
Introduction
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease characterized by chronic immune activation, the presence of a plethora of autoantibodies, and diverse clinical phenotypes [1]. Although abnormal immune responses with excessive release of pro-inflammatory cytokines, as well as genetic factors, have been implicated in the pathogenesis SLE, the mechanistic details are still unclear [2, 3]. Recent compelling evidence has shown that abnormal Th1, Th2 and Th17 cell immune responses are crucial in the pathogenesis of SLE [4].
Members of the Gq family of membrane-associated heterotrimeric guanine nucleotide-binding proteins (G proteins) include Gq, G11, G14, and G15/16. They mediate the canonical activation of phospholipase Cβ isozymes, and like all heterotrimeric G proteins, are composed of three subunits, Gα, Gβ and Gγ [5, 6]. Gαq, the α-subunit of the Gq protein, is encoded by the GNAQ gene and is widely expressed in various cells of the immune system, including T cells [7, 8]. In recent years, by using knockout (KO) mice and chemical inhibitors, the functions of G proteins in the immune system have been extensively reported; accumulated data indicates that G protein signaling systems control important aspects of innate and adaptive immunity [9].
Several studies from our group and others have explored the relationship between Gαq signaling and autoimmune disease. Abrahamsen et al. reported, for instance, that stimulation of T cells with anti-CD3/anti-CD28 antibodies recruits Gαq subunits to lipid rafts, indicating that Gαq is involved in T cell receptor signaling [10]. Our own studies showed that migration from the skin to the draining lymph nodes after fluorescein isothiocyanate sensitization is impaired in Gαq-deficient (Gnaq-/-) neutrophils and dendritic cells [11], and that Gnaq-/- bone marrow chimeras with immune-specific Gαq deficiency spontaneously develop manifestations of systemic autoimmune disease with high titer antinuclear antibody, multi-organ involvement and swelling of the joints [12]. We also reported that the protein and mRNA levels of Gαq in peripheral blood lymphocytes from rheumatoid arthritis (RA) patients were significantly lower compared with healthy controls, and that decreased Gαq expression was closely correlated with disease activity [13]. Furthermore, Gnaq-/- T cells showed significant survival advantages both in vitro and in vivo [14]. These studies supported a pivotal role of the Gαq subunit in the pathogenesis of autoimmune diseases. However, whether Gαq contributes to the pathogenesis of SLE is not known. To address this question, Gαq expression was measured in peripheral blood mononuclear cells (PBMCs) and T cells from SLE patients, and its relationship with SLE Disease Activity Index (SLEDAI), clinical laboratory indicators, Th1, Th2 and Th17 cytokines, and apoptosis-regulatory proteins was determined. Our results showed a significantly decreased Gαq expression in both PBMCs and T lymphocytes from SLE patients, is in comparison with healthy individuals. In addition, significant correlations were observed between T cell Gαq expression and SLEDAI, Complement 3 (C3), and urine protein and creatinine (CRE) in SLE patients. As expected, Gαq expression was correlated with enhanced Th1/Th2/Th17 differentiation and cytokine secretion, and distinctly associated with the expression of the apoptosis-related genes Bcl-2 and Bax. Altogether, our data suggest that decreased Gαq expression might contribute to T cell dysfunction and development of SLE.
Results
Decreased Gαq expression in PBMCs and T cells from patients with SLE
A contribution of Gαq to the pathogenesis of RA was reported by us previously [13]. To assess if Gαq signaling is also associated with SLE, we first measured Gαq mRNA expression in PBMCs from SLE patients and healthy controls by real time-PCR. Although mRNA expression of Gαq was significantly lower in PBMC from SLE patients (Figure 1A, top) no correlation with SLEDAI was found (Figure 1B, top). Because T cells have been specifically implicated in the development of SLE, we next analyzed Gαq expression in T cells. As expected, the levels of Gαq mRNA in CD3+ T cells were lower in SLE patients than in healthy controls (Figure 1A, bottom), and correlated negatively with SLEDAI (Figure 1B, bottom).
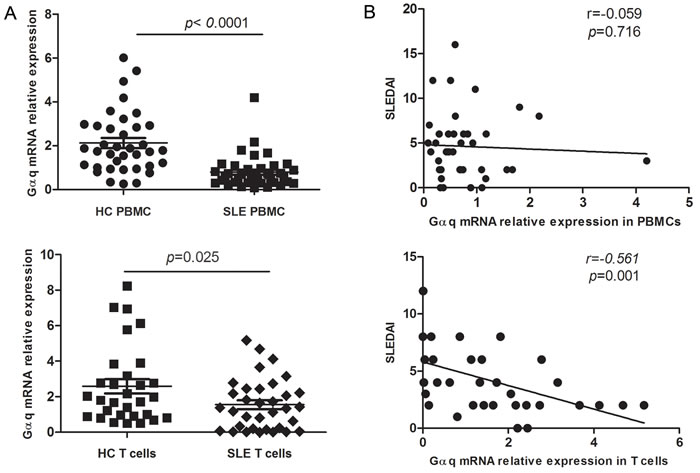
Figure 1: Decreased Gαq expression in PBMCs and T lymphocytes from SLE patients. A. Expression of Gαq mRNA in PBMCs and CD3+ T cells from SLE patients and healthy controls (HC), detected by real time-PCR (SLE, n = 40, HC, n = 37). Each symbol represents an individual sample; horizontal lines denote median values. The Mann-Whitney U test was used to evaluate statistical differences between SLE and HC data. B. Correlation between T cell Gαq mRNA expression and disease activity index (SLEDAI) in SLE patients, assessed using the Spearman’s rank correlation test.
Correlation between T cell Gαq levels and parameters of disease activity
To assess the correlation between Gαq expression in T lymphocytes and organ involvement in SLE, SLE patients were grouped based on the presence or absence of renal damage (lupus nephritis), rash, arthritis, hematological involvement, serositis, oral ulcer, and alopecia (Table 1). Although for several parameters Gαq expression was lower in patients exhibiting clinical symptoms, a significant decrease in Gαq levels was found to be associated only with symptomatic lupus nephritis (p = 0.002; Table 1).
Table 1: Gαq mRNA expression in T cells from SLE patients with or without clinical manifestations
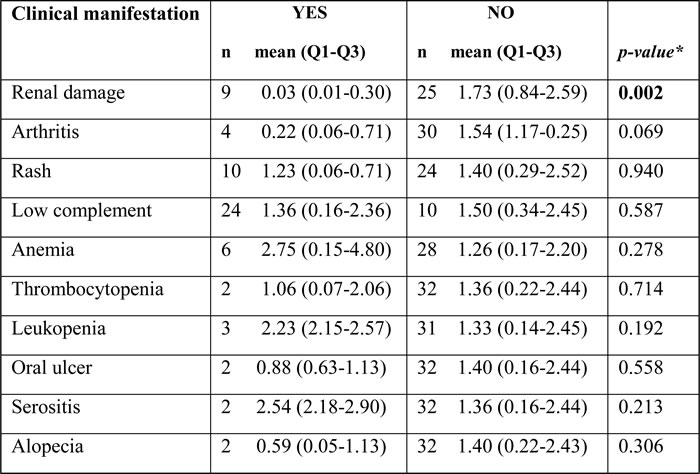
* Mann-Whitney U test was used to assess differences in Gαq expression in CD3+ T cells from SLE patients with or without clinical disease manifestations. p < 0.05 was considered statistically significant.
Next, we analyzed the relationship between T cell Gαq expression and laboratory parameters in SLE patients. Gαq levels were positively correlated with C3 levels (r = 0.390, p = 0.022; Figure 2A). In line with the correlation found for lupus nephritis, negative correlations were detected between Gαq levels and 24h urine protein (r = -0.379, p = 0.026) and CRE (r = -0507, p = 0.002; Figure 2B, 2C). In contrast, no correlation was found between Gαq expression in T cells of SLE patients and dsDNA antibodies (p = 0.532), IgG (p = 0.970), C4 (p = 0.239) or ANA (p = 0.241; data not shown).
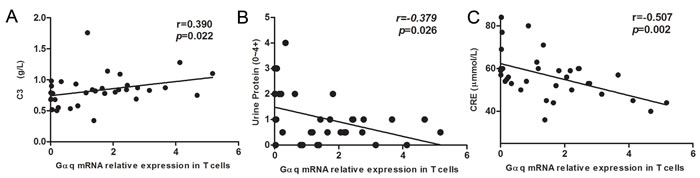
Figure 2: Correlation between Gαq mRNA levels in SLE T cells and laboratory values. The relationship between T cell Gαq mRNA expression levels and laboratory values in SLE patients is shown. Spearman’s correlation analysis was used to calculate significance.
Reduced Gαq expression is correlated with increased T cell cytokine expression and differentiation in SLE
Because abnormal T cell activation is an important pathological feature of SLE, we further measured IFN-γ, IL-4, IL-17 and Foxp3 in T cells from SLE patients and from healthy controls. In keeping with previous studies, IFN-γ, IL-17 and Foxp3 expression was significantly higher in SLE patients than in controls (p = 0.033, p = 0.012, and p = 0.001, respectively), while no significant difference was observed in the expression of IL-4 (Figure 3). On the other hand, in SLE T cells a negative correlation between Gαq and both IFN-γ and IL-17 levels (Figure 4) was determined. In contrast with the corresponding findings in SLE patients and healthy controls, Gαq was negatively correlated with IL-4, but not with Foxp3 (Figure 4). To infer the impact of Gαq expression on T cell differentiation, splenic Th1, Th2 and Th17 subsets were quantified in Gαq knockout mice. As expected, the frequencies of CD4+ Th1, Th2 and Th17 cells were increased in knockout mice compared with wild type mice (Figure 5).
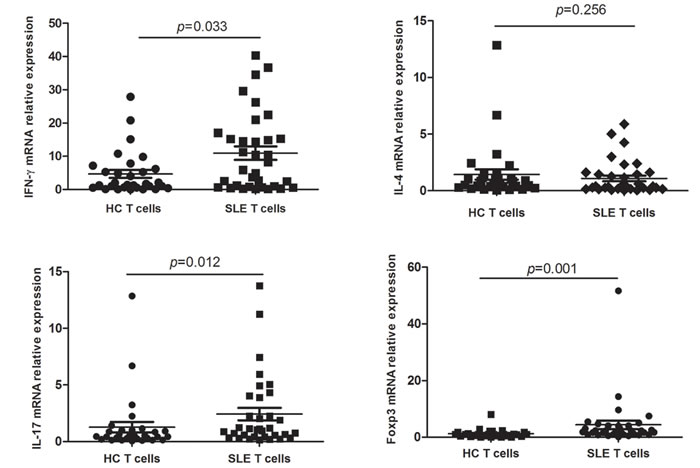
Figure 3: IFN-γ, IL-4, IL-17 and Foxp3 expression in T cells from SLE patients. Relative mRNA expression of IFN-γ, IL-4, IL-17 and Foxp3 in CD3+ T cells from SLE patients and controls was detected by real time-PCR (SLE, n = 34; HC, n = 30). IFN-γ, IL-17 and Foxp3, but not IL-4, were increased in SLE T cells. Mann-Whitney U test was used to assess expression differences between SLE and HC.
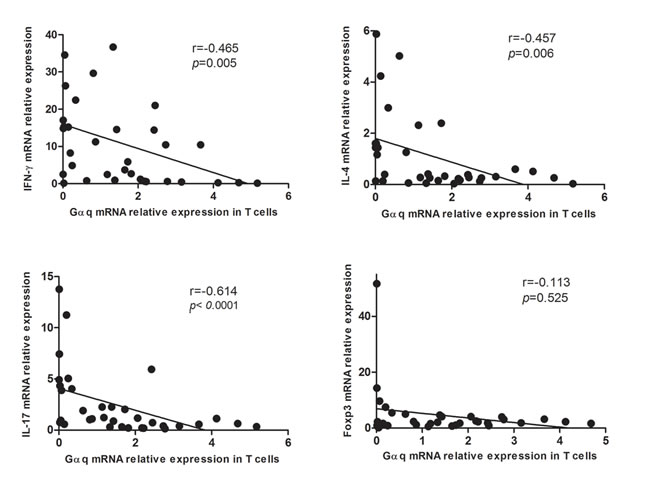
Figure 4: Correlation between Gαq and IFN-γ, IL-4, IL-17 and Foxp3 expression in T cells from SLE patients. Spearman’s correlation analysis showed that IFN-γ, IL-4, and IL-17, but not Foxp3, were inversely correlated with Gαq levels.
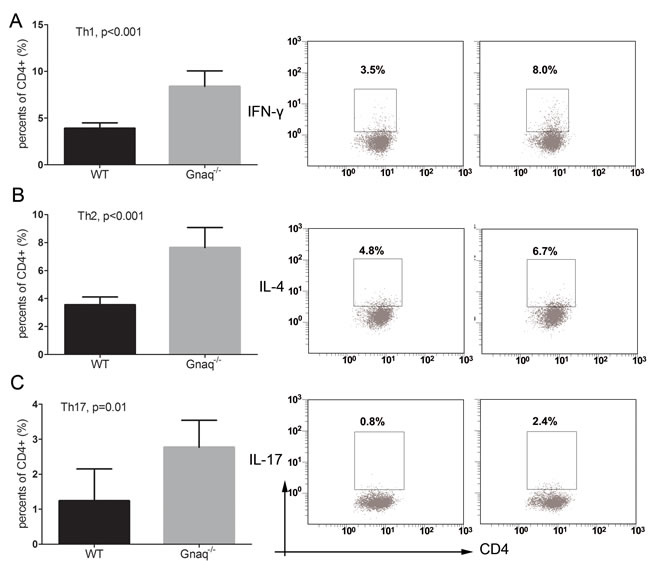
Figure 5: Gαq deletion promotes the differentiation of T-helper cells. The expression of IFN-γ A., IL-4 B., and IL-17 C. was studied by flow cytometry in Gαq knockout (Gnaq-/-) and wild type (WT) mice-derived splenic CD4+ T cells stimulated with PMA, ionomycin, and BFA. Data from three independent experiments are presented as mean ± SD.
Gαq levels and apoptosis-related gene expression in T cells from SLE
An important feature of T cell dysfunction in autoimmune disorders is the prolonged cell survival that results from the abnormal onset and progression of the apoptotic program. Therefore, we analyzed apoptosis-related genes in T lymphocytes from SLE patients. As expected, a negative correlation between Gαq levels and the expression of the anti-apoptotic gene Bcl-2 was observed (r = -0.365, p = 0.033). In contrast, the expression of Gαq correlated positively with the expression of the pro-apoptotic gene Bax (r = 0.542, p = 0.001; Figure 6). These data suggest that Gαq might contribute to the pathogenesis of SLE by prolonging T cell survival.
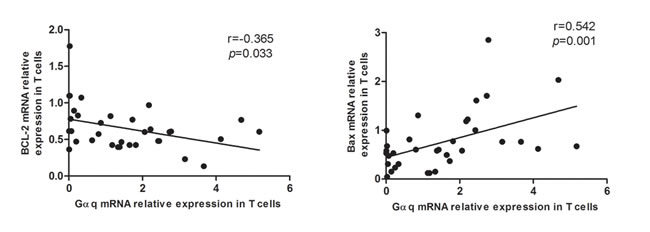
Figure 6: Correlation between Gαq and apoptosis-regulatory proteins in T cells from SLE patients. Gαq mRNA expression is positively correlated with anti-apoptotic Bcl-2, and negatively correlated with pro-apoptotic Bax mRNA levels in SLE T cells. Spearman’s correlation analysis was used to calculate significance.
Discussion
Gαq is abundantly expressed in the cells and tissues of the immune system [15]. Our previous work has demonstrated that Gnaq-/- chimeric mice possess several features of autoimmune disease, including production of autoreactive antibodies, deposition of IgG2a- and IgG2c-containing immune complexes in the kidney, thrombotic microangiopathy, a reduction in the number of red blood cells, synovitis, bone resorption, exostotic bone development, and osteolytic activity [12]. Furthermore, the involvement of Gαq in the pathogenesis of RA was confirmed by showing that Gαq prevented apoptosis in peripheral blood lymphocytes of RA patients by regulating the activity of Mcl-1 and caspase-3 [13]. Whether Gαq contributes to the pathogenesis of SLE, however, remains unknown. To address this question, the expression of Gαq was assessed in PBMCs and T lymphocytes from SLE patients. We found that Gαq expression was significantly decreased in both PBMCs and T cells from SLE patients, compared with healthy controls. However, only T cell Gαq mRNA expression was negatively correlated with SLEDAI, urine protein and CRE, and positively correlated with both lupus nephritis and C3 levels. These results suggest that decreased signaling through Gαq may be related to impaired kidney function in SLE.
The overproduction of Th2 cytokines typically promotes B-cell hyperactivity and humoral responses, while T cell hyperactivity and inflammation are frequently associated with an excess of Th1 cytokines [16]. The present data showed that the levels of IFN-γ (a Th1 cytokine), IL-17 (a Th17 cytokine) and Foxp3 (a marker of regulatory T cells) were higher in T cells from SLE patients, compared with controls, while low Gαq mRNA expression in T cells from SLE patients was associated with higher levels of IFN-γ, IL-4 (a Th2 cytokine), and IL-17. Moreover, intracellular staining showed that Th1, Th2 and Th17 helper cell subsets were over-represented in Gαq knockout mice, compared with wild-type controls. These data suggest that reduced Gαq signaling may contribute to augmented T cell activity in SLE.
Previous studies suggested that IL-17 contributes to the pathogenesis of kidney dysfunction in SLE patients [17-19]. We, on the other hand, have shown that Gαq inhibits the differentiation of Th17 cells by regulating the activity of extracellular signal-regulated kinase 1/2/ (ERK1/2) to control the expression of STAT3 and RORα [20]. As significantly lower Gαq levels were measured in LSE patients with lupus nephritis, compared with those without renal damage, and low Gαq expression correlated with higher IL-17 levels in SLE T cells, our data may underscore a link between reduced Gαq expression in T cells leading to overproduction of Th17 cells, enhanced IL-17 production, and subsequent kidney damage.
Abnormal signal transduction in T-lymphocytes is considered a potential cause of lupus [21], with decreased MAPK activity and impaired ERK signaling among the important alterations found in T cells from SLE patients [22]. While several studies indicated that Gαq is involved in the activation of ERK [23], Gαq-/- primary T cells also show reduced proximal LAT phosphorylation, and in line with the present cytokine expression results, augmented immune responses, including increased secretion of IL-2, IL-5, IL-12 and TNF-α [24]. Another important hallmark of T cell dysfunction in SLE is apoptosis, a critical process for immune tolerance and autoimmunity [25]. The PI3K-Akt signaling pathway regulates many normal cellular processes, including proliferation, motility, and survival [26]. Gαq was reported to inhibit PI3K activation, which prevented Akt signaling and promoted apoptosis [27]. Accordingly, in a previous study we showed that upon T-cell receptor ligation, Akt activity was increased in Gnaq-/- T cells in comparison with wild-type T cells, and the survival advantage of Gnaq-/- T cells was significantly attenuated by Akt inhibition. We also proved that Gαq deficiency promotes T cell survival via upregulation of Bcl-xL and downregulation of Fas and FasL expression [14]. Here, we further show that Gαq expression was inversely correlated anti-apoptotic Bcl-2, and directly correlated with pro-apoptotic Bax mRNAs levels in T cells from SLE patients. These data further suggest that reduced expression of Gαq contributes to the pathogenesis and progression of SLE through inhibition or impairment of apoptosis in T cells.
In summary, we demonstrated that Gαq expression is significantly decreased in T cells of patients with SLE, and this is correlated with SLE disease activity, increased differentiation of Th1, Th2 and Th17 cells and altered levels of apoptosis-related proteins. Taken together, our results suggest that therapeutic restoration of Gαq levels may correct the over-excitable T cell phenotype in SLE.
Materials and methods
Patients’ characteristics, disease activity and clinical features
40 adult patients (35 women and 5 men, aged 32.6 ± 9.4 years) with a diagnosis of SLE based on the American College of Rheumatology criteria [28] were consecutively enrolled in the study after providing informed consent. All patients were referred from the Department of Rheumatology and Clinical Immunology at the First Affiliated Hospital of Xiamen University. Patients with cancer, hematopathy, severe infections, hepatitis, tuberculosis or allergies were excluded. The control group consisted of 37 healthy volunteers (34 women and 3 men, aged 33.5 ± 9.5 years) that were enrolled after giving informed consent. 34 patients and 30 controls donated additional blood for CD3+ T cell isolation. Table 1 depicts the demographic and clinical characteristics of all patients and controls. All clinical manifestations and laboratory findings were recorded on the day of blood withdrawal. Clinical manifestations of disease in SLE patients were determined on the basis of the SLE Disease Activity Index (SLEDAI), which assesses the presence of malar rash, alopecia, proteinuria, cutaneous vasculitis, oral ulcers, arthritis, and serositis [29]. This study was approved by the institutional research board (IRB) of the First Affiliated Hospital of Xiamen University.
Animal experiments
All experimental procedures involving mice were approved by the Animal Care and Use Committee of Xiamen University. C57BL/6J (B6) and Gnaq-/- (backcrossed > 5 times to B6) mice were bred in the Xiamen University animal facilities and used between 6 and 8 weeks of age.
Flow cytometry analysis
Spleen lymphocytes were harvested for intracellular cytokine staining. The lymphocytes were stimulated with 50 ng/ml PMA, 500 ng/ml ionomycin, and 5 μg/ml BFA (Alexis Biochemicals) followed by surface anti-CD4 staining. The cells were then fixed, permeabilized and stained with intracellular antibodies against IL-4, IL-17, and IFN-γ. All antibodies were purchased from Biolegend.
Separation of peripheral blood mononuclear cells and purification of T cells
Peripheral blood samples from SLE patients and healthy volunteers were collected in anticoagulant tubes. Peripheral blood mononuclear cells (PBMCs) were isolated by standard Ficoll-Hypaque density-gradient centrifugation for 30 min, and washed twice with phosphate buffered saline before T cell purification by negative selection with the human T Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany). The purity of T cells was > 95% as determined by flow cytometry.
Reverse transcription-polymerase chain reaction (RT-PCR) and real-time PCR
Total RNA was extracted from PBMCs and CD3+ T cells by TRIzol™ Reagent (Invitrogen, Carlsbad, CA) and reverse-transcribed to cDNA with a Transcriptor First Strand cDNA Synthesis Kit, according to the manufacturer’s protocol (Roche); RT- PCR was performed with a Bio-Rad System. mRNA expression levels of Gαq, IL-4, IFN-γ, IL-17, Foxp3, Bax, Bcl-2 and GAPDH were determined using a real-time quantitative PCR System (ABI 7500). The SYBR Green master (ROX) was purchased from Roche. Cycling conditions were as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 15s and 60°C for 1min. Gene expression data was normalized to that of GAPDH and relative expression was calculated by the 2-ΔΔCt method. The following primer sequences were used in Table 2.
Table 2: primer sequences
Gene |
Sequence |
|
GAPDH |
F: 5'-AGCCACATCGCTCAGACAC-3' |
R:5'-GCCCAATACGACCAAATCC-3' |
Gαq |
F:5'-TGGTGTATCAGAACATCTTCACG-3' |
R:5'-CTCGAACTAATTGTGCATGAGC-3' |
IFN-γ |
F: 5'-AGCTCTG- CATCGTTTTGGGTT-3' |
R:5'-GTTCCATTATCCGCTACATCTGAA-3' |
Foxp3 |
F: 5'-CACTTACAGGCACTCCTCCAGG -3' |
R:5'-CCACCGTTGAGAGCTGGTGCAT-3' |
IL-4 |
F:5'-CACAAGCAGCTGATCCGATTC-3' |
R:5'-TCTGGTTGGCTTCCTTCACAG-3 |
IL-17 |
F:5'-AACCGATCCACCTCACCTTG-3' |
R:5'- TCTCTTGCTGGATGGGGACA -3' |
Bcl-2 |
F:5'-ATGTGT GTGGAGAGCGTCAACC -3' |
R:5'- GCATCCCAGCCTCCGTTATC -3' |
Bax |
F:5'-CCTTTTCTACTTTGCCAGCAAAC-3' |
R:5'- GAGGCCGTCCCAACCAC-3' |
Statistical analysis
All data were analyzed using GraphPad Prism 5. Mann-Whitney U-test and Spearman’s correlation analysis were used to calculate significance. P values < 0.05 were considered statistically significant.
Acknowledgments
We are extremely grateful to all the patients and volunteers who took part in this study, as well as the whole rheumatology team, including nurses, medical record system personnel and laboratory technicians.
Conflicts of Interest
The authors have declared that no competing interests exist.
Grant Support
This work was supported by The National Basic Research Program of China (973 Program) 2014CB541903 to Guixiu Shi, the National Natural Science Foundation of China (NSFC 81671544, 81302564, to Lihua Duan, NFSC 81471534 to Guixiu Shi), the Major disease research projects of Xiamen no.3502Z20149029 and Xiamen Science and Technology Bureau Project no.3502Z20144005 to Guixiu Shi, Fujian Province health planning of young outstanding talents training project no.2016-ZQN-82 to Lihua Duan
References
1. Comte D, Karampetsou MP, Tsokos GC. T cells as a therapeutic target in SLE. Lupus. 2015; 24: 351-63. doi: 10.1177/0961203314556139.
2. Su DL, Lu ZM, Shen MN, Li X, Sun LY. Roles of pro- and anti-inflammatory cytokines in the pathogenesis of SLE. J Biomed Biotechnol. 2012; 2012: 347141. doi: 10.1155/2012/347141.
3. Richez C, Blanco P, Rifkin I, Moreau JF, Schaeverbeke T. Role for toll-like receptors in autoimmune disease: the example of systemic lupus erythematosus. Joint Bone Spine. 2011; 78: 124-30. doi: 10.1016/j.jbspin.2010.09.005.
4. Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol. 2001; 1: 147-53. doi: 10.1038/35100573.
5. Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008; 9: 60-71. doi: 10.1038/nrm2299.
6. Taylor SJ, Chae HZ, Rhee SG, Exton JH. Activation of the beta 1 isozyme of phospholipase C by alpha subunits of the Gq class of G proteins. Nature. 1991; 350: 516-8. doi: 10.1038/350516a0.
7. Druey KM. Regulation of G-protein-coupled signaling pathways in allergic inflammation. Immunol Res. 2009; 43: 62-76. doi: 10.1007/s12026-008-8050-0.
8. Grant KR, Harnett W, Milligan G, Harnett MM. Differential G-protein expression during B- and T-cell development. Immunology. 1997; 90: 564-71.
9. Wang Y, Li Y, Shi G. The regulating function of heterotrimeric G proteins in the immune system. Arch Immunol Ther Exp (Warsz). 2013; 61: 309-19. doi: 10.1007/s00005-013-0230-5.
10. Abrahamsen H, Baillie G, Ngai J, Vang T, Nika K, Ruppelt A, Mustelin T, Zaccolo M, Houslay M, Tasken K. TCR- and CD28-Mediated Recruitment of Phosphodiesterase 4 to Lipid Rafts Potentiates TCR Signaling. The Journal of Immunology. 2004; 173: 4847-58. doi: 10.4049/jimmunol.173.8.4847.
11. Shi G, Partida-Sanchez S, Misra RS, Tighe M, Borchers MT, Lee JJ, Simon MI, Lund FE. Identification of an alternative G{alpha}q-dependent chemokine receptor signal transduction pathway in dendritic cells and granulocytes. J Exp Med. 2007; 204: 2705-18. doi: 10.1084/jem.20071267.
12. Misra RS, Shi G, Moreno-Garcia ME, Thankappan A, Tighe M, Mousseau B, Kusser K, Becker-Herman S, Hudkins KL, Dunn R, Kehry MR, Migone TS, Marshak-Rothstein A, et al. G alpha q-containing G proteins regulate B cell selection and survival and are required to prevent B cell-dependent autoimmunity. J Exp Med. 2010; 207: 1775-89. doi: 10.1084/jem.20092735.
13. Wang Y, Li Y, He Y, Sun Y, Sun W, Xie Q, Yin G, Du Y, Wang L, Shi G. Expression of G protein alphaq Subunit is Decreased in Lymphocytes from Patients with Rheumatoid Arthritis and is Correlated with Disease Activity. Scand J Immunol. 2012; 75: 203-9. doi: 10.1111/j.1365-3083.2011.02635.x.
14. Wang D, Zhang Y, He Y, Li Y, Lund FE, Shi G. The deficiency of Galphaq leads to enhanced T-cell survival. Immunol Cell Biol. 2014; 92: 781-90. doi: 10.1038/icb.2014.53.
15. Hubbard KB, Hepler JR. Cell signalling diversity of the Gqalpha family of heterotrimeric G proteins. Cell Signal. 2006; 18: 135-50. doi: 10.1016/j.cellsig.2005.08.004.
16. Lourenco EV, La Cava A. Cytokines in systemic lupus erythematosus. Curr Mol Med. 2009; 9: 242-54. doi:
17. Garrett-Sinha LA, John S, Gaffen SL. IL-17 and the Th17 lineage in systemic lupus erythematosus. Curr Opin Rheumatol. 2008; 20: 519-25. doi: 10.1097/BOR.0b013e328304b6b5.
18. Dolff S, Quandt D, Wilde B, Feldkamp T, Hua F, Cai X, Specker C, Kribben A, Kallenberg CG, Witzke O. Increased expression of costimulatory markers CD134 and CD80 on interleukin-17 producing T cells in patients with systemic lupus erythematosus. Arthritis Res Ther. 2010; 12: R150. doi: 10.1186/ar3100.
19. Mok MY, Wu HJ, Lo Y, Lau CS. The relation of interleukin 17 (IL-17) and IL-23 to Th1/Th2 cytokines and disease activity in systemic lupus erythematosus. J Rheumatol. 2010; 37: 2046-52. doi: 10.3899/jrheum.100293.
20. Liu Y, Wang D, Li F, Shi G. Galphaq controls rheumatoid arthritis via regulation of Th17 differentiation. Immunol Cell Biol. 2015; 93: 616-24. doi: 10.1038/icb.2015.13.
21. King PD. Lupus-like autoimmunity caused by defects in T-cell signal transduction. Curr Opin Investig Drugs. 2004; 5: 517-23.
22. Cedeno S, Cifarelli DF, Blasini AM, Paris M, Placeres F, Alonso G, Rodriguez MA. Defective activity of ERK-1 and ERK-2 mitogen-activated protein kinases in peripheral blood T lymphocytes from patients with systemic lupus erythematosus: potential role of altered coupling of Ras guanine nucleotide exchange factor hSos to adapter protein Grb2 in lupus T cells. Clin Immunol. 2003; 106: 41-9.
23. White CD, Coetsee M, Morgan K, Flanagan CA, Millar RP, Lu ZL. A crucial role for Galphaq/11, but not Galphai/o or Galphas, in gonadotropin-releasing hormone receptor-mediated cell growth inhibition. Mol Endocrinol. 2008; 22: 2520-30. doi: 10.1210/me.2008-0122.
24. Ngai J, Methi T, Andressen KW, Levy FO, Torgersen KM, Vang T, Wettschureck N, Tasken K. The heterotrimeric G-protein alpha-subunit Galphaq regulates TCR-mediated immune responses through an Lck-dependent pathway. Eur J Immunol. 2008; 38: 3208-18. doi: 10.1002/eji.200838195.
25. Xu G, Shi Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007; 17: 759-71. doi: 10.1038/cr.2007.52.
26. Browne CD, Del Nagro CJ, Cato MH, Dengler HS, Rickert RC. Suppression of phosphatidylinositol 3,4,5-trisphosphate production is a key determinant of B cell anergy. Immunity. 2009; 31: 749-60. doi: 10.1016/j.immuni.2009.08.026.
27. Ballou LM, Lin HY, Fan G, Jiang YP, Lin RZ. Activated G alpha q inhibits p110 alpha phosphatidylinositol 3-kinase and Akt. J Biol Chem. 2003; 278: 23472-9. doi: 10.1074/jbc.M212232200.
28. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997; 40: 1725. doi: 10.1002/1529-0131(199709)40:9<1725::AID-ART29>3.0.CO;2-Y.
29. Gladman DD, Ibanez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002; 29: 288-91.