
Introduction
The treatment of cancer is evolving from toxic, broad chemotherapeutic approaches to therapies targeted towards specific molecular abnormalities that drive tumor growth. In order to continue this evolution, robust and accurate assessment of molecular aberrations within tumors is required to determine which patients are suitable for these targeted therapeutics, and, in turn, which therapies are appropriate in different settings. Generally, molecular testing has been performed using tumor tissue obtained by surgery or biopsy. Adequate tumor (tissue or cytology) samples taken in a suitable form are clinically important for a complete pathological diagnosis including tumor typing and subtyping, and analysis of predictive markers [1]. It can be challenging to obtain sufficient tumor tissue for molecular testing (particularly where biopsy samples are small or are prioritized for disease diagnosis) and invasive biopsy procedures may present with too great a health risk for some patients. Indeed, 27–31%
of patients with non-small-cell lung cancer (NSCLC) may be unable to provide a biopsy sample suitable for epidermal growth factor receptor (EGFR) mutation analysis at diagnosis [2] or following disease progression. Analysis of circulating free tumor-derived DNA (ctDNA) has been proposed as an alternative, minimally invasive method for the detection of EGFR mutations [3, 4].
Genetic mutations in DNA found in the serum of patients with NSCLC were first observed in 1998 [5]. In 2006, the potential clinical value of ctDNA was uncovered when EGFR mutations detected in serum were associated with response to gefitinib in a small, retrospective study [6]. As the link between EGFR mutations in NSCLC and response to EGFR tyrosine kinase inhibitor (EGFR TKI) therapy was further elucidated [7], the clinical utility of ctDNA was more robustly investigated via preplanned analyses in large-scale studies. The presence of EGFR mutations in ctDNA was shown to predict response to the EGFR TKIs gefitinib and erlotinib in the first-line setting [8, 9]. Moreover, objective response rates and progression-free survival were found to be similar in patients with EGFR mutations detected in their ctDNA and in those who were identified as EGFR mutation-positive in their tumor sample [10]. Detection of resistance mutations such as T790M and C797S in the plasma of patients with NSCLC who progress on EGFR TKIs also has promising utility in identifying which patients are suitable for subsequent therapies, both following initial diagnosis and following disease progression with first-line TKI therapy [11-14].
The process by which tumor DNA enters the bloodstream is not fully understood. Diehl et al reported a correlation between the amount of mutant ctDNA in colorectal cancer and the invasiveness of the tumor [15]. This finding, along with the highly fragmented nature of ctDNA, led the authors to propose that the DNA came from necrotic neoplastic cells that had been phagocytized by macrophages [15]. It has also been proposed that ctDNA is released by apoptotic cells [16, 17], based on the observation that ctDNA forms a ladder pattern during electrophoresis, which is reminiscent of that produced by apoptosis [18]. Another possible source of ctDNA is the breakdown of circulating tumor cells; however, in patients with a range of malignancies (excluding lung), levels of ctDNA have been observed to be higher than that of circulating tumor cells, suggesting that these cells are not the source of ctDNA [19]. Finally, it has been suggested that tumor cells may actively secrete DNA fragments, as has been observed for lymphocytes and seen recently in patients with NSCLC via extracellular vesicles (exosomes) [20, 21].
Meta-analyses have indicated that detection of EGFR mutations using ctDNA may be effective for diagnostic purposes [22-24]. However, results from individual studies are variable, with many indicating that detection of EGFR mutations in ctDNA is more difficult in plasma samples than in tumor tissue, with an average sensitivity of 65–70% (see Table 1 for definition of concordance parameters) [25]. Indeed, ctDNA is frequently highly diluted and, therefore, highly sensitive techniques are required in order to detect somatic mutations in blood samples. Recent real-world studies [26, 27] have shown that the performance of ctDNA testing varies significantly between different geographic regions and different laboratories, demonstrating the need for standardized and well-validated ctDNA methodology. With recommendations based upon methods for ctDNA mutation analysis that have been employed in clinical trials, in this review, we look at the key steps required to ensure accurate ctDNA test results and propose options to optimize sensitivity.
Table 1: Definitions of specificity, sensitivity, positive predictive value, and negative predictive value
True positive |
Patient’s tumor carries the mutation and patient tests positive for the mutation |
True negative |
Patient’s tumor does not carry the mutation and patient tests negative for the mutation |
False positive |
Patient’s tumor does not have the mutation, yet tests positive for the mutation |
False negative |
Patient’s tumor carries the mutation, yet tests negative for the mutation |
Sensitivity |
True positive / (true positive + false negative) |
Specificity |
True negative / (true negative + false positive) |
Positive predictive value |
True positive / (true positive + false positive) |
Negative predictive value |
True negative / (true negative + false negative) |
Pre-analytical factors
Plasma versus serum
In order to measure any biomarker, optimization of sample processing, handling, and storage is needed to ensure that the biomarker is neither degraded nor masked by other constituents of the sample. For example, it is important to reduce contamination of ctDNA samples by wild-type DNA originating from circulating leukocytes, which has the potential to mask the small (low ng/mL) quantities of tumor DNA in the sample. For this reason, plasma is preferable to serum because the clotting process in serum leads to the release of genomic DNA from white blood cells (reviewed by El Messaoudi et al 2013 [28]). Furthermore, a head-to-head comparison of analysis of plasma and serum ctDNA samples from the same patients demonstrated higher sensitivity with plasma versus serum [29], thus confirming the assertion that the use of plasma is preferable for EGFR mutation analysis of ctDNA. Common anticoagulants such as ethylenediaminetetraacetic acid (EDTA) and citrate are both suitable for processing of blood samples for ctDNA analysis [30], but it is recommended that heparin should be avoided since it may interfere with downstream polymerase chain reaction (PCR) applications [31].
Plasma processing and storage
Aside from the EGFR mutation testing method selected for analysis of ctDNA samples, the quality of the test is also dependent on the laboratory successfully completing the pre-analytical steps. The recommended workflow for plasma collection, processing, and storage is summarized in Figure 1. The timing from blood draw to plasma isolation is crucial: blood should be processed within 4 hours of drawing, as an increase in total DNA (indicative of white cell degradation) occurs if blood remains unprocessed for longer periods. There is no difference in DNA yield between samples stored at room temperature or at 4°C within this 4-hour time frame [28]. If processing within 4 hours of blood draw is possible, then EDTA tubes should be used. In clinical practice however, processing an individual blood sample to obtain plasma within a 4-hour time frame may be challenging. An alternative option is to stabilize blood in the collection tube, using specific fixatives that allow the blood to be processed later at a more convenient time. Several commercial options are available (eg cell-free DNA BCT® [Streck, Omaha, NE, USA] and cfDTM [Ariosa Diagnostics, San Jose, CA, USA]), which claim to stabilize blood for up to 7 days without compromising downstream DNA analysis. A comparison of ctDNA testing in samples from patients with advanced breast cancer (for mutations in four known drivers in breast cancer: PIK3CA exon 9 and 20, ESR1 ligand binding domain, AKT1, and ERBB2) showed no differences in samples taken in Streck cell-free DNA BCT® tubes that were processed after 48–72 hours compared with samples that were processed immediately [32]. Streck cell-free DNA BCT® tubes have also been shown to prevent lysis and cellular release of genomic DNA from ctDNA samples of patients with metastatic breast cancer, compared with PAXgene tubes (PreAnalytiX, Feldbachstrasse, Hombrechtikon, Switzerland) [33]. Furthermore, blood samples from patients with melanoma stored in Streck cell-free DNA BCT® tubes at room temperature were found to remain serviceable for ctDNA testing, even after long-term storage (up to 10 days at room temperature) [34].
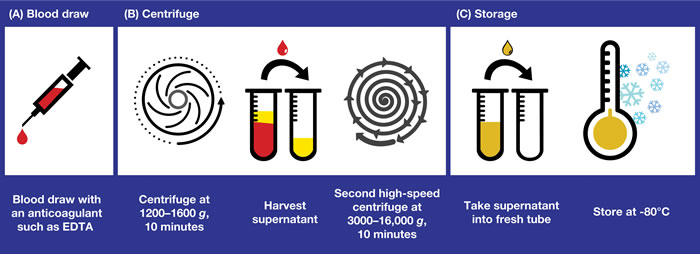
Figure 1: Obtaining plasma samples for ctDNA analysis: recommended workflow for plasma collection, processing, and storage. The key pre-analytical steps involved in collecting plasma samples that are suitable for ctDNA analysis are shown. A. The timing from blood draw to plasma isolation is crucial for success of the test. EDTA tubes should be used only if the time from blood draw to delivery of the sample to the testing laboratory is within 4 hours. Alternatively, tubes containing specific fixatives that prevent the lysis of white blood cells should be used. B. Plasma is obtained by centrifugation of the blood sample at 1200–1600 g for 10 minutes and harvesting of the supernatant. A second, higher-speed centrifugation (eg 3000–16,000 g) in a microcentrifuge is recommended to remove residual cellular contamination and to generate a clean sample for further analysis. This second centrifuge may be performed either before or after freeze/thaw. C. Fresh plasma should be stored at -80°C in the long term (-20°C acceptable for ~1 month) and shipping, if required, should be on dry ice. Repeated freeze/thaw should be avoided. ctDNA, circulating free tumor-derived DNA; EDTA, ethylenediaminetetraacetic acid.
Plasma is obtained by centrifugation of the blood sample at 1200–1600 g for (typically) 10 minutes and harvesting of the supernatant. This process does not remove all cellular contamination; therefore, a second, higher-speed centrifugation (eg 3000–16,000 g) in a microcentrifuge or filtration through a 0.2 µM filter is recommended to remove residual cellular contamination and to generate a clean sample for further analysis [35]. This second spin can be performed either before or after the freeze/thaw [36]. Fresh plasma should be stored at -80°C in the long-term (-20°C is acceptable for ~1 month), and shipping, if required, should be in dry ice to avoid sample degradation. Repeated freeze/thaw should be avoided, although 2–3 cycles is considered acceptable [28]. Long-term stability of DNA in plasma is best demonstrated at -80°C, a temperature at which DNA may be stable for up to 9 months [28]; however, median plasma DNA concentrations are at risk of significantly decreasing over time [37].
Plasma processing as outlined above is relatively straightforward. However, stabilization tubes, although more expensive, could be used in regions where infrastructure is poor or to support testing when samples cannot be processed locally, and instead need to be shipped to external expert laboratories for analysis.
DNA extraction
Many methods exist for DNA extraction, including individual laboratory protocols, company in-house procedures, and several commercial kits. However, there is a lack of consensus on optimal ctDNA extraction methods, with guideline recommendations tending to focus on tissue/cytology samples for EGFR mutation testing [38]. It should not be assumed that extraction methods validated for tumor tissue or other matrices are suitable for ctDNA, which is fragmented and present at such low concentrations that specialized extraction protocols are required. Using the wrong extraction method can significantly reduce the ability to detect mutations in ctDNA. Although a number of kits for ctDNA extraction from different commercial sources are available, few comparative data are available. One study compared the ability of three methods specifically designed for extraction of ctDNA (QIAamp® circulating nucleic acid kit [QIAGEN, Manchester, UK], polymer mediated enrichment (PME) free-circulating DNA extraction kit [Analytik Jena, Jena, Germany], and DSP virus/pathogen midi kit performed on QIAsymphony® [QIAGEN, Hilden, Germany]) to detect KRAS mutations in plasma samples from patients with NSCLC (n = 10). All methods demonstrated successful DNA extraction, although the DNA yields differed significantly. As was found in earlier research [39], the different yields, (measured by quantitative PCR [qPCR]), may have been due to isolation of fragments of differing size distributions, in turn due to the varying nucleic acid size exclusion limits in a given kit [40]. In the large, multicenter, non-interventional, diagnostic ASSESS study – which investigated the utility of ctDNA for EGFR mutation testing in a real-world diagnostic setting (EU/Japan) – an initial plasma sensitivity of only 17% was seen in a subset of 92 patients using the QIAamp® MinElute® virus spin kit for DNA extraction (QIAGEN, Manchester, UK) and a peptide nucleic acid (PNA) clamp-based detection method. However, this DNA extraction kit can isolate high molecular weight DNA, but not fragmented DNA. Indeed, sensitivity increased to 51% when the same PNA clamp-based detection method was used on DNA extracted from a higher volume of plasma using the QIAamp® circulating nucleic acid kit, which has been designed to isolate fragmented DNA; this highlights the importance of using a method specifically developed for ctDNA [27, 41]. The QIAamp® circulating nucleic acid kit was used in the IFUM study, in which data from ctDNA analysis were associated with response to gefitinib [10]. Other kits may also be adequate for ctDNA extraction, but we are not currently aware of extensive data supporting their use for this purpose.
Since it is likely that only a limited number of DNA copies in plasma will have arisen from a tumor, the need for a large volume of plasma is important: 2 mL has been shown to yield accurate results using various methods [29], advocating this as an appropriate volume for use in clinical practice.
MUTATION DETECTION METHODS
ctDNA is present in blood at low levels amidst a significant background of wild-type DNA; therefore, a sensitive mutation detection method with the ability to detect mutant alleles that comprise < 1% of the total DNA is required. However, highly sensitive assays are at risk of yielding false-positive results (Table 1) unless robustly validated, and so a trade-off must occur between the sensitivity and specificity of a chosen assay. Clinically, it may be better to favor an assay with high specificity, at the slight expense of sensitivity, in order to ensure that targeted therapies are administered appropriately [42, 43] (see Table 2 [8,9,27,29,43-60] for sensitivity and specificity of selected assays). The quality of EGFR mutation testing methods may only be warranted by participating in External Quality Assessment (EQA) schemes. EQA schemes for liquid biopsy for EGFR mutations are not currently available (at time of writing), but have been announced for 2017.
Table 2: Specificity and sensitivity of selected assays
Assay |
Sensitivity, n/N (%) |
Specificity, n/N (%) |
Reference (no. of matched tissue/cytology and plasma samples) |
||
EGFR sensitizing mutations* |
T790M mutations (after EGFR TKI progression) |
EGFR sensitizing mutations* |
T790M mutations (after EGFR TKI progression) |
||
BEAMing |
186/244 (76.2) |
N/A (95.8) |
IMPRESS study [44] |
||
49/60 (81.7) |
33/45 (73.3) |
2/3 (66.7) |
9/18 (50) |
Karlovich et al 2016 [45] |
|
43/51 (84.3) |
33/41 (80.5) |
65/67 (97.0) |
14/24 (58.3) |
Thress et al 2015 [43] |
|
Cycleave® |
29/57 (50.9) |
132/133 (99.2) |
ASSESS study [46] |
||
ddPCR |
30/37 (81.1) |
N/A (97.0) |
Zhu et al 2015 [47] |
||
DHPLC |
188/269 (69.9) |
445/553 (80.5) |
Huang et al 2012 [48] |
||
High-resolution melting |
29/45 (66.4) |
73/75 (97.3) |
Jing et al 2013 [49] |
||
Mass spectrometry genotyping |
8/18 (44.4) |
11/13 (84.6) |
Brevet et al 2011 [50] |
||
Mutation-based PCR-quenching probe Exon 19 L858R |
2/23 (8.7) |
23/23 (100) 47/47 (100) |
Nakamura et al 2012 [51] |
||
Mutant-enriched PCR |
7/18 (38.9) |
11/13 (84.6) |
Brevet et al 2011 [50] |
||
16/45 (35.6) |
63/66 (95.5) |
Zhao et al 2013 [52] |
|||
NGS-based deep sequencing Exon 19 L858R |
N/A (50.9) N/A (51.9) |
N/A (98.0) N/A (94.1) |
Uchida et al 2015 [53] |
||
L858R |
N/A (70.6) |
N/A |
Yao et al 2016 [54] (n=39) |
||
PNA-LNA PCR clamp |
15/29 (51.7) |
61/62 (98.4) |
ASSESS study [46] |
||
16/26 (61.5) |
70/70 (100) |
Pasquale et al 2015 [55] |
|||
97/164 (59.1) |
N/A |
Rosell et al 2009 [56] |
|||
58/109 (53.2) |
N/A |
Rosell et al 2012 [9] |
|||
PNA-PCR clamp with AS-APEX assay |
32/32 (100) |
4/5 (80.0) |
Yam et al 2012 [57] |
||
SARMS |
22/51 (43.1) |
35/35 (100) |
IPASS study [58] |
||
27/40 (67.5) |
46/46 (100) |
Liu et al 2013 [59] |
|||
SARMS-based QIAGEN therascreen® EGFR RGQ PCR kit |
16/22 (72.7) |
115/116 (99.1) |
ASSESS study [27] |
||
69/105 (65.7) |
546/547 (99.8) |
IFUM study [8] |
|||
17/26 (65.4) |
70/70 (100) |
Pasquale et al 2015 [55] |
|||
18/19 (94.7) |
66/66 (100) |
Vallée et al 2013 [29] |
|||
Roche cobas® AS-PCR |
43/51 (84.3) |
30/41 (73.2) |
65/67 (97.0) |
16/24 (66.7) |
Thress et al 2015 [43] |
17/28 (60.7) |
162/168 (96.4) |
Weber et al 2014 [60] |
|||
55/75 (73.3) |
21/33 (63.6) |
24/24 (100) |
61/62 (98.4) |
Karlovich et al 2016 [45] (n=55) |
|
*Exon 19 deletion/L858R mutation.
AS, allele-specific; BEAMing, beads, emulsions, amplification, and magnetics; ddPCR, digital droplet polymerase chain reaction; DHPLC, denaturing high-pressure liquid chromatography; EGFR, epidermal growth factor receptor; N/A, not available; NGS, next-generation sequencing; PCR, polymerase chain reaction; PNA-LNA, peptide nucleic acid-locked nucleic acid; RGQ, Rotor-Gene Q; SARMS, Scorpion amplified-refractory mutation system; TKI, tyrosine kinase inhibitor.
PCR-based methods
Traditional methods such as Sanger sequencing (which are unable to detect mutant DNA present at levels < 10–20% of total DNA) or pyrosequencing, which has a sensitivity of approximately 5%, are not suitable for ctDNA mutation analysis because they can lead to a high proportion of false negatives (Table 1) [61]. To date, most ctDNA data have been generated with allele-specific qPCR-based methods adapted to enrich for mutant DNA; these methods are generally able to detect 1–5% of mutant DNA. Adaptations to improve sensitivity include the use of mutation-specific primers. For example, the amplified-refractory mutation system (ARMS) [62] combined with Scorpion molecules (SARMS) [62, 63] that link specific primers with fluorescent probes, selectively amplifies only DNA containing the target allele. SARMS detection methods targeted to EGFR mutations are used in two commercial assays (QIAGEN therascreen® EGFR Rotor-Gene Q [RGQ] PCR kit [QIAGEN, Manchester, UK] and the Roche cobas® EGFR mutation test v2 [Roche Molecular Systems, Inc., CA, USA]) that were approved by the US Food and Drug Administration (FDA) in 2013 as companion diagnostics to select patients suitable for EGFR TKI therapy [64]. In June 2016, the FDA approved the use of the Roche cobas® EGFR mutation test v2 with plasma samples. Silencing of non-mutant DNA amplification using non-amplifiable PNA sequences, a synthetic DNA analog [65], directed against wild-type sequences (PNA clamping) has also been used to improve sensitivity. Such methods for ctDNA mutation analysis have demonstrated sensitivity relative to tumor averaging 65% [25]. Importantly, many of these methods have been shown to have high specificity ( > 88%), such that a positive result in plasma is highly likely to be linked to a positive result in the tumor [10, 25]. For example, a head-to-head comparison of the ability of the QIAGEN therascreen® EGFR RGQ PCR kit versus PNA clamping to detect EGFR mutations in 96 patients with NSCLC revealed that specificity (100% for both) and sensitivity (65% for therascreen® and 62% for PNA clamping) were high, and similar between assays [55]. In-house, laboratory-developed test solutions have also shown similar performance metrics and can be used, provided the appropriate level of performance has been demonstrated during assay validation.
Newer technologies with even greater sensitivity than existing assays may provide a means of reducing the false-negative rates observed with qPCR-based ctDNA testing methods. Methods based upon emulsion PCR, such as digital droplet PCR (ddPCR) and BEAMing [66] (Sysmex Inostics, Chou-Ku, Kobe, Japan) are enhancements of traditional PCR. These methodologies partition the sample into individual DNA fragments that are amplified in microdroplets, allowing scientists to detect mutant DNA even if only a few copies are present amongst a majority of wild-type DNA sequences. Such technologies have an analytical threshold as low as 0.001% of mutant DNA in a background of wild-type DNA [67]. However, as it becomes possible to detect extremely low levels of specific mutations, signals may be detected from small, subclinical clones that may not be relevant to treatment decisions at the time the sample is analyzed. This raises the question of whether the level of mutant DNA identified in the blood reflects the presence of a specific driver mutation within the primary tumor at a particular time. Whereas the threshold of detection of mutant DNA in liquid biopsy will lower with improvements in technology, it will not be easy to establish the clinical correlation between low levels of mutant DNA in plasma and the probability of response to targeted agents – further research is required to establish this. The fraction of mutant DNA in plasma depends on several variables, including: disease burden; the levels of expression of the mutation in the primary tumor; the rate of shedding of tumor DNA into the bloodstream; and the levels of DNA derived from non-transformed cells (inflammation of normal tissue surrounding the tumor, lysis of leukocytes after blood drawing etc). Such considerations are particularly relevant for detection of T790M, the most common resistance mechanism for first-line EGFR TKIs [68]. T790M mutations have sometimes been detected well in advance of any clinical progression using sensitive methods [11, 12]. However, determining when emergence of T790M mutations should be considered as indicators of ineffective TKI therapy, versus symptomatic progression, still remains to be resolved. Nevertheless, detection of T790M mutation at clinical progression of the disease using highly sensitive methods will enable patients to access a highly effective treatment.
While some PCR-based detection methods do not allow quantification of mutated gene copies, methods such as ddPCR and BEAMing enable absolute quantification of mutant EGFR levels in plasma and could be used for monitoring treatment response, disease progression, and early treatment failure associated with acquired drug resistance [69]. Robust data that link EGFR mutation levels detected in ctDNA to clinical endpoints are required to clarify what level of sensitivity is clinically relevant. With cut-offs set at 0.02% of mutant DNA, sensitivity of up to 86% and specificity of 98% versus tumor-based detection has been demonstrated for common activating EGFR mutations using the BEAMing platform [44, 70]. It is interesting to note that even these highly sensitive methods do not appear to detect EGFR mutations in the blood of every patient with a mutation in their primary tumor, indicating that there is a proportion of patients with NSCLC whose tumors do not shed sufficient amounts of DNA into the blood for molecular testing [32, 71]. It is likely that the most important factor when defining thresholds of sensitivity for technologies is the number of mutated ctDNA copies per volume of blood or plasma. Finally, it is important to underscore that different results were obtained by Thress et al when testing for the presence of T790M mutations in the plasma of patients experiencing disease progression following prior EGFR TKI therapy using the BEAMing and Roche cobas® platforms [43]. These methods yielded a sensitivity of 73% and 81%, respectively, for the T790M mutation, although the corresponding specificities were lower at 67% and 58%, respectively. Of note, among 72 cases evaluated, 9 “false-positive” plasma results were detected by BEAMing, of which 7 tested positive using the Roche cobas® test. Interestingly, the response rate to the “third-generation” TKI osimertinib (TAGRISSOTM [AZD9291]) in patients with T790M-positive plasma samples (59%), was almost identical to that in patients with T790M-positive tissue samples (61%). Similar results were more recently obtained by Oxnard et al, in a large cohort of patients treated with osimertinib who underwent plasma EGFR mutation testing via BEAMing [70]. Altogether, these data suggest that plasma T790M mutation testing can be used to confirm whether the mutation is actually present in those patients whose tissue biopsy samples test negative for the mutation (likely due to heterogeneous disease), yet may actually benefit from TKI treatment targeting this resistance mutation.
In addition to the PCR-based method selected, it is the responsibility of the testing laboratory to implement basic measures to prevent contamination that may lead to false-positive results. The PCR reaction should be prepared in a laminar flow hood, with the pre- and post-PCR areas physically separated. Every PCR reaction must include the appropriate positive and negative controls, and it is recommended that negative samples are regularly (monthly, if possible) run in the laboratory from DNA extraction to PCR analysis, in order to ensure that there are no steps in the process in which contamination may occur.
Next-generation sequencing and post-PCR methods
Next-generation sequencing (NGS) (otherwise known as massive parallel sequencing) describes a large group of technologies that can sequence millions of small DNA fragments in parallel. NGS shares some similarities with ddPCR, in that individual DNA fragments are isolated prior to sequencing, enabling detection of a small fraction of a mutated sequence. As such, NGS is a highly sensitive ctDNA solution providing a relative quantification (allelic frequency); with an accuracy comparable to that of ddPCR for ctDNA detection (99% accuracy) [72]. Of note, NGS techniques that include labelling of ctDNA fragments with barcode sequences (discriminating between reads of individual molecules to allow grouping of reads from each) have demonstrated near-complete elimination of false-positive results that can be applied to EGFR mutation analysis [73]. To date, limited ctDNA data are available on NGS platforms in NSCLC. A proof-of-concept study, on the use of NGS to screen ctDNA for a range of tumor biomarkers, found that between 68 matched tumor and ctDNA samples of patients with newly diagnosed metastatic NSCLC, overall mutation status concordance (including BRAF, EGFR, ERBB2, KRAS, and PI3KCA amplicons) was 68%, with a sensitivity of 58% and specificity of 87% [74]. Uchida et al conducted a prospective study to evaluate biopsy samples from 288 treatment-naïve patients with NSCLC using an NGS EGFR mutation detection system, utilizing genotyping results from biopsy samples as a comparator. Diagnostic sensitivity and specificity results were as follows: exon 19 deletion, 50.9% and 98.0%, respectively; L858R mutation, 51.9% and 94.1%, respectively. Uchida et al concluded that NGS “deep sequencing” of plasma DNA can be used for genotyping of EGFR in lung cancer patients, with the high specificity results in particular indicating that the system could enable a direct EGFR TKI recommendation on the basis of plasma-positive results [53]. In another study that aimed to detect sensitizing and resistance EGFR mutations in NSCLC using NGS of plasma ctDNA samples, results from the first five patients tested (from a cohort of 31 who had a repeat biopsy) indicated that multiplexed NGS is feasible and shows a high concordance with results from tumor biopsy [75]. Thress et al also utilized NGS to analyze ctDNA from seven patients with NSCLC who had developed resistance to osimertinib, and had an acquired EGFR C797S mutation detected in one sample [14]. A commercial laboratory based in the USA has used NGS to screen for a panel of mutations in the ctDNA of 1000 patients with cancer (including lung cancer), and detected ctDNA in 78% of these patients (of these, 74% had an actionable genetic aberration) [76, 77]. More recently, Rachiglio et al performed NGS analysis of ctDNA sampled from 44 patients with metastatic NSCLC. EGFR mutations were detected in 17/22 samples of patients with EGFR mutation-positive tumors (sensitivity 77%), as well as in samples of two patients whose tumors were not known to harbor EGFR mutations (both confirmed via ddPCR in plasma and tissue samples) [78]. Villaflor et al evaluated a targeted ctDNA NGS gene panel in 31 patients with NSCLC who provided matching tissue and blood samples: of 7 samples with an activating EGFR mutation detected in either tissue or blood, a concordance rate of 71% was calculated [79]. Yao et al performed targeted DNA sequencing to screen for driver gene mutations in matched ctDNA and tissue samples from 39 Chinese patients with NSCLC; the overall gene mutation concordance was 78.2% between matched samples, with sensitivity of EGFR, KRAS, PIK3CA mutations, and gene rearrangements detected in ctDNA at 70.6%, 75%, 50%, and 60%, respectively [54]. Chabon et al employed NGS (CAPP-Seq) to study resistance mechanisms of ctDNA samples of 43 patients with NSCLC treated with rocelitinib. In 28 patients, one or more putative resistance mechanisms were identified (of which the most common was MET copy-number gain as observed in 11 patients) [80].
The ability of NGS to analyze several genes in parallel has the potential to allow a single plasma sample to be used to determine eligibility for different specific therapies.
Methods employed after PCR amplification that have less commonly been used for EGFR mutation analysis in ctDNA samples of patients with NSCLC, and so are not discussed in detail here, include denaturing high-pressure liquid chromatography, mass spectrometry, and high-resolution melting analysis (reviewed by Huang et al [3]).
Table 3: Key considerations for successful detection of EGFR mutations in ctDNA of patients with advanced NSCLC
Pre-analytical factors |
|
Plasma versus serum |
Plasma (vs serum) should be used for ctDNA mutation analysis |
Plasma processing and storage |
• Blood should be processed to plasma within 4 hours of draw; alternatively, use of stabilization collection tubes containing fixatives should be considered to allow blood processing at a later time • Plasma is obtained via centrifugation of the blood sample; a second, high-speed spin (before or after freeze/thaw [3000‒16,000 g]) in a microcentrifuge is necessary to generate clean samples for mutation analysis • Fresh plasma should be stored at -20°C or -80°C (on dry ice for shipping), with long-term stability of DNA in plasma best demonstrated at -80°C |
DNA extraction |
• Use of DNA extraction methods specifically developed for ctDNA – which is fragmented and only present at low concentrations – is essential |
Mutation detections methods |
|
• Traditional methods (Sanger, pyrosequencing) are not suitable for ctDNA mutation analysis due to low sensitivity • PCR methods that increase the proportion of mutant to wild-type DNA (mutant enriched-PCR, SARMS, PNA clamping) provide greater sensitivity than traditional sequencing methods • Enhancements of traditional PCR (ddPCR, BEAMing) demonstrate increased sensitivity ddPCR and NGS enable quantification of mutant EGFR levels and may be used to monitor treatment response and disease progression • Alternative methods (mass spectrometry genotyping, DHPLC, and high-resolution melting) represent potentially efficient and reliable methods for routine diagnostic use |
|
BEAMing, beads, emulsions, amplification, and magnetics; ctDNA, circulating free tumor-derived DNA; ddPCR, digital droplet polymerase chain reaction; EGFR, epidermal growth factor receptor; DHPLC, denaturing high-pressure liquid chromatography; NGS, next-generation sequencing; NSCLC, non-small-cell lung cancer; PCR, polymerase chain reaction; SARMS, Scorpion amplified-refractory mutation system.
Conclusions and recommendations
Molecular testing for actionable mutation status information, including use of targeted treatments, has become routine practice across the field of clinical oncology [81]. Use of ctDNA to detect activating mutations provides a promising diagnostic approach if tumor tissue is not accessible. Another advantage of ctDNA is that mutant DNA from all tumors within a patient (including metastases) can be sampled, which may reduce the risk of missing a mutation due to tumor heterogeneity or sampling issues. The ASSESS study suggested that ctDNA use could detect EGFR mutations that may have been missed in the tumor due to use of cytology and small biopsies that can be problematic for EGFR analysis [27]. Of interest, additional analyses of ASSESS study data found that detection of EGFR mutations in plasma was significantly more likely when patients had a higher metastatic tumor burden and distant metastases [82]. In this respect, in patients with progressive disease the liquid biopsy may change to become positive over time with the increase in tumor burden. However, current evidence suggests that, despite using the latest, highly sensitive technologies, ctDNA does not allow detection of the common EGFR activating mutations in all patients with NSCLC and mutations in their tumors. For this reason, tumor tissue, where available, should be the first sample of choice for EGFR testing to provide an initial molecular diagnosis, to determine if patients are suitable for TKI therapy. However, in some cases, tissue test results are not evaluable (for example, due to low tumor cell count or degraded DNA) or tissue is not available [46, 83, 84]. ctDNA provides a viable alternative in this setting (Figure 2A). A recent clinical study of patients with NSCLC whose tissue samples could not be evaluated in mutation testing (n = 3) found that upon EGFR mutation detection in ctDNA samples, all presented with significant partial response to EGFR TKI therapy [85]. Interestingly, emerging data are demonstrating the feasibility of detecting EGFR mutations in ctDNA from urine samples, potentially providing a completely non-invasive alternative sample type in the absence of the preferred tumor tissue [86-88]. Furthermore, cerebrospinal fluid is also under investigation as a source of ctDNA for mutation analysis [89-92]; notably, EGFR mutations have been detected in ctDNA sampled from the cerebrospinal fluid of patients with lung adenocarcinoma and brain metastases [93]. Finally, the sensitivity of liquid biopsy could be increased by coupling the analysis of ctDNA and circulating tumor cells (CTCs) [94]. However, methods for CTC detection are complex, costly, and currently limited to the research setting.
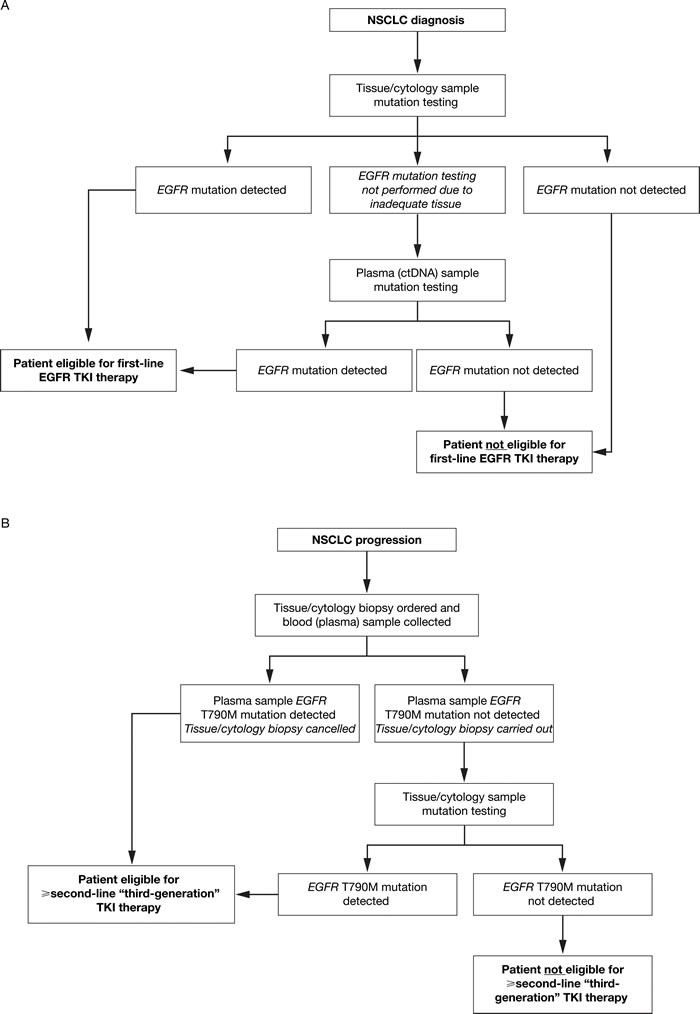
Figure 2: ctDNA mutation testing: recommended use to inform treatment selection at A. NSCLC diagnosis and B. NSCLC progression following first-line TKI therapy. At diagnosis of NSCLC A., plasma (ctDNA) sample testing is recommended when it is not possible to obtain adequate or suitable tissue at biopsy. In patients who have progressed following first-line TKI therapy B., plasma (ctDNA) and tissue/cytology sample testing is recommended to determine whether the T790M mutation is present, which informs eligibility for subsequent TKI therapy. ctDNA, circulating free tumor-derived DNA; EGFR, epidermal growth factor receptor; NSCLC, non-small-cell lung cancer; TKI, tyrosine kinase inhibitor.
When patients progress on first-line EGFR TKI therapy, and a new biopsy would otherwise be required to determine the molecular mechanism (eg T790M mutations) underlying the disease progression, ctDNA may have advantages over tissue. This is a particularly relevant subject, with a TKI targeting resistance mutations such as T790M [95, 96] recently approved in the USA, EU, and Japan (at time of writing). In fact, increasing evidence suggests that tumors are more heterogeneous following TKI therapy than at initial diagnosis [97]. In heterogeneous tumors, ctDNA samples may provide a better way to determine overall tumor mutation status than a small biopsy, which in most cases represents the available tissue sample at disease progression [94, 98]. Since ctDNA is much less invasive than a tissue biopsy, ctDNA may become the preferred option in this setting. However, due to the limited sensitivity of liquid biopsy, a new tissue sample should be obtained if the plasma test is negative for EGFR mutations (Figure 2B). Another advantage of using ctDNA is that it allows repeated sampling (monitoring) over time [99-104], with a faster mutation test turnaround time (TAT) compared with tissue [105] – something that could not be achieved using tumor biopsies. TAT is a relevant issue as it will directly impact upon the possibility of the patient to receive a targeted therapy. TAT may be influenced by several variables: the organization of the testing workflow; communication within the multidisciplinary team; the level of automation in the testing laboratory; and the mutation testing method employed. In our clinical experience, TAT tends to be quite short (3–4 days) for SARMS detection methods such as the QIAGEN therascreen® EGFR RGQ PCR kit and the Roche cobas® EGFR mutation test v2, and longer in more complex methods such as BEAMing (7–8 working days). Even greater TAT may be observed with NGS, perhaps between 10 and 14 working days, with bioinformatics needed for interpretation of the results.
Such studies can yield interesting information about how tumors evolve in response to treatment [11, 12, 106]; however, care must be taken not to over-interpret such findings. With the emergence of highly sensitive methods such as NGS and ddPCR, resistance mutations have been observed far in advance of clinical signs or radiographic progression. For T790M analysis, we recommend that ctDNA analysis should be performed at the same time as a tissue biopsy is ordered if available/feasible as each provides complementary information, and that monitoring should be restricted to clinical trials. Research should look to address remaining questions as to whether blood samples should be drawn at specific times of day, potentially based upon periods of fasting or exercise for example [107].
In this review, we have outlined the critical steps for obtaining an accurate result using ctDNA, including recommendations regarding standardization of plasma processing and handling, DNA extraction, and appropriate sensitive detection methods. In expert hands, good results can be obtained. In the real world, sensitivity and specificity have been observed to be lower than that achieved in centrally controlled clinical studies [41, 108], highlighting that standardization and training are vital to ensuring high-quality ctDNA testing in the clinic. Implementation of quality assurance schemes will be important to increase confidence in the quality of ctDNA testing outside of specialist laboratories. However, if well implemented, ctDNA has the potential to allow a great number of patients to benefit from targeted therapies.
Abbreviations
AKT1, v-akt murine thymoma viral oncogene homolog 1; ARMS, amplified-refractory mutation system; AS, allele-specific; BRAF, B-Raf proto-oncogene, serine/threonine kinase; ctDNA, circulating free tumor-derived DNA; ddPCR, digital droplet polymerase chain reaction; EDTA; ethylenediaminetetraacetic acid; EGFR, epidermal growth factor receptor; ERBB2, erb-b2 receptor tyrosine kinase 2; ESR1, estrogen receptor 1; EQA, External Quality Assessment; FDA, US Food and Drug Administration; KRAS, Kirsten rat sarcoma viral oncogene homolog; LNA, locked nucleic acid; N/A, not available; NGS, next-generation sequencing; NSCLC, non-small-cell lung cancer; PCR, polymerase chain reaction; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PME, polymer mediated enrichment; PNA, peptide nucleic acid; qPCR, quantitative polymerase chain reaction; RGQ, Rotor-Gene Q; SARMS, Scorpion amplified-refractory mutation system; TAT, turnaround time; TKI, tyrosine kinase inhibitor.
Acknowledgments
We thank Complete Medical Communications, who provided editorial support funded by AstraZeneca.
N. Normanno is supported by a grant from the Associazione Italiana Ricerca sul Cancro (AIRC; grant n. IG17135).
Conflicts of interests
N Normanno – grants/research support/consultant: AstraZeneca, Qiagen, and Roche Diagnostics.
MG Denis – grants/research support/consultant: AstraZeneca, Qiagen, Roche Pharma, and Boehringer Ingelheim.
M Reck – speakers’ bureau: F. Hoffmann-La Roche,
Lilly, BMS, AstraZeneca, Pfizer, Daiichi-Sankyo, and Boehringer Ingelheim; consultant: F. Hoffmann-La Roche,
Lilly, BMS, AstraZeneca, Pfizer, Daiichi-Sankyo, Boehringer Ingelheim, and MSD.
KS Thress and M Ratcliffe are employees of AstraZeneca and hold shares in AstraZeneca.
References
1. National Institute for Health and Care Excellence. Diagnosis and staging of lung cancer. Available at: http://pathways.nice.org.uk/pathways/lung-cancer#path = view%3A/pathways/lung-cancer/diagnosis-and-staging-of-lung-cancer.xml&content = view-quality-statement%3Aquality-statements-tissue-diagnosis. Last updated 2016. Accessed 4 July 2016.
2. Royal College of Physicians. National Lung Cancer Audit annual report 2015 (for the audit period 2014). Available at: https://www.rcplondon.ac.uk/projects/outputs/nlca-annual-report-2015. Last updated 2 December 2015. Accessed 4 July 2016.
3. Huang WL, Wei F, Wong DT, Lin CC and Su WC. The emergent landscape of detecting EGFR mutations using circulating tumor DNA in lung cancer. Biomed Res Int. 2015; 2015: 340732.
4. Sun W, Yuan X, Tian Y, Wu H, Xu H, Hu G and Wu K. Non-invasive approaches to monitor EGFR-TKI treatment in non-small-cell lung cancer. J Hematol Oncol. 2015; 8: 95.
5. Sánchez-Céspedes M, Monzó M, Rosell R, Pifarré A, Calvo R, López-Cabrerizo MP and Astudillo J. Detection of chromosome 3p alterations in serum DNA of non-small-cell lung cancer patients. Ann Oncol. 1998; 9: 113-116.
6. Kimura H, Fujiwara Y, Sone T, Kunitoh H, Tamura T, Kasahara K and Nishio K. High sensitivity detection of epidermal growth factor receptor mutations in the pleural effusion of non-small cell lung cancer patients. Cancer Sci. 2006; 97: 642-648.
7. Mok TS, Wu Y-L, Thongprasert S, Yang C-H, Chu D-T, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang J-J et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009; 361: 947-957.
8. Douillard J-Y, Ostoros G, Cobo M, Ciuleanu T, McCormack R, Webster A and Milenkova T. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open label, single arm study. Br J Cancer. 2014; 110: 55-62.
9. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012; 13: 239-246.
10. Douillard JY, Ostoros G, Cobo M, Ciuleanu T, Cole R, McWalter G, Walker J, Dearden S, Webster A, Milenkova T and McCormack R. Gefitinib treatment in EGFR mutated caucasian NSCLC: circulating-free tumor DNA as a surrogate for determination of EGFR status. J Thorac Oncol. 2014; 9: 1345-1353.
11. Oxnard GR, Paweletz CP, Kuang Y, Mach SL, O’Connell A, Messineo MM, Luke JJ, Butaney M, Kirschmeier PT, Jackman DM and Jänne PA. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014; 20: 1698-1705.
12. Sorensen BS, Wu L, Wei W, Tsai J, Weber B, Nexo E and Meldgaard P. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-sensitizing and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib. Cancer. 2014; 120: 3896-3901.
13. Wang Z, Chen R, Wang S, Zhong J, Wu M, Zhao J, Duan J, Zhuo M, An T, Wang Y, Bai H and Wang J. Quantification and dynamic monitoring of EGFR T790M in plasma cell-free DNA by digital PCR for prognosis of EGFR-TKI treatment in advanced NSCLC. PLoS One. 2014; 9: e110780.
14. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015; 21: 560-562.
15. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz Jr LA, Goodman SN, David KA, Juhl H, Kinzler KW and Vogelstein B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005; 102: 16368-16373.
16. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo Jr BW, Alizadeh AA et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014; 20: 548-554.
17. Diaz Jr LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014; 32: 579-586.
18. Giacona MB, Ruben GC, Iczkowski KA, Roos TB, Porter DM and Sorenson GD. Cell-free DNA in human blood plasma: length measurements in patients with pancreatic cancer and healthy controls. Pancreas. 1998; 17: 89-97.
19. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014; 6: 224ra24.
20. Anker P, Stroun M and Maurice PA. Spontaneous extracellular synthesis of DNA released by human blood lymphocytes. Cancer Res. 1976; 36: 2832-2839.
21. Krug AK, Karlovich C, Koestler T, Bentink S, Brinkmann K, Spiel A, Emenegger J, Noerholm M, O’Neill V, Sequist LV, Soria J, Golmand JW, Camidge DR et al. Plasma EGFR mutation detection using a combined exosomal RNA and circulating tumor DNA approach in patients with acquired resistance to first-generation EGFR-TKIs. Poster presented at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, Boston, MA, USA, 5-9 November, 2015.
22. Qiu M, Wang J, Xu Y, Ding X, Li M, Jiang F, Xu L and Yin R. Circulating tumor DNA is effective for the detection of EGFR mutation in non-small cell lung cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2015; 24: 206-212.
23. Qian X, Liu J, Sun Y, Wang M, Lei H, Luo G, Liu X, Xiong C, Liu D, Liu J and Tang Y. Circulating cell-free DNA has a high degree of specificity to detect exon 19 deletions and the single-point substitution mutation L858R in non-small cell lung cancer. Oncotarget. 2016; 7: 29154-29165. doi: 10.18632/oncotarget.8684.
24. Luo J, Shen L and Zheng D. Diagnostic value of circulating free DNA for the detection of EGFR mutation status in NSCLC: a systematic review and meta-analysis. Sci Rep. 2014; 4: 6269.
25. Li Z, Zhang Y, Bao W and Jiang C. Insufficiency of peripheral blood as a substitute tissue for detecting EGFR mutations in lung cancer: a meta-analysis. Target Oncol. 2014; 9: 381-388.
26. Han B, Tjulandin S, Hagiwara K, Normanno N, Wulundari L, Konstantin Konstantinovich L, Hudoyo A, Ratcliffe M, McCormack R and Reck M. Determining the prevalence of EGFR mutations in Asian and Russian patients (pts) with advanced non-small-cell lung cancer (aNSCLC) of adenocarcinoma (ADC) or non-ADC histology: IGNITE study. Ann Oncol. 2015; 26 (Suppl 1): i29-i30.
27. Reck M, Hagiwara K, Han B, Tjulandin S, Grohe C, Yokoi T, Morabito A, McCormack R, Ratcliffe M and Normanno N. Investigating the utility of circulating-free tumour-derived DNA (ctDNA) in plasma for the detection of epidermal growth factor receptor (EGFR) mutation status in European and Japanese patients (pts) with advanced non-small-cell lung cancer (aNSCLC). Ann Oncol. 2015; 26 (Suppl 1): i58-i59.
28. El Messaoudi S, Rolet F, Mouliere F and Thierry AR. Circulating cell free DNA: preanalytical considerations. Clin Chim Acta. 2013; 424: 222-230.
29. Vallée A, Marcq M, Bizieux A, Kouri CE, Lacroix H, Bennouna J, Douillard JY and Denis MG. Plasma is a better source of tumor-derived circulating cell-free DNA than serum for the detection of EGFR alterations in lung tumor patients. Lung Cancer. 2013; 82: 373-374.
30. Lam NY, Rainer TH, Chiu RW and Lo YM. EDTA is a better anticoagulant than heparin or citrate for delayed blood processing for plasma DNA analysis. Clin Chem. 2004; 50: 256-257.
31. Beutler E, Gelbart T and Kuhl W. Interference of heparin with the polymerase chain reaction. Biotechniques. 1990; 9: 166.
32. Garcia-Murillas I, Shiavon G, Hrebien S, Oleary B, Morden J, Smith IE and Turner N. Development and validation of multiplex digital PCR assays on circulating tumor DNA in advanced breast cancer: implementation for clinical routine use. Ann Oncol. 2015; 26 (Suppl 3): iii16.
33. Toro PV, Erlanger B, Beaver JA, Cochran RL, VanDenBerg DA, Yakim E, Cravero K, Chu D, Zabransky DJ, Wong HY, Croessmann S, Parsons H, Hurley PJ et al. Comparison of cell stabilizing blood collection tubes for circulating plasma tumor DNA. Clin Biochem. 2015; 48: 993-998.
34. Denis MG, Knol AC, Théoleyre S, Vallée A and Dréno B. Efficient detection of BRAF mutation in plasma of patients after long-term storage of blood in cell-free DNA blood collection tubes. Clin Chem. 2015; 61: 886-888.
35. Chiu RW, Poon LL, Lau TK, Leung TN, Wong EM and Lo YM. Effects of blood-processing protocols on fetal and total DNA quantification in maternal plasma. Clin Chem. 2001; 47: 1607-1613.
36. Swinkels DW, Wiegerinck E, Steegers EA and de Kok JB. Effects of blood-processing protocols on cell-free DNA quantification in plasma. Clin Chem. 2003; 49: 525-526.
37. Sozzi G, Roz L, Conte D, Mariani L, Andriani F, Verderio P and Pastorino U. Effects of prolonged storage of whole plasma or isolated plasma DNA on the results of circulating DNA quantification assays. J Natl Cancer Inst. 2005; 97: 1848-1850.
38. Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J, Thunnissen E and Ladanyi M. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013; 8: 823-859.
39. Fleischhacker M, Schmidt B, Weickmann S, Fersching DM, Leszinski GS, Siegele B, Stotzer OJ, Nagel D and Holdenrieder S. Methods for isolation of cell-free plasma DNA strongly affect DNA yield. Clin Chim Acta. 2011; 412: 2085-2088.
40. Sherwood JL, Corcoran C, Brown H, Sharpe AD, Musilova M and Kohlmann A. Optimised pre-analytical methods improve KRAS mutation detection in circulating tumour DNA (ctDNA) from patients with non-small cell lung cancer (NSCLC). PLoS One. 2016; 11: e0150197.
41. Reck M, Hagiwara K, Tjulandin S, Han B, McWalter G, McCormack R and Normanno N. Investigating the utility of plasma derived circulating free DNA for the detection of epidermal growth factor receptor (EGFR) mutations in European and Japanese patients with advanced non-small-cell lung cancer (NSCLC): ASSESS study design. J Thorac Oncol. 2013; 8 (Suppl 2): 482.
42. Luke JJ, Oxnard GR, Paweletz CP, Camidge DR, Heymach JV, Solit DB and Johnson BE. Realizing the potential of plasma genotyping in an age of genotype-directed therapies. J Natl Cancer Inst. 2014; 106: dju214.
43. Thress KS, Brant R, Carr TH, Dearden S, Jenkins S, Brown H, Hammett T, Cantarini M and Barrett JC. EGFR mutation detection in ctDNA from NSCLC patient plasma: a cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer. 2015; 90: 509-515.
44. Kim S-W, Wu Y-L, Nakgawa K, Yang J-J, Ahn M-J, Wang J, Yang JC-H, Lu Y, Atagi S, Ponce S, Soria J-C, Mok T, Shi X et al. Retrospective analysis of ctDNA EGFR mutations in the Phase III, randomized IMPRESS study. J Thorac Oncol. 2015; 10 (Suppl 2): S204-S205.
45. Karlovich CA, Goldman J, Sun JM, Mann E, Sequist LV, Konopa K, Wen W, Angenendt P, Horn L, Spigel DR, Soria JC, Solomon B, Camidge DR et al. Assessment of EGFR mutation status in matched plasma and tumor tissue of NSCLC patients from a phase 1 study of rociletinib (CO-1686). Clin Cancer Res. 2016; 22: 2386-2395.
46. Normanno N, Hagiwara K, Han B, Tjulandin S, Grohé C, Yokoi T, Morabito A, Novello S, Arriola E, Molinier O, McCormack RM, Ratcliffe M and Reck M. Local diagnostic practices for advanced non-small-cell lung cancer in Europe and Japan: ASSESS study. J Thorac Oncol. 2015; 10 (Suppl 2): 197-198.
47. Zhu G, Ye X, Dong Z, Lu YC, Sun Y, Liu Y, McCormack R, Gu Y and Liu X. Highly sensitive droplet digital PCR method for detection of EGFR-activating mutations in plasma cell-free DNA from patients with advanced non-small cell lung cancer. J Mol Diagn. 2015; 17: 265-272.
48. Huang Z, Wang Z, Bai H, Wu M, An T, Zhao J, Yang L, Duan J, Zhuo M, Wang Y, Wang S and Wang J. The detection of EGFR mutation status in plasma is reproducible and can dynamically predict the efficacy of EGFR-TKI. Thoracic Caner. 2012; 3: 334-340.
49. Jing CW, Wang Z, Cao HX, Ma R and Wu JZ. High resolution melting analysis for epidermal growth factor receptor mutations in formalin-fixed paraffin-embedded tissue and plasma free DNA from non-small cell lung cancer patients. Asian Pac J Cancer Prev. 2014; 14: 6619-6623.
50. Brevet M, Johnson ML, Azzoli CG and Ladanyi M. Detection of EGFR mutations in plasma DNA from lung cancer patients by mass spectrometry genotyping is predictive of tumor EGFR status and response to EGFR inhibitors. Lung Cancer. 2011; 73: 96-102.
51. Nakamura T, Sueoka-Aragane N, Iwanaga K, Sato A, Komiya K, Kobayashi N, Hayashi S, Hosomi T, Hirai M, Sueoka E and Kimura S. Application of a highly sensitive detection system for epidermal growth factor receptor mutations in plasma DNA. J Thorac Oncol. 2012; 7: 1369-1381.
52. Zhao X, Han RB, Zhao J, Wang J, Yang F, Zhong W, Zhang L, Li LY and Wang MZ. Comparison of epidermal growth factor receptor mutation statuses in tissue and plasma in stage I-IV non-small cell lung cancer patients. Respiration. 2013; 85: 119-125.
53. Uchida J, Kato K, Kukita Y, Kumagai T, Nishino K, Daga H, Nagatomo I, Inoue T, Kimura M, Oba S, Ito Y, Takeda K and Imamura F. Diagnostic accuracy of noninvasive genotyping of EGFR in lung cancer patients by deep sequencing of plasma cell-free DNA. Clin Chem. 2015; 61: 1191-1196.
54. Yao Y, Liu J, Li L, Yuan Y, Nan K, Wu X, Zhang Z, Wu Y, Li X, Zhu J, Meng X, Wei L, Chen J et al. Detection of circulating tumor DNA in patients with advanced non-small cell lung cancer. Oncotarget. 2016; 8:2130-214. doi: 10.18632/oncotarget.12883.
55. Pasquale R, Fenizia F, Esposito Abate R, Sacco A, Esposito C, Forgione L, Rachiglio AM, Bevilacqua S, Montanino A, Franco R, Rocco G, Botti G, Denis MG et al. Assessment of high-sensitive methods for the detection of EGFR mutations in circulating free tumor DNA from NSCLC patients. Pharmacogenomics. 2015; 16: 1135-1148.
56. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, Insa A, Massuti B, Gonzalez-Larriba JL et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009; 361: 958-967.
57. Yam I, Lam DC, Chan K, Chung-Man HJ, Ip M, Lam WK, Chan TK and Chan V. EGFR array: uses in the detection of plasma EGFR mutations in non-small cell lung cancer patients. J Thorac Oncol. 2012; 7: 1131-1140.
58. Goto K, Ichinose Y, Ohe Y, Yamamoto N, Negoro S, Nishio K, Itoh Y, Jiang H, Duffield E, McCormack R, Saijo N, Mok T and Fukuoka M. Epidermal growth factor receptor mutation status in circulating free DNA in serum: from IPASS, a phase III study of gefitinib or carboplatin/paclitaxel in non-small cell lung cancer. J Thorac Oncol. 2012; 7: 115-121.
59. Liu X, Lu Y, Zhu G, Lei Y, Zheng L, Qin H, Tang C, Ellison G, McCormack R and Ji Q. The diagnostic accuracy of pleural effusion and plasma samples versus tumour tissue for detection of EGFR mutation in patients with advanced non-small cell lung cancer: comparison of methodologies. J Clin Pathol. 2013; 66: 1065-1069.
60. Weber B, Meldgaard P, Hager H, Wu L, Wei W, Tsai J, Khalil A, Nexo E and Sorensen BS. Detection of EGFR mutations in plasma and biopsies from non-small cell lung cancer patients by allele-specific PCR assays. BMC Cancer. 2014; 14: 294.
61. Liu Y, Liu B, Li XY, Li JJ, Qin HF, Tang CH, Guo WF, Hu HX, Li S, Chen CJ, Liu B, Gao HJ and Liu XQ. A comparison of ARMS and direct sequencing for EGFR mutation analysis and tyrosine kinase inhibitors treatment prediction in body fluid samples of non-small-cell lung cancer patients. J Exp Clin Cancer Res. 2011; 30: 111.
62. Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC and Markham AF. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989; 17: 2503-2516.
63. Whitcombe D, Theaker J, Guy SP, Brown T and Little S. Detection of PCR products using self-probing amplicons and fluorescence. Nat Biotechnol. 1999; 17: 804-807.
64. Hsiue EH, Lee JH, Lin CC and Yang JC. Profile of the therascreen® EGFR RGQ PCR Kit as a companion diagnostic for gefitinib in non-small cell lung cancer. Expert Rev Mol Diagn. 2016; Epub ahead of print.
65. Wittung P, Nielsen PE, Buchardt O, Egholm M and Norden B. DNA-like double helix formed by peptide nucleic acid. Nature. 1994; 368: 561-563.
66. Dressman D, Yan H, Traverso G, Kinzler KW and Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003; 100: 8817-8822.
67. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011; 83: 8604-8610.
68. Ma C, Wei S and Song Y. T790M and acquired resistance of EGFR TKI: a literature review of clinical reports. J Thorac Dis. 2011; 3: 10-18.
69. Yung TK, Chan KC, Mok TS, Tong J, To KF and Lo YM. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin Cancer Res. 2009; 15: 2076-2084.
70. Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, Yang JC, Barrett JC and Jänne PA. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J Clin Oncol. 2016; 34: 3375-3382.
71. Thress K, Yang J, Ahn M, Kim D, Kim S, Ohe Y, Planchard D, Brant R, Carr H, Dearden S, Jenkins S, Cantarini M, Ghiorghiu S et al. Levels of EGFR T790M in plasma DNA as a predictive biomarker for response to AZD9291, a mutant-selective EGFR kinase inhibitor. Ann Oncol. 2014; 25 (Suppl 4): iv426-iv470.
72. Ohira T, Sakai K, Matsubayashi J, Kajiwara N, Kakihana M, Hagiwara M, Hibi M, Yoshida K, Maeda J, Ohtani K, Nagao T, Nishio K and Ikeda N. Tumor volume determines the feasibility of cell-free DNA sequencing for mutation detection in non-small-cell lung cancer. Cancer Sci. 2016; 107: 1660-1666.
73. Kukita Y, Matoba R, Uchida J, Hamakawa T, Doki Y, Imamura F and Kato K. High-fidelity target sequencing of individual molecules identified using barcode sequences: de novo detection and absolute quantitation of mutations in plasma cell-free DNA from cancer patients. DNA Res. 2015; 22: 269-277.
74. Couraud S, Vaca-Paniagua F, Villar S, Oliver J, Schuster T, Blanché H, Girard N, Trédaniel J, Guilleminault L, Gervais R, Prim N, Vincent M, Margery J et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: a proof-of-concept study from BioCAST/IFCT-1002. Clin Cancer Res. 2014; 20: 4613-4624.
75. Goldberg SB, Narayan A, Carriero NJ, Nemati R, Bommakanti A, Wurtz A, Boffa DJ, Decker RH, Herbst RS, Juergensmeier JM, Politi KA, Gettinger SN and Patel A. Detection of sensitizing and resistance EGFR mutations from circulating tumor DNA (ctDNA) in blood using multiplexed next-generation sequencing in patients with advanced EGFR-mutant lung adenocarcinoma. J Clin Oncol (Meeting Abstracts). 2014; 32 (15 Suppl):
76. Collisson E, Mortimer S, Sebisanovic D, Mokhtari R, Bakhtiari S, Lopez R, Gadde DM, Vidamo MM, Patel H, Bahram GK, Eltoukhy H, Lanman RB and Talasaz A. Biopsy-free comprehensive tumor profiling of 1,000+ consecutive cancer patients using CLIA-certified commercial test and its clinical utility. Poster presented at the 106th Annual Meeting of the American Association for Cancer Research, Philadelphia, PA, USA, 20 April, 2015.
77. Kidess E, Heirich K, Wiggin M, Vysotskaia V, Visser BC, Marziali A, Wiedenmann B, Norton JA, Lee M, Jeffrey SS and Poultsides GA. Mutation profiling of tumor DNA from plasma and tumor tissue of colorectal cancer patients with a novel, high-sensitivity multiplexed mutation detection platform. Oncotarget. 2015; 6: 2549-2561. doi: 10.18632/oncotarget.3041.
78. Rachiglio AM, Abate RE, Sacco A, Pasquale R, Fenizia F, Lambiase M, Morabito A, Montanino A, Rocco G, Romano C, Nappi A, Iaffaioli RV, Tatangelo F et al. Limits and potential of targeted sequencing analysis of liquid biopsy in patients with lung and colon carcinoma. Oncotarget. 2016; doi: 10.18632/oncotarget.10704.
79. Villaflor V, Won B, Nagy R, Banks K, Lanman RB, Talasaz A and Salgia R. Biopsy-free circulating tumor DNA assay identifies actionable mutations in lung cancer. Oncotarget. 2016; doi: 10.18632/oncotarget.11801.
80. Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, Kurtz DM, Stehr H, Scherer F, Karlovich CA, Harding TC, Durkin KA, Otterson GA et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016; 7: 11815.
81. Khoo C, Rogers TM, Fellowes A, Bell A and Fox S. Molecular methods for somatic mutation testing in lung adenocarcinoma: EGFR and beyond. Transl Lung Cancer Res. 2015; 4: 126-141.
82. Normanno N, Brown H, Haddad V, Ratcliffe M, McCormack R, Tjulandin S, Hagiwara K, Han B and Reck M. 580_PR: Clinical and demographic features that influence EGFR mutation detection in plasma from patients (pts) with aNSCLC: The ASSESS experience. Journal of Thoracic Oncology 2016; 11: S151.
83. Tjulandin S, Han B, Hagiwara K, Normanno N, Wulandari L, Laktionov K, Hudoyo A, He Y, Zhang YP, Wang M-Z, Liu CY, Ratcliffe M, McCormack R et al. Local diagnostic practices for advanced non-small-cell lung cancer in Asia-Pacific and Russia: IGNITE study. J Thorac Oncol. 2015; 10 (Suppl 2): 401-402.
84. Spicer J, Tischer B and Peters M. EGFR mutation testing and oncologist treatment choice in advanced NSCLC: global trends and differences. Ann Oncol. 2015; 26 (Suppl 1): i60.
85. Denis MG, Masson P, Vallée A, Pigeanne T and Lacroix H. EGFR mutation detection in plasma of lung tumor patients in the absence of contributive tissue is a relevant alternative for prescription of tyrosine kinase inhibitors in a routine clinical setting. Annal Oncol. 2015; 26 (Suppl 1): i33.
86. Husain H, Kosco K, Guerrero S, Lu TT, Vibat CRT, Erlander MG and Melnikova V. Detection of EGFR T790M mutation in urinary circulating tumor DNA from metastatic non-small-cell lung cancer patients. Ann Oncol. 2015; 26 (Suppl 1): i10.
87. Chen S, Zhao J, Cui L and Liu Y. Urinary circulating DNA detection for dynamic tracking of EGFR mutations for NSCLC patients treated with EGFR-TKIs. Clin Transl Oncol. 2016;
88. Reckamp KL, Melnikova VO, Karlovich C, Sequist LV, Camidge DR, Wakelee H, Perol M, Oxnard GR, Kosco K, Croucher P, Samuelsz E, Vibat CR, Guerrero S et al. A highly sensitive and quantitative test platform for detection of NSCLC EGFR mutations in urine and plasma. J Thorac Oncol. 2016; 11: 1690-1700
89. Pan W, Gu W, Nagpal S, Gephart MH and Quake SR. Brain tumor mutations detected in cerebral spinal fluid. Clin Chem. 2015; 61: 514-522.
90. Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, Dobbyn L, Ptak J, Brem H, Chaichana K, Gallia GL, Gokaslan ZL, Groves ML, Jallo GI et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci U S A. 2015; 112: 9704-9709.
91. Shingyoji M, Kageyama H, Sakaida T, Nakajima T, Matsui Y, Itakura M, Iuchi T, Yokoi S, Kimura H and Iizasa T. Detection of epithelial growth factor receptor mutations in cerebrospinal fluid from patients with lung adenocarcinoma suspected of neoplastic meningitis. J Thorac Oncol. 2011; 6: 1215-1220.
92. Zhao J, Ye X, Xu Y, Chen M, Zhong W, Sun Y, Yang Z, Zhu G, Gu Y and Wang M. EGFR mutation status of paired cerebrospinal fluid and plasma samples in EGFR mutant non-small cell lung cancer with leptomeningeal metastases. Cancer Chemother Pharmacol. 2016; Epub ahead of print.
93. Yang H, Cai L, Zhang Y, Tan H, Deng Q, Zhao M and Xu X. Sensitive detection of EGFR mutations in cerebrospinal fluid from lung adenocarcinoma patients with brain metastases. J Mol Diagn. 2014; 16: 558-563.
94. Sundaresan TK, Sequist LV, Heymach JV, Riely GJ, Janne PA, Koch WH, Sullivan JP, Fox DB, Maher R, Muzikansky A, Webb A, Tran HT, Giri U et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clin Cancer Res. 2016; 22: 1103-1110.
95. Sequist LV, Goldman JW, Wakelee HA, Camidge DR, Yu HA, Varga A, Solomon B, Oxnard GR, Ou S-HI, Papadimitrakopoulou V, Chao BH, Liu SV, Reckamp KL et al. Efficacy of rociletinib (CO-1686) in plasma-genotyped T790M-positive non-small cell lung cancer (NSCLC) patients (pts). J Clin Oncol (Meeting Abstracts). 2015; 33 (15 Suppl): 8001.
96. Ramalingam SS, Yang JC-H, Lee CK, Kurata T, Kim D-W, John T, Nogami N, Ohe Y, Rukazenkov Y, Frewer P, Cantarini M, Ghiorghiu S and Janne AP. AZD9291, a mutant-selective EGFR inhibitor, as first-line treatment for EGFR mutation-positive advanced non-small cell lung cancer (NSCLC): results from a phase I expansion cohort. J Clin Oncol (Meeting Abstracts). 2015; 33 (15 Suppl): 8000.
97. Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, Fulton L, Hata AN, Lockerman EL, Kalsy A, Digumarthy S, Muzikansky A, Raponi M et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015; 5: 713-722.
98. Thress K, Brant R, Carr H, Brown H, Dearden SP, Jenkins S, Hammett T, Cantarini M, Ghiorghiu S and Barrett JC. EGFR mutation detection in ctDNA from NSCLC patient plasma: a cross-platform comparison of technologies to support the clinical development of AZD9291. Lung Cancer. 2015; 90: 509-515:
99. Imamura F, Uchida J, Kukita Y, Kumagai T, Nishino K, Inoue T, Kimura M, Oba S and Kato K. Monitoring of treatment responses and clonal evolution of tumor cells by circulating tumor DNA of heterogeneous mutant EGFR genes in lung cancer. Lung Cancer. 2016; 94: 68-73.
100. Marchetti A, Palma JF, Felicioni L, De Pas TM, Chiari R, Del GM, Filice G, Ludovini V, Brandes AA, Chella A, Malorgio F, Guglielmi F, De TM et al. Early prediction of response to tyrosine kinase inhibitors by quantification of EGFR mutations in plasma of NSCLC patients. J Thorac Oncol. 2015; 10: 1437-1443.
101. Marcq M, Vallee A, Bizieux A and Denis MG. Detection of EGFR mutations in the plasma of patients with lung adenocarcinoma for real-time monitoring of therapeutic response to tyrosine kinase inhibitors? J Thorac Oncol. 2014; 9: e49-e50.
102. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B and Diaz LA, Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14: 985-990.
103. Imamura F, Uchida J, Kukita Y, Kumagai T, Nishino K, Inoue T, Kimura M and Kato K. Early responses of EGFR circulating tumor DNA to EGFR tyrosine kinase inhibitors in lung cancer treatment. Oncotarget. 2016; doi: 10.18632/oncotarget.12373.
104. Riediger AL, Dietz S, Schirmer U, Meister M, Heinzmann-Groth I, Schneider M, Muley T, Thomas M and Sültmann H. Mutation analysis of circulating plasma DNA to determine response to EGFR tyrosine kinase inhibitor therapy of lung adenocarcinoma patients. Sci Rep. 2016; 6: 33505.
105. Sacher AG, Paweletz C, Dahlberg SE, Alden RS, O’Connell A, Feeney N, Mach SL, Janne PA and Oxnard GR. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol. 2016; 2: 1014-1022.
106. Valleé A, Audigier-Valette C, Herbreteau G, Merrien J, Tessonnier L, Théoleyre S and Denis MG. Rapid clearance of circulating tumor DNA during treatment with AZD9291 of a lung cancer patient presenting the resistance EGFR T790M mutation. Lung Cancer. 2016; 91: 73-74.
107. Beiter T, Fragasso A, Hudemann J, Niess AM and Simon P. Short-term treadmill running as a model for studying cell-free DNA kinetics in vivo. Clin Chem. 2011; 57: 633-636.
108. Han B, Tjulandin S, Normanno N, Hagiwara K, Reck M, McWalter G and McCormack R. A diagnostic study to determine the prevelance of epidermal growth factor receptor (EGFR) mutations in Asian and Russian patients with non-small-cell lung cancer (NSCLC) of adenocarcinoma and non-adenocarcinoma histology: IGNITE study design. J Thorac Oncol. 2013; 8 (Suppl 2): 1097.