
INTRODUCTION
Despite the many advances in biomedical sciences, the productivity of research and development programs in the pharmaceutical industry is on the decline [1, 2]. The failure rates in clinical trials approach 90% for all disease categories with oncology among the categories with the lowest (5.1%) likelihood of approval (LOA) after Phase I [1, 3, 4]. One of the reasons this high failure rate is an inefficient early lead discovery process, which mostly relies on the screening of large compound libraries to identify potential leads for further preclinical development. Despite exhaustive efforts, such as modification and combination of compounds screening libraries in order to place them in relevant druggable space [5], such screening remains a blind search.
Despite many prior failures, in silico based approaches promise an attractive alternative to empower the industry with more efficient screening methods able to provide more reliable results at a reduced cost and timescale [6]. Although, the use of computational methods within the pharmaceutical industry is now well established, the development of new mathematical methods coupled with the availability of more powerful and cheaper computational resources, contribute to the continuous improvement and development of new techniques. Among them, Machine Learning (ML) algorithms and specifically Deep Learning (DL) methods offer a great potential for further significant advances within the industry.
In recent years DL methods demonstrated surprising results surpassing human accuracy in many tasks including image and voice recognition [7] and managed to overcome many limitations of more traditional ML approaches. From a technical point of view, modern DL techniques are structured as deep architectures, called Deep Neural Networks (DNNs). Because of this flexibility and adaptability of DNN for learning from large range of data, DNNs are now considered as an increasingly important area in the biomedical field that shows significant potential in comprehensive -omics analysis and could be useful for tackling many current issues [1, 8, 9]. Most DL-based methods require a massive amount of data for their training, optimization and validation and are often applied in most data-rich fields of biomedical sciences [10].
While the range of DL applications is diverse, two common uses are classification and prediction. One can find DL methods involving discriminative models used for classification tasks [11, 12]. These algorithms are based on the well-established backpropagation and dropout algorithms and make use of piecewise linear units [13–15] as activation functions, as they are known for having a well-behaved gradient descent.
On the other hand, there is also an increasing demand and research developed for using DL to directly generate a model that could be successfully applied, for example, to compression, denoising, inpainting, texture synthesis, semi-supervised learning, unsupervised feature learning and other tasks. However, designing deep generative models is a bigger challenge than discriminative models. This is due to the fact that initial generative models, such as restricted Boltzmann machines, Denoising autoencoders or deep Boltzmann machines [16] are probabilistic and based on a parametric specification of a probability distribution function. Training of such models requires the maximization of the log-likelihood, a function that is usually computationally intractable. As a consequence, many difficulties arise when trying to approximate the associated probabilistic computations (strategies involving the use of Markov chains (MCMC algorithm) or unrolled approximate inference networks during either training or generation of samples). Furthermore, supplementary complications may also appear when trying to leverage the benefits of piecewise linear units, the most commonly used activation function, in the generative context. In order to overcome the limitations of parametric methods, several alternative generative models have been suggested. Generative stochastic networks are an example of models that do not require the explicit representation of the likelihood while being able to generate samples from the desired distribution. As a result of the investigations performed so far, one can identify different classes of deep generative models: Deep directed graphical models, deep undirected graphical models and generative autoencoders. It is important to notice that each of these methods have advantages and disadvantages with respect to different computational and modeling steps that can be classified into five categories, that is, training, inference, sampling, likelihood evaluation and model design. As any in silico based approaches, these methods must be subject to validation and performance assessment. Depending on the goals, properties and specificities of the application, the global evaluation and interpretation of a generative model can be done using three independent criteria: average log-likelihood, Parzen window estimates, and visual fidelity of samples [16, 17].
The latest class of non-parametric approaches for deep generative models is known as generative adversarial network (GAN). In this new framework, initially proposed by Goodfellow et al. [18], generative models are estimated via an adversarial process. In practice, two models are simultaneously trained: a generative model G that captures the data distribution, and a discriminative model D that estimates the probability that a sample came from the training data rather than G. The training procedure for G is to maximize the probability of D making an error [19]. Thus, this framework does not correspond to the standard optimization problem as it is based on a value function that one model seeks to maximize and the other seeks to minimize. The process terminates at a saddle point that is a minimum with respect to one model’s strategy and a maximum with respect to the other model’s strategy [18]. Because GANs do not require an explicit representation of the likelihood, neither approximate inference nor Markov chains are necessary. Consequently, GANs provide an attractive alternative to maximum likelihood techniques.
As recently reviewed in [20, 21], the application in oncology of various types of knowledge-based in silico methods for predicting drug responses that require multiple kinds of -omics data for training has lead to the development and maintenance of large public databases containing curated data sets of molecular profiles of cell lines treated with the variety of small molecules. There are currently three major publicly available databases that can be used for the training of drug response prediction models. First among these is the Cancer Cell Line Encyclopedia (CCLE) [22] that contains data from more than 1000 cell lines from 36 tumor sites and drug sensitivity data from more than 11000 experiments obtained altogether from 24 anticancer drugs tested on overall 500 cell lines. Second is the Genomics of Drug Sensitivity in Cancer (GDSC) project [23], which contains data obtained from different measurements of drug sensitivity in cancer cell lines. More precisely, the GDSC contains more than 75 000 experiments that tested 138 anticancer drugs on 1000 cell lines from various cancer types. Furthermore, the GDSC also contains baseline data, that is, data obtained from untreated samples, which include gene copy number, expression data and somatic mutations in 75 genes relevant to cancer. Finally, another resource of interest is the NCI-60 cancer cell line collection [24], that provides drug screening data for thousands of drugs with potential applications in cancer therapy and 60 cell lines from nine different cancers.
In this work, we propose a deep adversarial model, specifically adversarial autoencoder, for identification and generation of new compounds that make a use of available biological and chemical data. We demonstrate that this purely insilico-based approach is capable of providing biologically relevant predictions and consequently could contribute to speed up the drug discovery process and ultimately increase the success rate within the field of anticancer therapy.
RESULTS
The adversarial autoencoder (AAE) architecture used in this study is depicted on Figure 1. As an input AAE uses a vector of binary fingerprints and log concentration (LCONC) of the molecule. AAE outputs concentration and a vector, consisting of probabilities assigned to each bit of the fingerprint. In the latent layer we also introduced a neuron responsible for Growth Inhibition percentage (GI), where negative values indicate the reduction in the number of tumor cells after drug treatment.
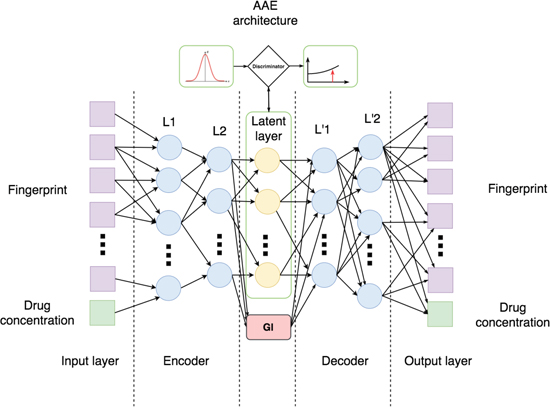
Figure 1: Architecture of Adversarial Autoencoder (AAE) used in this study. Encoder consists of two consequent layers L1 and L2 with 128 and 64 neurons, respectively. In turn, decoder consists of layers L’1 and L’2 comprising 64 and 128 neurons. Latent layer consists of 5 neurons one of which is Growth Inhibition percentage (GI) and the other 4 are discriminated with normal distribution.
AAE was trained on fingerprint, LCONC and GI data for 6252 compounds profiled on MCF-7 cell line. After that we sampled 640 vectors from prior distribution in latent layer with 640 GI values from normal distribution N (5,1). Based on this data, we used decoder to generate 640 probability vectors with corresponding LCONC values. Then we extracted the set of probability vectors with LCONC < -5.0 M. In total, we obtained 32 vectors.
We screened 32 vectors them against a library of 72 million compounds derived from Pubchem [25] (Figure 2). We used the maximum likelihood function to select top 10 hits for each of the 32 vectors. This amounted for a set of 69 unique compounds (Supplementary Table 1). In order to assess the biological relevance of the our results, Pubchem BioAssay database [26] was used to identify the compounds for which anticancer activities and other relevant biomedical properties of interest have been either tested or demonstrated. This also includes several patented compounds. Although information about potential mode of action and more precisely anticancer activity is not available for all compounds, several of them are already known as anticancer agents of various kinds. Most of these compounds are related to anthracyclines (or anthracycline antibiotics). Anthracyclines are used in cancer chemotherapy to treat many cancers, including leukemias, lymphomas, stomach, uterine, ovarian, breast cancer, and lung cancers. The anthracyclines are among the most effective anticancer treatments currently available. Daunorubicinol, is an anticancer agent previously tested for treating infant with leukemia. CHEMBL519482 is another potential anticancer agent whose cytotoxicity against human KB cells has been tested using squamous cell carcinoma. Epi-daunomycin (CID:153753), also belongs to the class of anthracyclines and is classified as an antitumor antibiotic, in the treatment of neoplastic disease and blood cancers, (leukaemia and lymphoma), and many types of carcinoma and soft tissue sarcomas. Another compound, Idarubicin (CID:42890) is often traded under the denomination idamycin and idamycin PFS. This compound is classified as an antitumor antibiotic and orally administered anthracycline antineoplastic used in treatment against various types of cancers including leukemia, breast cancer and multiple myeloma. Similar to other anthracyclines, it induces histone eviction from chromatin and inhibits the activity of DNA topoisomerase II. It has been patented for numerous clinical purposes. (7R,9R)-Idarubicin (CID:151582) is another anticancer agent tested for small cell lung cancer therapies. CID:53304462 has undergone several testing phase and available bioassays demonstrate its activity for various functions including inhibitor of protein arginine methyltransferase 1 (PRMT1). Epi-daunorubicin (CID:125250) has been the subject of several studies to assess its effects against cancer such as nasopharyngeal carcinoma. CID:57620448 was patented in 2009 (patent ID US7893023) as prodrug activated by plasmin that can be used in cancer chemotherapy. CID:44398799 was tested regarding cytotoxicity properties against K562 leukemia cell line and SW620 colon cancer cell line. CID:59835410 is the subject of two patents (US2010022467and US7452901) for its activity as anticancer phosphonate analog. CID:21563452 is a synthetic compound patented (patent ID US3933827) for its ability to actively inhibit the growth of transplantable tumors and is therefore useful as cytostatic agent. CID:15573184 is also patented (patent ID US6838469) as it exhibits reduced gastrointestinal side-effects comprising a known active substance having antitumor effects. CID:59283582 is patented small molecule (patent ID US2010098691) as a composition for the treatment of cancer as several benzimidazole based anticancer agents can be used in combination with a second anti-cancer agent to obtain positive therapeutic outcomes.
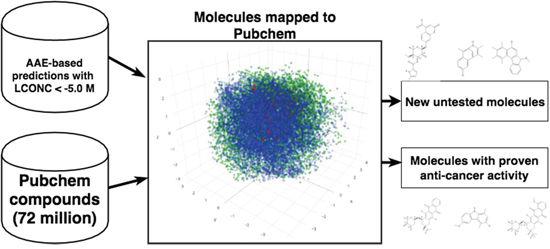
Figure 2: Mapping generated molecules to chemical space of Pubchem. Pubchem compounds are depicted in green, training set is shown in blue and mapped predictions in red.
Other compounds identified have been the subject of test phases and patent applications for other biomedical purposes. CID:54706490 has been tested as having antibacterial activity against ofloxacin, oxacillin, erythromycin resistant Staphylococcus aureus. Both compounds, CID:44329845 and CID:44329846, were tested in vitro and demonstrated antibacterial activity against Staphylococcus aureus. CID:58771432 is involved in a patent (patent ID US2006286103) for a stable antibody formulation. CID:58076509 is patented (patent ID US2012029167) as a drug using the magnetic properties of a metal salen complex. CID:15573192 is patented (patent ID US2015093398) as treatment indicates an inhibitory effect of that small molecule on the Zika virus induced caspase-3 activity that may correlate with its effect on reduction of Zika virus induced cell death. Finally, CID:57077355 has been patented as part of a method for treating migraine headaches (patent ID WO9506468).
DISCUSSION AND PERSPECTIVES
While the use of DL methods in the biomedical field is still in its infancy and most of the applications are restricted to pure classification tasks, these techniques may transform drug discovery and biomarker development. In this work, we demonstrated how DNNs can be used not only for classification tasks but for biologically relevant generating models. The new conceptual architecture of AAE was used to develop and validate a complex DL-based work-flow capable of generating models of new compounds in cancer and oncology using drug concentrations and fingerprints as sole inputs. As a result, we predicted 69 compounds belonging to various chemical classes. The anticancer activity for our prediction have already been identified and in some cases these molecules are already used as anticancer agents for treating various cancer types including leukemia and breast cancer. This confirms the ability of this approach to provide biologically relevant results. To the best of our knowledge, this is the first application of GAN techniques within the field of cancer drug discovery. Further experimental validation is in order to assess whether the remaining predicted compounds show anticancer activity. One of the ways to evaluate the effects of the small molecule in multiple human cancers and validate the predictions could be transcriptional response analysis using signaling pathway activation analysis algorithms [27, 28] in PDX models, where human tissue is grafted into immunodeficient mice [29].
Generative capabilities of deep adversarial network techniques open the doors to new perspectives as it could contribute to overcome several limitations of current data driven computational methods. For example, we can apply GANs on transcriptomics data for the generation of new samples for a desired phenotypic groups and in chemoinformatics for the prediction of the physical, chemical, or biological properties and structures of molecules. Quantitative structure–activity relationships (QSAR) and quantitative structure–property relationships (QSPR) are still considered as the modern standard for predicting properties of novel molecules [30]. To that end, many ML-based approaches have been developed to tackle such problems, but recent results show that the DL-based methods match or outperform other state-of-the-art methods and demonstrate better predictive performance, parsimony and interpretability and web-based predictors are available on some cases [31]. Furthermore, new methods based on convolutional neural networks are able to perform predictions by directly using graphs of arbitrary size and shape as inputs rather than fixed feature vectors [32] and one can expect to see the development of more flexible deep generative architectures that can be applied directly to other structured data such as sequences, trees, graphs, and 3D structures [31, 33]. Thus, the deep adversarial network techniques could be used to improve accuracy, generative capabilities and predictive power and address several issues including computational cost, limited computation at each layer and limited information propagation across the graph [32].
Finally, target prediction and mapping of bioactive small compounds and molecules by analyzing binding affinities and chemical properties is another area of research that makes extensive use of data-driven computational methods in order to optimize the use of data available in existing repositories [34, 35]. Despite promising results and the availability of web-platforms to computationally identify new targets for uncharacterized molecules or secondary targets for known molecules such as SwissTargetPrediction [34], in general, the available methods remain too inaccurate for systematic binding predictions and physical experiments remain the state of the art for binding determination. In this field, DL-based methods, such as the recently released methods AtomNet based on deep convolutional neural networks [36] have allowed to circumvent several limitations and outperform more traditional computational methods including RFs, SVMs for QSAR and ligand-based virtual screening [37–39]. One can expect that the development of DL-methods making use of the GAN framework will also lead to significant improvement with respect to prediction accuracy and power.
MATERIALS AND METHODS
Data set selection
In this work, we used NCI-60 cell line assay full dose response data (released on September 2014) available at the Developmental Therapeutics Program (DTP) website of NCI/NIH (http://dtp.nci.nih.gov/index.html). The SMILES annotation for compounds generated using program CACTVS v. 3.2 was also downloaded from the DTP website. We utilised the Open Babel chemistry toolbox [40] to convert SMILES string into 166-bit Molecular ACCess System (MACCS) chemical fingerprints. In total, we generated MACCS fingerprints for a total of 6252 molecules with known growth inhibition percentage (GIPRCNT or GI) in NCI-60 assay. MACCS fingerprints were also generated from 72200431 molecules derived from Pubchem database [25].
Design and training of the GAN
The architecture of the GAN used in this study was inspired by recent work in this field [18, 41]. According to original studies, the adversarial network and the autoencoder are trained jointly with SGD in two phases – the reconstruction phase and the regularization phase – executed on each mini-batch. In the reconstruction phase, the autoencoder updates the encoder and the decoder to minimize the reconstruction error of the inputs. In the regularization phase, the adversarial network first updates its discriminative network to tell apart the true samples (generated using the prior) from the generated samples (the hidden codes computed by the autoencoder). The adversarial network then updates its generator (which is also the encoder of the autoencoder) to confuse the discriminative network. Once the training procedure is done, the decoder of the autoencoder will define a generative model that maps the imposed prior of p(z) to the data distribution.
We divided the input layer into a fingerprint part and a concentration input neuron. So, our AAE was trained to encode and reconstruct not only molecular fingerprints, but also experimental concentrations. The Encoder consists of two consequent layers L1 and L2 with 128 and 64 neurons, respectively. The decoder consists of the two layers L’1 and L’2, comprising 64 and 128 neurons respectively. The latent layer consists of 5 neurons, one of which is the GI and the four others are discriminated with normal distribution. Since we train an encoder net to predict ‘efficiency’ against ‘cancer’ in a single neuron of latent layer, we divided the latent vector in two parts - ‘GI’ and ‘representation’. So we added a regression term to the encoder cost function. Furthermore, we restrict our encoder to map the same fingerprint to the same latent vector independently from input concentration by additional ‘manifold’ cost. Here we compute mean and variance of the concentrations through all dataset and then use them to sample concentrations for ‘manifold’ step. On each step we sample fingerprint from trainset and batch of concentration from normal distribution with given mean and variance. The training net with ‘manifold’ loss is performed by maximization of cosine similarity between ‘representations’ of similar fingerprints with different concentrations
All these changes resulted in a 5-step train iteration instead of a 3-step in AAE basic model:
a) Discriminator trained to distinguish between given latent distribution and encoded ‘representation’; b) Encoder trained to confuse Discriminator with generated ‘representations’; c) Encoder and Decoder trained jointly as Autoencoder; d) Encoder trained to fit ‘score’ part of latent vector; e) Encoder trained with ‘manifold’ cost.
The two first steps (a,b) are trained as usual adversarial networks. The Autoencoder cost function was computed as a sum of logloss [42] of fingerprint part and MSE of concentration parts and MSE was also used as a regression cost function.
The code for the AAE implemented in this paper is available at https://github.com/spoilt333/onco-aae.
ACKNOWLEDGMENTS
We would like to thank NVIDIA and Mark Berger and Andrea de Souza personally for providing samples of the Tesla K80 and early access to Titan X GPUs. We would like to thank Franco Cortese of the Biogerontology Research Foundation for suggestions and edits to the manuscript, and Michael Palovich of GlaxoSmithKline for the insightful advice and valuable comments.
Authors would also like to acknowledge the many valuable biologically relevant and technical comments from reviewers during all phases of review that substantially improved the paper.
CONFLICTS OF INTEREST
The authors are affiliated with Insilico Medicine, Inc, which is applying deep learning techniques, including generative adversarial networks to drug discovery for internal research, providing services to the pharmaceutical companies and identifying novel geroprotectors. The authors have vested interest in demonstrating and popularizing the successful applications of the deep learning techniques for drug discovery and biomarker development.
FUNDING
The work of Arthur Kadurin was performed according to the Russian Government Program of Competitive Growth of Kazan Federal University and partially supported by the Government of the Russian Federation (grant 14.Z50.31.0030).
Insilico Medicine’s Pharma.AI project is supported by a grant from the Life Extension Foundation 2016-LEF-AA- INSIL.
REFERENCES
1. Munos BH, Chin WW. How to revive breakthrough innovation in the pharmaceutical industry. Sci Transl Med. 2011; 3:89cm16.
2. Mignani S, Huber S, Tomás H, Rodrigues J, Majoral J-P. Why and how have drug discovery strategies in pharma changed? What are the new mindsets? Drug Discov Today. 2016; 21:239-49.
3. Kola I, Ismail K, John L. Opinion: Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004; 3:711-6.
4. Thomas DW, Burns J, Audette J, Carrol A, Dow-Hygelund C, Hay M. Clinical development success rates 2006-2015. San Diego: Biomedtracker/Washington, DC: BIO/Bend: Amplion; 2016.
5. Yu MJ. Druggable chemical space and enumerative combinatorics. J Cheminform. Springer; 2013; 5:19.
6. Vanhaelen Q, Mamoshina P, Aliper AM, Artemov A, Lezhnina K, Ozerov I, Labat I, Zhavoronkov A. Design of efficient computational workflows for in silico drug repurposing. Drug Discov Today. 2016;. doi: 10.1016/j.drudis.2016.09.019.
7. Oquab M, Bottou L, Laptev I, Sivic J. Learning and Transferring Mid-level Image Representations Using Convolutional Neural Networks. 2014 IEEE Conference on Computer Vision and Pattern Recognition. IEEE; p. 1717-24.
8. Putin E, Mamoshina P, Aliper A, Korzinkin M, Moskalev A, Kolosov A, Ostrovskiy A, Cantor C, Vijg J, Zhavoronkov A. Deep biomarkers of human aging: Application of deep neural networks to biomarker development. Aging (Albany, NY). 2016; 8 :1021-33. doi: 10.18632/aging.100968.
9. Aliper A, Plis S, Artemov A, Ulloa A, Mamoshina P, Zhavoronkov A. Deep Learning Applications for Predicting Pharmacological Properties of Drugs and Drug Repurposing Using Transcriptomic Data. Mol Pharm. 2016; 13:2524-30.
10. Mamoshina P, Polina M, Armando V, Evgeny P, Alex Z. Applications of Deep Learning in Biomedicine. Mol Pharm. 2016; 13:1445-54.
11. Krizhevsky, A, Sutskever, I, Hinton, G. ImageNet classification with deep convolutional neural networks. NIPS’2012.
12. Hinton G, Deng L, Yu D, Dahl G, Mohamed A-R, Jaitly N, Senior A, Vanhoucke V, Nguyen P, Sainath T, Kingsbury B. Deep Neural Networks for Acoustic Modeling in Speech Recognition: The Shared Views of Four Research Groups. IEEE Signal Process Mag. 29:82-97.
13. Huanhuan M, Yue Z. Classification of Electrocardiogram Signals with Deep Belief Networks. 2014 IEEE 17th International Conference on Computational Science and Engineering. IEEE; p. 7-12.
14. Jarrett K, Kavukcuoglu K, Ranzato MA, LeCun Y. What is the best multi-stage architecture for object recognition? 2009 IEEE 12th International Conference on Computer Vision. IEEE; p. 2146-53.
15. Goodfellow IJ, Warde-Farley D, Mirza M, Courville A, Bengio Y. Maxout Networks. arXiv:1302.4389v4 [stat. ML]. 2013.
16. Denton, Emily L., Soumith Chintala, Rob Fergus. Deep Generative Image Models using a Laplacian Pyramid of Adversarial Networks. Adv Neural Inf Process Syst. 2015; : 1486-94.
17. van den Oord Matthias Bethge LTA. A note on the evaluation of the generative models. arXiv:1511.01844v3 [stat.ML]. 2016.
18. Goodfellow, Ian, Jean Pouget-Abadie, Mehdi Mirza, Bing Xu, David Warde-Farley, Sherjil Ozair, Aaron Courville, Yoshua Bengio. Generative adversarial nets. Adv Neural Inf Process Syst. 2014; : 2672-80.
19. Daniel Jiwoong Im, Chris Dongjoo Kim, Hui Jiang, Roland Memisevic. Generating images with recurrent adversarial Networks. arXiv:1602.05110v4 [cs.LG]. 2016.
20. Costello JC, Heiser LM, Georgii E, Gönen M, Menden MP, Wang NJ, Bansal M, Ammad-ud-din M, Hintsanen P, Khan SA, Mpindi J-P, Kallioniemi O, Honkela A, et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol. 2014; 32: 1202-12.
21. Azuaje F. Computational models for predicting drug responses in cancer research. Brief Bioinform. 2016; doi: 10.1093/bib/bbw065.
22. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603-7.
23. Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012; 483:570-5.
24. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006; 6:813-23.
25. Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, Wang J, Yu B, Zhang J, et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016; 44:D1202-13.
26. Wang Y, Suzek T, Zhang J, Wang J, He S, Cheng T, Shoemaker BA, Gindulyte A, Bryant SH. PubChem BioAssay: 2014 update. Nucleic Acids Res. 2013; 42:D1075-82.
27. Lezhnina K, Kovalchuk O, Zhavoronkov AA, Korzinkin MB, Zabolotneva AA, Shegay PV, Sokov DG, Gaifullin NM, Rusakov IG, Aliper AM, Roumiantsev SA, Alekseev BY, Borisov NM, et al. Novel robust biomarkers for human bladder cancer based on activation of intracellular signaling pathways. Oncotarget. 2014; 5:9022-32. doi: 10.18632/oncotarget.2493.
28. Ozerov IV, Lezhnina KV, Izumchenko E, Artemov AV, Medintsev S, Vanhaelen Q, Aliper A, Vijg J, Osipov AN, Labat I, West MD, Buzdin A, Cantor CR, et al. In silico Pathway Activation Network Decomposition Analysis (iPANDA) as a method for biomarker development. Nat Commun. 2016; 7:13427.
29. Zhu Q, Izumchenko E, Aliper AM, Makarev E, Paz K, Buzdin AA, Zhavoronkov AA, Sidransky D. Pathway activation strength is a novel independent prognostic biomarker for cetuximab sensitivity in colorectal cancer patients. Hum Genome Var. 2015; 2:15009.
30. Mitchell JBO. Machine learning methods in chemoinformatics. Wiley Interdiscip Rev Comput Mol Sci. 2014; 4:468-81.
31. Lusci A, Pollastri G, Baldi P. Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. J Chem Inf Model. 2013; 53:1563-75.
32. David Duvenaud, Dougal Maclaurin, Jorge Aguilera-Iparraguirre Rafael Gomez-Bombarelli, Timothy Hirzel, Alan Aspuru-Guzik, Ryan P. Adams. Convolutional Networks on Graphs for Learning Molecular Fingerprints. arXiv:1509.09292v2 [cs.LG]. 2015.
33. Steven Kearnes, Kevin McCloskey, Marc Berndl, Vijay Pande, Patrick Riley. Molecular Graph Convolutions: Moving Beyond Fingerprints. arXiv:1603.00856v3 [stat.ML]. 2016.
34. Gfeller D, Grosdidier A, Wirth M, Daina A, Michielin O, Zoete V. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014; 42:W32-8.
35. Mervin LH, Afzal AM, Drakakis G, Lewis R, Engkvist O, Bender A. Target prediction utilising negative bioactivity data covering large chemical space. J Cheminform. 2015; 7:51.
36. Izharwallach Michael Dzamba. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. arXiv:1510.02855v1 [cs.LG]. 2015.
37. George E. Dahl, Navdeep Jaitly, Ruslan Salakhutdinov. Multi-task Neural Networks for QSAR Predictions. arXiv:1406.1231v1 [stat.ML]. 2014.
38. Thomas Unterthiner, Andreas Mayr, Günter Klambauer, Sepp Hochreiter. Toxicity Prediction using Deep Learning. arXiv:1503.01445v1 [stat.ML]. 2015.
39. Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V. Deep neural nets as a method for quantitative structure-activity relationships. J Chem Inf Model. 2015; 55:263-74.
40. O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011; 3:33.
41. Alireza Makhzani, Jonathon Shlens, Navdeep Jaitly, Ian Goodfellow, Brendan Frey. Adversarial Autoencoders. arXiv:1511.05644v2 [cs.LG]. 2016.
42. Bishop C. Pattern Recognition and Machine Learning. Springer-Verlag New York; 2006.