
INTRODUCTION
The myeloproliferative neoplasms (MPN) are chronic myeloid cancers characterized by the overproduction of mature blood cells and risk of transformation into acute myeloid leukaemia (AML) [1, 2]. In addition to chronic myeloid leukaemia (CML) characterized by the BCR-ABL fusion gene, the three most common myeloproliferative neoplasms are essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF). In 2005 the JAK2 V617F mutation has been identified in patients with PV, ET and PMF [3, 4]. Mutations of JAK2 exon 12 [5, 6] and MPL exon 10 [7] were subsequently detected in a subsets of patients. Recently, several studies identified additional mutations present in smaller fraction of patients [8–12]. In 2013, somatic mutations in the CALR gene, encoding calreticulin, were detected in most patients with ET or PMF without mutations in JAK2 and MPL genes [13, 14].
Calreticulin is a protein with different functions. In the endoplasmic reticulum the protein modulates calcium homeostasis and regulates the folding of glycoproteins. [15] Calreticulin is also localized in other cellular compartments where it regulates many biological functions including apoptosis and proliferation. [16]. All mutant CALR proteins discovered until now share a novel amino acid sequence at the C-terminal. The non-mutant CALR C-terminal is mainly negatively charged, whereas the mutant CALR C-terminal is largely positively charged [17]. The mutant CALR protein lacks the endoplasmic reticulum retention signal (KDEL) and therefore it may have an altered subcellular localization. The role of CALR in the pathogenesis of MPN is largely unknown. Klampfl and colleagues and Nangalia and colleagues [13, 14] elegantly demonstrated that CALR mutations are acquired early in the major clone. Several studies are trying to shed light on how mutant CALR can sustain the uncontrolled proliferation. It was demonstrated that mutant CALR activates JAK/STAT signaling thus generating a proliferative signaling [13, 14]. Recently, it was shown that the forced expression of mutant CALR in hematopoietic cell lines resulted in MPL-dependent activation of MAPK signaling. In line whit this finding, patients with CALR mutations showed increased MAPK activity in blood cells and in CD34+ cells leading to enhance megakaryopoiesis and pro-platelet production. [18] Recently it was also shown the IL3 independent growth of CALR mutant cells [17].
CALR mutations have been reported as mutually exclusive with JAK2 and MPL mutations and are present in a percentage of patients ranging from 56 to 88% of JAK2/MPL-negative cases [13, 14]. A recent paper reported a patient who carried simultaneously both mutations, JAK2 V617F and CALR exon 9 [19]. Many different CALR mutations have been described in exon 9 but the most frequent is type-1 [L367fs*46; deletion of 52 base pairs (bp)] and type-2 [K385fs*47; insertion of 5 bp] [13, 14]. Currently, the method commonly exploited for the detection of CARL mutations is sanger sequencing.
Peptide nucleic acid (PNA) is able to hybridize very specifically to DNA. It is recognized that the binding PNA/DNA is more stable than DNA/DNA or DNA/RNA duplexes [20, 21], but PNA sequences cannot be extended by polymerase [22]. Consequently, PNA/DNA duplex suppresses DNA amplification. This is the principle that allowed to design PNA directed PCR clamping to discriminate wild type from mutated sequences [22–24].
The goal of the study was to make the diagnosis of CALR mutations more rapid, easy and accessible.
Based on this premise, we developed a novel and sensitive assay based on PNA directed PCR clamping for the detection of CALR mutations (type-1 and type-2) in MPN patients.
RESULTS
As show in Figure 1, the method described here forecasts that both PNA probe and PCR primer competitor target sites overlap, thus leading to a direct competition towards complementary DNA. In case CALR sequence is wild type, a perfect matching occurs between PNA (designed on wild sequence) and DNA. As a consequence, the PNA-template hybridization is favoured and DNA amplification is strongly suppressed. This occurs in patients with CALR wild-type (WT) or CALR type-1 mutation (DEL), producing weak bands at different lengths.
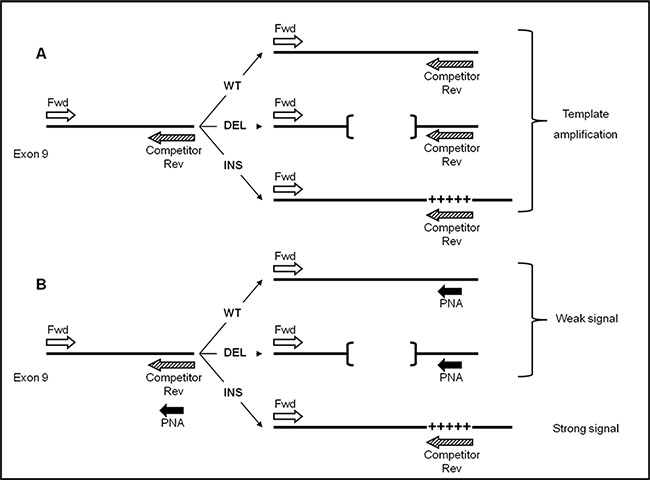
Figure 1: Step II: PNA clamping experimental design. Template amplification of CALR wild-type (WT), type-1 (DEL) and type-2 (INS) mutations (A) in absence of PNA probe (B) and with PNA probe. Perfect PNA/DNA hybridization occurs when CALR template sequence is wild-type (WT) or type-1 mutation (DEL, indicated by [¨¨]), thus leading to a significant reduction of PCR amplification. By contrast, in presence of type-2 mutation (INS, indicated by +++++), PNA/DNA duplex is highly destabilized, on behalf of primer Competitor Rev, allowing strong template amplification.
The lengths of the bands allow to distinguish between CALR WT and DEL (Figure 2B). Conversely, a non-perfect matching ( as in case of a mutation) destabilizes the PNA hybridization thus favouring the hybridization between template and primer competitor. In this case an amplification product is evident. (as in the case of patients with CALR type-2 mutation (INS)) (Figure 2B).
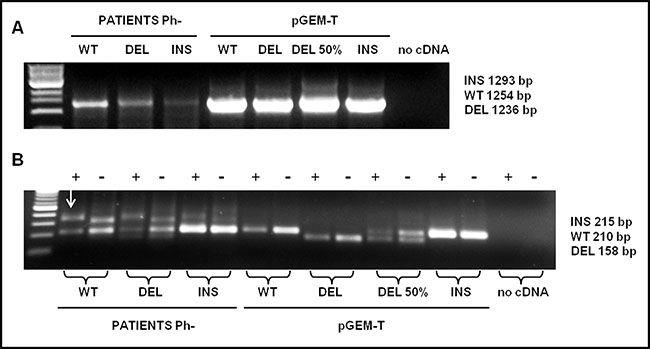
Figure 2: Detection analysis by PNA directed PCR clamping of CALR type-1 (DEL) and type-2 (INS) mutations. (A) Step I: cDNA amplification of patients with CALR wild-type (WT), type-1 (DEL) and type-2 (INS) mutations. pGEM-T-CALR wild-type (WT), pGEM-T-CALR type-1 mut (DEL), pGEM-T-CALR type-1 mut 50% vs. wild-type (DEL 50%) and pGEM-CALR type-2 mut (INS) were used as PCR positive control. 5 μL of each amplifier were loaded on 1% Agarose-TBE 1x gel with 5 μg/mL ethidium bromide (EtBr) and run at 120 V for 30 minutes. (B) Step II: PCR amplification of a small area of CALR gene, containing type-1 and type-2 mutations, was carried-out in absence (−) or in presence (+) of PNA probe. The plasmids amplified in the step I were used, in a dilution of 1:100, in the step II in order to interpret the results, 10 μL of each amplifier were loaded on 2% Agarose-TBE 1x gel with 5μg/mL EtBr and run at 100 V for 30 minutes and than at 65 V for 15 minutes. The arrow indicates the non-specific band present in the amplified of patients.
There are two PCR steps. Through the first step, the selected CALR sequence is amplified (Figure 2A). The second step is carried-out in duplicate, in the presence (+) or in the absence (-) of the PNA probe. The final result should be interpreted by reading the double loading for each individual patient (Figure 2B).
As shown in Figure 2B a non-specific band appears in patients approximately 100 bp higher than the WT band; however, it will not compromise the interpretation of the result. As shown in Figure 3, sensitivity was assessed mixing, at different ratio, mutated (type-1 and type-2) and WT template. Dilutions were as follow: 100, 50, 20, 10, and 5 % type-1 deletion (52 bp) mutated versus WT template and 100, 50, 20, 10, 5, 1, 0.1, 0.01, 0.001% type-2 insertion (5 bp) mutated versus WT template. The method displays a quite high sensitivity, allowing to detect an amount of mutated template as low as 10% for CALR type-1 and 0,1% for CALR type-2 mutations, which cannot be identified by sanger sequencing. Our diagnostic test shows a sensitivity of 100% (CI 79.41% to 100.00%) and a specificity of 98,5% (CI 91.96% to 99.96%) with an AUC corresponding to 0.99.
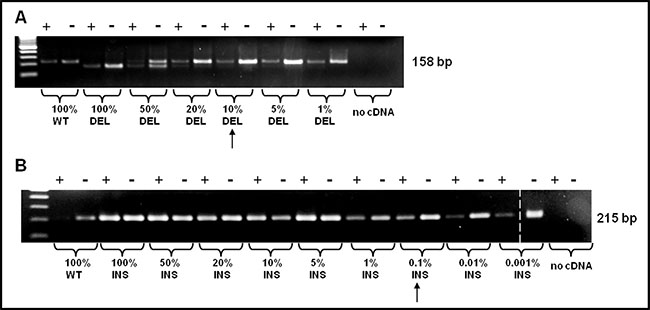
Figure 3: PNA directed PCR clamping sensitivity was assessed mixing, at different ratio, mutated (type-1 and type-2) and WT template. The arrows indicate the sensitivity of the method. (A) Dilutions were as follow: 100, 50, 20, 10, 5, 1% CALR type-1 mutation vs. WT template and (B) 100, 50, 20, 10, 5, 1, 0,1, 0,01, 0,001% CARL type-2 mutation vs. WT template. The dashed vertical bar separates different parts of the same gel.
Seventy-five patients affected by MPN and seven controls were examined in double blind either by direct DNA sequencing or by PNA directed PCR clamping, displaying identical readout except for two samples: mutated (type-1 mutation) in PNA directed PCR clamping, but wild-type by sanger sequencing. For these patients, the experiment was repeated and the shorter bands, resulting by step II of PNA directed PCR clamping, were purified and cloned in pGEM-T Easy Vector as previously described. Then, they were sequenced by sanger method using T7 and SP6 primers (respectively #Q5021 e #Q5011, Promega, Milan, Italy): one of these resulted wild-type and represents the only false positive sample tested; unexpectedly, the second one was found to have both the CALR type-1 and 2 mutations (Figure 4).
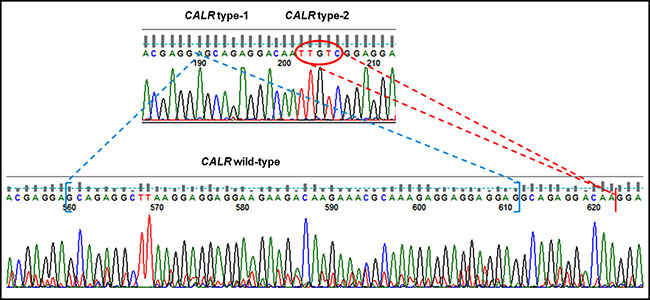
Figure 4: Sequencing chromatogram of patient with both CARL type-1 and 2 mutations compared with WT sequence. CALR type-1 mutation is indicated in blue, while CALR type-2 is highlighted in red.
DISCUSSION
The majority of Ph- myeloproliferative neoplasms shows mutations in JAK2 or MPL genes. Recently, additional mutations were identified in ET and PMF wild-type for JAK2 and MPL. We developed a novel molecular assay, based on PNA direct PCR clamping, for a rapid diagnosis of CALR type-1 and 2 mutations in patients affected by MPN. The assay shows high efficiency, sensitivity and specificity. Our method appears to be faster, cheaper and easily interpretable compared with DNA sanger sequencing, the method most commonly used for the detections of these mutations. In clinical practice, the evaluation of CARL mutations increases the diagnostic accuracy of ET and PMF and allows a more accurate diagnosis in patients without other molecular markers. Greater accuracy of diagnosis can result in a more precise prediction of prognosis and in a more effective therapeutic choice [25, 26]. Furthermore, the mutated protein CALR could represent a new therapeutic target for molecular drugs [25]. Contrary to JAK2 V617F mutation, a correlation between CALR mutant allele quantification and clinical presentation has not yet been demonstrated [27]. Despite the sanger sequencing is the most commonly method used to diagnose these mutations, many laboratories in the world are not equipped with such a facility or cannot effort the cost of this procedure. In conclusion, considering the diagnostic relevance of the CALR mutations, the PNA direct PCR clamping assay might represent a good alternative method for the detection of CALR type-1 and type-2 mutations, especially considering the costs and the lower sensitivity of the DNA sequencing method. Furthermore, because this assay is faster, more sensitive, and less expensive than sanger sequencing, it might be applied also in laboratory not equipped for sophisticated analysis, reducing costs and time for an advanced molecular diagnosis.
MATERIALS AND METHODS
Cells sources and RNA extraction
After informed consent, peripheral blood were collected at diagnosis from 82 patients: 75 patients affected by myeloproliferative disease and 7 controls (5 healthy donors and 2 patients affected by myelodysplastic syndromes). The study was approved by the ethic committee on December 16th 2015 (number of approval 212/2015). Table 1 summarizes clinical and molecular characteristics of patients.
Table 1: Clinical and molecular characteristics of patients
Disease |
n° of pts |
% of JAK2 mut pts |
% of CALR mut pts |
|---|---|---|---|
PV |
25 |
28 |
8 |
ET |
38 |
13 |
40 |
PMF |
12 |
17 |
25 |
Controls |
7 |
0 |
0 |
PV: polycythemia vera; ET: essential thrombocythemia; PMF: primary myelofibrosis
Total RNA was extracted from buffy-coat fraction using TRIzol Reagent (Ambion, Thermo Fisher Scientific, Massachusetts, USA), following the manufacturer’s instructions, and 1 μg was reverse transcribed using random hexamers as primers in a final volume of 25 μL.
Cloning PCR controls with pGEM®-T easy vector
CALR WT, CALR DEL and CALR INS were amplified and purified by QIAgenquick Gel Extraction Kit (Qiagen, Hildem, Germany) and cloned in pGEM-T Easy Vector (Promega, Milan, Italy). All reactions were performed following the manufacturer’s instructions. Plasmids were used as positive control in PCR steps. Primers sequences are listed in Table 2.
Table 2: Primers sequences used for cloning and for sanger sequencing
Cloning |
CALR Fwd |
5'–ATGCTGCTATCCGTGCCGCTGCT–3' |
CALR Rev WT |
5'–CTACAGCTCGTCCTTGGCCTGGC–3' |
|
CALR Rev Mut |
5'–TCAGGCCTCAGTCCAGCCCTGGA–3' |
|
Sequencing |
CALR Fwd |
5'–ACAACTTCCTCATCACCAACG–3' |
CALR Rev |
5'–GGCCTCAGTCCAGCCCTG–3' |
Capillary sanger sequence method for the detection of CALR
Detection of CALR mutations was performed by capillary sanger sequence method. DNA was extracted by Maxwell 16 (Promega, Milan, Italy) following the manufacturer’s protocol. CALR was amplified and analyzed by sequencing with BigDye terminator v3.1 (Applied Biosystem, Foster City, California) and capillary electrophoresis on ABI PRISM 3130XL (Applied Biosystem, Foster City, California). Primers sequences are listed in Table 2. The sensitivity was previously estimated by serial dilutions experiments to be 20%.
PNA directed PCR clamping
Step I: 3 μL of cDNA were used to amplify the CALR mRNA sequence (acc. # NM_004343) and ~10 ng of pGEM-T-CALR wild-type (WT), pGEM-T-CALR type-1 mut (DEL), pGEM-T-CALR type-1 mut 50% vs. wild-type (DEL 50%) and pGEM-CALR type-2 mut (INS) were used as PCR positive control.
Primers sequences were as follow (step I):
Fwd: 5'–ATGCTGCTATCCGTGCCGCTGCT–3'
Rev: 5'–TCAGGCCTCAGTCCAGCCCTGGA–3'
Reaction volume was 50 μL and the final concentrations of the reagents were the following: MgCl2 [2, 5 μM], 10X PCR Buffer [1X], dNTPs [0,4 μM], AmpliTaq 2U, Primers [0, 6 μM], DMSO 10% , H2O MilliQ.
Step II: 1μL of step I PCR product was used to amplify a small area of CALR gene, containing type-1 and type-2 mutations. PCR amplification was carried-out in absence (−) or in presence (+) of PNA probe, at a concentration 6 × greater than primer Competitor Rev. The amplification performed without (−) PNA probe represents an internal positive control displaying the PCR efficiency.
Primers and PNA probe sequences were as follow (step II):
Fwd: 5'–CCTCTGGCAGGTCAAGTCTG–3'
Rev Competitor: 5'– ATCCTCCGACAATTGTCCT CTG–3'
PNA probe: 5'–OO–CATCCTCCTTGTCCTC–3'
Reaction volume was 50 μL and he final concentrations of the reagents were the following:
MgCl2 [1, 25 μM], 10X PCR Buffer [1X], dNTPs [0, 2 μM], AmpliTaq 1U, Primers [0, 2 μM], PNA probe [1, 2 μM].
PNA probe, designed on the human CALR cDNA (acc. # NM_004343), encompasses a very short sequence in exon 9, common to both the wild-type and type-1 mutation sequence. PNA-PCR clamping conditions for CALR mutations detection were as follow: step I PCR: 94°C 3 min, (94°C 30 s, 65°C 40 s, 72°C 1 min 30 s) for 35 cycles, 72°C 5 min; step II PCR: 94°C 3 min, (94°C 30 s, 55°C 27 s, 72°C 45 s) for 30 cycles.
Diagnostic test valuation with a confidence interval (CI) of 95% and the area under the curve (AUC) were calculated with MedCalc software (MedCalc, Osten, Belgium).
The second step of this assay has been patented by the authors and by the University of Turin (patent pending, number 102016000042586).
CONFLICTS OF INTEREST
The authors have no conflicts of interest.
GRANT SUPPORT
DC was funded by grants from: AIRC, MURST-COFIN,. The study was partially funded by a special grant from “AIRC 5 per mille” to the AGIMM group (AIRC-Gruppo Italiano Malattie Mileoproliferative); for a complete list of AGIMM investigators see at http://www.progettoagimm.it.
VR is a fellow of Compagnia di San Paolo.
JP is a fellow of Gigi Ghirotti Foundation.
REFERENCES
1. Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008; 112:2190–8.
2. Campbell PJ, Green AR. The myeloproliferative disorders. N Engl J Med. 2006; 355:2452–66.
3. James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005; 434:1144–8.
4. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005; 352:1779–1790.
5. Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007; 356:459–468.
6. Passamonti F, Elena C, Schnittger S, Skoda RC, Green AR, Girodon F, Kiladjian JJ, McMullin MF, Ruggeri M, Besses C, Vannucchi AM, Lippert E, Gisslinger H, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood. 2011; 117:2813–2816.
7. Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, DeAngelo DJ, Clark JJ, Lee SJ, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006; 3:e270.
8. Rumi E, Pietra D, Guglielmelli P, Bordoni R, Casetti I, Milanesi C, Sant’Antonio E, Ferretti V, Pancrazzi A, Rotunno G, Severgnini M, Pietrelli A, Astori C, et al. Acquired copyneutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood. 2013; 121:4388–4395.
9. Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adélaïde J, Rey J, Vainchenker W, Bernard OA, Chaffanet M, Vey N, Birnbaum D, Mozziconacci MJ. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009; 23:2183–2186.
10. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010; 42:722–726.
11. Harutyunyan A, Klampfl T, Cazzola M, Kralovics R. p53 lesions in leukemic transformation. N Engl J Med. 2011; 364:488–490.
12. Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, Finke C, Score J, Gangat N, Mannarelli C, Ketterling RP, Rotunno G, Knudson RA, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013; 27:1861–1869.
13. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013; 369:2379–2390.
14. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG, Aziz A, Godfrey AL, Hinton J, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013; 369:2391–2405.
15. Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J. 2009; 417:651–666.
16. Gold LI, Eggleton P, Sweetwyne MT, Van Duyn LB, Greives MR, Naylor SM, Michalak M, Murphy-Ullrich JE. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010; 24:665–83.
17. Lavi N. Calreticulin Mutations in Myeloproliferative Neoplasms. Rambam Maimonides Medical Journal. 2014; 5:e0035. doi:10.5041/RMMJ.10169.
18. Kollmann K, Warsch W, Gonzalez-Arias C, Nice FL, Avezov E, Milburn J, Li J, Dimitropoulou D, Biddie S, Wang M, Poynton E, Colzani M, Tijssen MR, et al. A novel signalling screen demonstrates that CALR mutations activate essential MAPK signalling and facilitate megakaryocyte differentiation. Leukemia. 2016; doi: 10.1038/leu.2016.280.
19. McGaffin G, Harper K, Stirling D, McLintock L. JAK2 V617 F and CALR mutations are not mutually exclusive; findings from retrospective analysis of a small patient cohort. Br J Haematol. 2014; 167:276–8.
20. Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, Nielsen PE. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature. 1993; 13:566–568.
21. Sugimoto N, Yamamoto K, Satoh N. Positional effect of single bulge nucleotide on PNA(peptide nucleic acid)/DNA hybrid stability. Nucleic Acids Symp Ser. 1999; 13:95–96.
22. Sugimoto N, Satoh N, Yamamoto K. Comparison of thermodynamic stabilities between PNA (peptide nucleic acid)/DNA hybrid duplexes and DNA/DNA duplexes. Nucleic Acids Symp Ser. 1999; 13:93–94.
23. Pellestor F, Paulasova P, Macek M, Hamamah S. The peptide nucleic acids: a new way for chromosomal investigation on isolated cells? Hum Reprod. 2004; 19:1946–51.
24. Lundin KE, Good L, Strömberg R, Gräslund A, Smith CI. Biological activity and biotechnological aspects of peptide nucleic acid. Adv Genet. 2006; 56:1–51.
25. Tefferi A, Thiele J, Vannucchi AM, Barbui T. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia. 2014; 28:1407–1413.
26. Park JH, Sevin M, Ramla S, Truffot A, Verrier T, Bouchot D, Courtois M, Bas M, Benali S, Bailly F, Favre B, Guy J, Martin L, et al. Calreticulin Mutations in Myeloproliferative Neoplasms: Comparison of Three Diagnostic Methods. PLoS One. 2015; 10:e0141010.
27. Malysz J, Crisan D. Correlation of JAK2 V617F Mutant Allele Quantitation with Clinical Presentation and Type of Chronic Myeloproliferative Neoplasm. Ann Clin Lab Sci. 2009 Fall; 39:345–50.