomic platforms?
We found several somatic hemizygous mutations in del(5q) cases, including G3BP1 and CSNK1A1. Mutations in CSNK1A1 were found in a canonical E98 position as recently reported [16, 25]; only hemizygous mutations were found with del(5q) in the context of various clinical subtypes, including aggressive diseases RAEB-1 or therapy-related MDS. CSNK1A1 E98 mutations increase β-catenin activity thereby providing selective growth advantage. G3BP1 is another gene encoded within the CDR, and unlike CSNK1A1 mutations those in G3BP1 occurred both in heterozygous and hemizygous configuration. G3BP1 is known to control p53 activity through a dual pathway involving direct protein interaction of G3BP1-p53 and deubiquitination by regulating the ubiquitine specific peptidase USP10 [26].
Several other hematopoiesis-related genes and tumor suppressor genes are located in the CDR (e.g., CTNNA1, PPP2CA, EGR1, SPARC, RPS14 and CDC25C) with previously reported [14, 27–31], however, we were unable to detect hemi- or heterozygous mutations in these genes. It is possible that they contribute to clinical heterogeneity, shape the clinical phenotype or modulate the growth advantage of the del(5q). For the purpose of our investigations we hypothesized that haploinsufficient genes in del(5q) may also be affected by loss of function/hypomorphic mutations in diploid cases, and we have identified several genes fitting this profile. They were affected only in a minority of patients, and most did not recapitulate the clinical features of del(5q). Of note is that even the haploinsufficient expression showed variability among del(5q) cases: while average expression values may be decreased in del(5q) cohorts, specific expression is indeed haploinsufficient only in a portion of cases, and may in part reflect the relative percentage of the malignant clone in each patient. These differences in the degree of haploinsufficiency may explain, in addition to the size of deleted region, the intrinsic diversity of del(5q). Epigenetic regulation also affects the expression of each genes on del(5q) whereby deletion of unsilenced allele could even lead to gain of silencing. However, there would be no impact if silenced allele is deleted and thus may not be a key determining factor for the degree of haploinsufficiency. Because del(5q) occurred in one allele but epigenetic regulation (hyper- or hypo methylation) occurred in both allele, most likely at random. Nevertheless, several genes were found to be haploinsufficient and affected by somatic mutations, including HDAC3, CSNK1A1, G3BP1 and DDX41. Moreover, the functional role of 5q genes in hematopoiesis has been shown using a murine model.
The number of somatic mutations on other chromosomes increased with the increasing length of the 5q deletion. Co-occurrence of a TP53 mutation was particularly prominent in this del(5q) cohort, as reported in other studies [19, 21, 22]. It is still unclear why TP53 mutations selectively coincide with del(5q), one could speculate that loss of p53 function might overcome p53 tumor suppressor effects and foster leukemia evolution. Of note is that there are several gene clusters of negative regulators of TP53 on chr5q, such as, PPP2CA, RPS14, CSNK1A1 and G3BP1.
Among del(5q) patients we found that inferior survival was associated with patients with both deletion of CRR1 and CRR2. This relationship became evident in the TP53-wild type cohort, whereas there was no survival difference in existence of TP53 mutation. The larger deletions were frequently associated with other chromosomal abnormalities, which associated with inferior survival [32, 33]. However, the reasons are still unclear. One possibility is that there are several tumor suppressor genes in this location and long deletion causes multifunctional loss of these genes. Alternatively, other gene mutations or loss of function of tumor suppressor gene may result in different clinical phenotype of extended version of del(5q). We also analyzed impact of a cohort of individual genes with low expression, which indicated recurrent somatic mutations. Low expression of G3BP1 or DDX41 correlated with inferior survival but there was no prognostic impact in CSNK1A1, CDC25C and EGR1 (data not shown). These results indicate that the presence of low expression did not always correlate with survival, and the loss of tumorsupressive function may affect their outcome. Thus, a loss of tumor suppressive function of G3BP1 or DDX41 may lead to leukemic evolution in del(5q).
To define the position of del(5q) within the clonal hierarchy, we have compared the clonal size of somatic mutations with that of del(5q) using a novel approach focusing on allelic imbalance. While it has been reported that del(5q) occurs in stem cells as an ancestral event in patients with the 5q- syndrome [23], our results indicate that the mutation of TP53 or other driver gene mutations such as DNMT3A occurred as initial events followed by deletion of 5q in a majority of case. Previously, co-occurrence of a TP53 mutation was described in various del(5q) cohorts [19, 21, 22], but the position of del(5q) and TP53 mutation within subclonal hierarchy could not be precisely established using Sanger sequencing. In this report we were able to overcome this shortcoming using NGS. We also found somatic mutation of APC (5q22) as initial event in del(5q) case, a role for low expression of APC in the pathogenesis of myeloid neoplasms [34].
In summary, comprehensive molecular analyses using SNP-A karyotyping, WES and targeted sequencing revealed recurrent somatic mutations involving CSNK1A1 and G3BP1 in the CDR and DDX41 in the CRR in myeloid neoplasms with the del(5q). These genes showed haploinsufficiency in deleted cases and low expression of G3BP1 or DDX41 is associated with poor survival, which may be due to loss of function. In addition, in assessing allelic imbalance in del(5q), our results suggested that del(5q) is not an universal ancestral event. Mutation of TP53 is the most common mutation in del(5q) cases and may serve as an ancestral event. These data illuminate the impact of the del(5q) in myeloid malignancy, providing deep insights into the identity and role of key genes.
MATERIALS AND METHODS
Samples
Paired bone marrow and germ line (GL, CD3+ lymphocytes) DNA was obtained from 389 patients with various myeloid neoplasms and additional 631 DNA samples were included for further targeted resequencing, including a total of 178 cases of -5/del(5q) (Table 1). All samples were obtained following written informed consent approved by the institutional review boards at Cleveland Clinic and the University of Tokyo. The Cancer Genome Atlas (TCGA) AML data set was obtained from http://cancergenome.nih.gov/.
Sequencing, SNP array analysis and gene expression
Single-nucleotide polymorphisms (SNP) array analysis and WES was performed by HiSeq 2000 (Illumina) and result analyzed as previously described [5]. All the selected observations were validated by targeted Sanger sequencing or PCR-amplified deep sequencing using MySeq (Illumina). Targeted sequencing was performed by TruSeq custom amplicon (Illumina). Previously published microarray expression data were obtained on a cohort of 183 MDS patients (-5/del(5q), N = 41) and healthy controls (N = 17) (Supplementary Figure S1) [5, 17].
Determination of clonal burden
The detection of clonal size of del(5q) was accomplished by calculation of allelic imbalance for informative SNPs present within deleted region in heterozygous configuration in GL. For all heterozygous SNPs in the region, label the lost allele A and the retained allele B. For reads covering a heterozygous site in the sample, the probability that the read will carry the B allele is: P[reading B allele] = P[reading B allele | read from 5q−] × P[read from 5q−]+P[reading B allele/read from non-5q−] × P[read from non-5q−]. The terms in this expression depend on clonal abundance c of the deletion, and may be computed as: P[reading B allele/read from 5q−] = 1, P[reading B allele/read from non-5q] = 0.5, P[read from 5q−] = c/(2 × (1–c)+c)=c/(2–c), P[read from non- 5q−] = 2 × (1–c)/(2 × (1–c)+c)=(2–2c)/(2–c). Substituting these values into the expression and algebraically simplifying gives the expected proportion of reads harboring the B allele as p = 1/(2–c). Solving for c, the proportion for cells harboring the deletion is approximately c≈2–(1/p). To combine information from all n (GL) heterozygous SNPs in the region, we estimated p across these SNPs as  , where the allele Bi for each SNPi is that with the highest read frequency.
, where the allele Bi for each SNPi is that with the highest read frequency.
Statistical analysis
Comparisons of proportions and ranks of variables between groups were performed by the χ2 test, Fisher exact test, Student t test or Mann-Whitney U test, as appropriate. We used the Kaplan-Meier and the Cox method to analyze overall survival (OS) with a 2-sided P less than or equal to .05 determining significance.
CONFLICTS OF INTEREST
The authors have declared that no conflicts of interest exists.
FUNDING
This work was supported by National Institutes of Health (R01 HL082983, U54 RR019391 and K24 HL077522 to JPM, U54 RR019391 to MAS); AA & MDS International Foundation to HM; American Cancer Society Research Scholar Grant 123436-RSG-12-159-01-DMC (to TL); and the Edward P. Evans Foundation (to MAS and JPM).
Authors’ contributions
N.H, H.M., A.J.,M.S., J.B. and J.M. designed research; N.H., R.M., C.P., L.Z., M.C. and J.T. performed research; B.P., K.Y., T.L., Y.S., K.C., H.T., S.M., E.C., M.S., and S.O. analyzed data; N.H., H.M., A.V., M.M., A.L., Y.S., M.S., J.B., S.O. and J.M. wrote the paper.
REFERENCES
1. Sole F, Espinet B, Sanz GF, Cervera J, Calasanz MJ, Luno E, Prieto F, Granada I, Hernandez JM, Cigudosa JC, Diez JL, Bureo E, Marques ML, et al. Incidence, characterization and prognostic significance of chromosomal abnormalities in 640 patients with primary myelodysplastic syndromes. Grupo Cooperativo Espanol de Citogenetica Hematologica. Br J Haematol. 2000; 108:346–56.
2. Boultwood J, Fidler C, Strickson AJ, Watkins F, Gama S, Kearney L, Tosi S, Kasprzyk A, Cheng JF, Jaju RJ, Wainscoat JS. Narrowing and genomic annotation of the commonly deleted region of the 5q- syndrome. Blood. 2002; 99:4638–41.
3. Boultwood J, Perry J, Pellagatti A, Fernandez-Mercado M, Fernandez-Santamaria C, Calasanz MJ, Larrayoz MJ, Garcia-Delgado M, Giagounidis A, Malcovati L, Della Porta MG, Jadersten M, Killick S, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010; 24:1062–5. doi:10.1038/leu.2010.20.
4. Boultwood J, Pellagatti A, McKenzie AN, Wainscoat JS. Advances in the 5q- syndrome. Blood. 2010; 116:5803–11. doi:10.1182/blood-2010–04–273771.
5. Jerez A, Gondek LP, Jankowska AM, Makishima H, Przychodzen B, Tiu RV, O’Keefe CL, Mohamedali AM, Batista D, Sekeres MA, McDevitt MA, Mufti GJ, Maciejewski JP. Topography, clinical, and genomic correlates of 5q myeloid malignancies revisited. J Clin Oncol. 2012; 30:1343–9. doi:10.1200/JCO.2011.36.1824.
6. Grimwade D, Walker H, Harrison G, Oliver F, Chatters S, Harrison CJ, Wheatley K, Burnett AK, Goldstone AH, Medical Research Council Adult Leukemia Working P. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001; 98:1312–20.
7. Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A, Paietta E, Willman CL, Head DR, Rowe JM, Forman SJ, Appelbaum FR. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000; 96:4075–83.
8. Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, Vardiman JW, Rowley JD, Larson RA. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003; 102:43–52. doi:10.1182/blood-2002–11–3343.
9. Patnaik MM, Lasho TL, Finke CM, Gangat N, Caramazza D, Holtan SG, Pardanani A, Knudson RA, Ketterling RP, Chen D, Hoyer JD, Hanson CA, Tefferi A. WHO-defined ‘myelodysplastic syndrome with isolated del(5q)’ in 88 consecutive patients: survival data, leukemic transformation rates and prevalence of JAK2, MPL and IDH mutations. Leukemia. 2010; 24:1283–9. doi:10.1038/leu.2010.105.
10. Nimer SD. Clinical management of myelodysplastic syndromes with interstitial deletion of chromosome 5q. J Clin Oncol. 2006; 24:2576–82. doi:10.1200/JCO.2005.03.6715.
11. Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, Goldstone A. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998; 92:2322–33.
12. Byrd JC, Mrozek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, Pettenati MJ, Patil SR, Rao KW, Watson MS, Koduru PR, Moore JO, Stone RM, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood. 2002; 100:4325–36. doi:10.1182/blood-2002–03–0772.
13. Lai F, Godley LA, Joslin J, Fernald AA, Liu J, Espinosa R, 3rd, Zhao N, Pamintuan L, Till BG, Larson RA, Qian Z, Le Beau MM. Transcript map and comparative analysis of the 1.5-Mb commonly deleted segment of human 5q31 in malignant myeloid diseases with a del(5q). Genomics. 2001; 71:235–45. doi:10.1006/geno.2000.6414.
14. Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, Golub TR. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. 2008; 451:335–9. doi:10.1038/nature06494.
15. Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, Hirst M, Hogge D, Marra M, Wells RA, Buckstein R, Lam W, Humphries RK, et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med. 2010; 16:49–58. doi:10.1038/nm.2054.
16. Schneider RK, Adema V, Heckl D, Jaras M, Mallo M, Lord AM, Chu LP, McConkey ME, Kramann R, Mullally A, Bejar R, Sole F, Ebert BL. Role of Casein Kinase 1A1 in the Biology and Targeted Therapy of del(5q) MDS. Cancer Cell. 2014. doi:10.1016/j.ccr.2014.08.001.
17. Pellagatti A, Cazzola M, Giagounidis A, Perry J, Malcovati L, Della Porta MG, Jadersten M, Killick S, Verma A, Norbury CJ, Hellstrom-Lindberg E, Wainscoat JS, Boultwood J. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia. 2010; 24:756–64. doi:10.1038/leu.2010.31.
18. Gomez-Segui I, Makishima H, Jerez A, Yoshida K, Przychodzen B, Miyano S, Shiraishi Y, Husseinzadeh HD, Guinta K, Clemente M, Hosono N, McDevitt MA, Moliterno AR, et al. Novel recurrent mutations in the RAS-like GTP-binding gene RIT1 in myeloid malignancies. Leukemia. 2013; 27:1943–6. doi:10.1038/leu.2013.179.
19. Jasek M, Gondek LP, Bejanyan N, Tiu R, Huh J, Theil KS, O’Keefe C, McDevitt MA, Maciejewski JP. TP53 mutations in myeloid malignancies are either homozygous or hemizygous due to copy number-neutral loss of heterozygosity or deletion of 17p. Leukemia. 2010; 24:216–9. doi:10.1038/leu.2009.189.
20. Jadersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Gohring G, Hedlund A, Hast R, Schlegelberger B, Porwit A, Hellstrom-Lindberg E, Mufti GJ. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol. 2011; 29:1971–9. doi:10.1200/JCO.2010.31.8576.
21. Kulasekararaj AG, Smith AE, Mian SA, Mohamedali AM, Krishnamurthy P, Lea NC, Gaken J, Pennaneach C, Ireland R, Czepulkowski B, Pomplun S, Marsh JC, Mufti GJ. TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br J Haematol. 2013; 160:660–72. doi:10.1111/bjh.12203.
22. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, Shlien A, Groves MJ, Forbes SA, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013; 122:3616–27; quiz 99. doi:10.1182/blood-2013–08–518886.
23. Tehranchi R, Woll PS, Anderson K, Buza-Vidas N, Mizukami T, Mead AJ, Astrand-Grundstrom I, Strombeck B, Horvat A, Ferry H, Dhanda RS, Hast R, Ryden T, et al. Persistent malignant stem cells in del(5q) myelodysplasia in remission. N Engl J Med. 2010; 363:1025–37. doi:10.1056/NEJMoa0912228.
24. Shen W, Clemente MJ, Hosono N, Yoshida K, Przychodzen B, Yoshizato T, Shiraishi Y, Miyano S, Ogawa S, Maciejewski JP, Makishima H. Deep sequencing reveals stepwise mutation acquisition in paroxysmal nocturnal hemoglobinuria. J Clin Invest. 2014; 124:4529–38. doi:10.1172/JCI74747.
25. Heuser M, Meggendorfer M, Cruz MM, Fabisch J, Klesse S, Kohler L, Gohring G, Ganster C, Shirneshan K, Gutermuth A, Cerny-Reiterer S, Kronke J, Panagiota V, et al. Frequency and prognostic impact of casein kinase 1A1 mutations in MDS patients with deletion of chromosome 5q. Leukemia. doi:leu201549 [pii] 10.1038/leu.2015.49.
26. Soncini C, Berdo I, Draetta G. Ras-GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene. 2001; 20:3869–79. doi:10.1038/sj.onc.1204553.
27. Liu TX, Becker MW, Jelinek J, Wu WS, Deng M, Mikhalkevich N, Hsu K, Bloomfield CD, Stone RM, DeAngelo DJ, Galinsky IA, Issa JP, Clarke MF, et al. Chromosome 5q deletion and epigenetic suppression of the gene encoding alpha-catenin (CTNNA1) in myeloid cell transformation. Nat Med. 2007; 13:78–83. doi:10.1038/nm1512.
28. Wei S, Chen X, Rocha K, Epling-Burnette PK, Djeu JY, Liu Q, Byrd J, Sokol L, Lawrence N, Pireddu R, Dewald G, Williams A, Maciejewski J, et al. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc Natl Acad Sci USA. 2009; 106:12974–9. doi:10.1073/pnas.0811267106.
29. Joslin JM, Fernald AA, Tennant TR, Davis EM, Kogan SC, Anastasi J, Crispino JD, Le Beau MM. Haploinsufficiency of EGR1, a candidate gene in the del(5q), leads to the development of myeloid disorders. Blood. 2007; 110:719–26. doi:10.1182/blood-2007–01–068809.
30. Pellagatti A, Jadersten M, Forsblom AM, Cattan H, Christensson B, Emanuelsson EK, Merup M, Nilsson L, Samuelsson J, Sander B, Wainscoat JS, Boultwood J, Hellstrom-Lindberg E. Lenalidomide inhibits the malignant clone and up-regulates the SPARC gene mapping to the commonly deleted region in 5q- syndrome patients. Proc Natl Acad Sci U S A. 2007; 104:11406–11. doi:10.1073/pnas.0610477104.
31. Ye Y, McDevitt MA, Guo M, Zhang W, Galm O, Gore SD, Karp JE, Maciejewski JP, Kowalski J, Tsai HL, Gondek LP, Tsai HC, Wang X, et al. Progressive chromatin repression and promoter methylation of CTNNA1 associated with advanced myeloid malignancies. Cancer Res. 2009; 69:8482–90. doi:10.1158/0008–5472.CAN-09–1153.
32. Mallo M, Cervera J, Schanz J, Such E, Garcia-Manero G, Luno E, Steidl C, Espinet B, Vallespi T, Germing U, Blum S, Ohyashiki K, Grau J, et al. Impact of adjunct cytogenetic abnormalities for prognostic stratification in patients with myelodysplastic syndrome and deletion 5q. Leukemia. 2011; 25:110–20. doi:10.1038/leu.2010.231.
33. Volkert S, Kohlmann A, Schnittger S, Kern W, Haferlach T, Haferlach C. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes Chromosomes Cancer. 2014; 53:402–10. doi:10.1002/gcc.22151.
34. Stoddart A, Fernald AA, Wang J, Davis EM, Karrison T, Anastasi J, Le Beau MM. Haploinsufficiency of del(5q) genes, Egr1 and Apc, cooperate with Tp53 loss to induce acute myeloid leukemia in mice. Blood. 2014; 123:1069–78. doi:10.1182/blood-2013–07–517953.