
INTRODUCTION
Mammalian spermatogenesis is a highly organized process that requires precise progressive differentiation of spermatogonia. Both somatic and germ cell-specific factors play critical roles in this process. As the primary somatic cells in seminiferous epithelium, Sertoli cells interact directly with germ cells at different stages, assuring normal spermatogonia differentiation [1]. Sertoli cell-specific factors are necessary for spermatogonia differentiation and tesitcular cord maintenance [2–4]. However, few studies have focused on the roles of germ cell-specific factors in these processes.
Mouse Rad9a is an evolutionarily conserved gene that helps maintain genome integrity [5]. RAD9A functions as part of the heterotrimeric 9-1-1 complex composed of RAD9A, RAD1 and HUS1 [6]. RAD9A participates in the cellular response to exogenous DNA damage via the ataxia telangiectasia and Rad3-related (ATR) and ataxia telangiectasia mutated (ATM) signaling pathways [7–9]. It also influences DNA repair directly by physically interacting with proteins involved in DNA replication and homologous recombination [10–17]. In addition to its well-known function in DNA damage repair, mammalian RAD9A regulates cell cycle checkpoints and apoptosis. In vitro, Rad9a knockdown increased spontaneous, as well as topoisomerase poison-induced death, spontaneous chromosomal aberrations, and radio-resistant DNA synthesis [6, 18–21]. RAD9A deficiency also impairs cell proliferation, migration and invasion [21, 22]. In vivo, embryos with homozygous Rad9a deletions died nine days post coitum [6]. While Rad9a null embryonic stem cells are viable, mouse embryonic fibroblasts devoid of Rad9a are not [6].
In a recent report, Rad9a was conditionally knocked out in undifferentiated spermatogonia in a mouse model via Cre recombinase expressed under the Stra8 promoter (Stra8-Cre) [23]. Spermatogenesis was only interrupted at the late zygotene or early pachytene stage of meiotic prophase I, indicating that Rad9a deletion did not fully inhibit spermatogonia differentiation. This may have been due to the fact that the deletion was carried out in later spermatogonia. Moreover, deletion of Rad9a via Stra8-Cre may not be entirely effective, as spermatocytes in mutant testes can escape floxed allele excision [23]. To investigate whether RAD9A functional disruption wholly blocks spermatogonia differentiation, and to assess the impacts of this disruption on testicular cord development, we generated mice in which Rad9a was conditionally deleted in spermatogonial stem cells via Cre recombinase driven by the Vasa promoter. While mutant spermatogonia were viable and proliferated normally, differentiation was completely blocked. Additionally, Rad9a deletion led to malformation of testicular cords. Our results show for the first time that RAD9A is indispensable for spermatogonia differentiation and testicular cord maintenance.
RESULTS
Rad9a deletion in spermatogonial stem cells leads to male infertility
Because conventional Rad9a deletion results in embryonic lethality, we constructed a mouse model with Rad9a deleted specifically in spermatogonia [6]. We crossed Rad9aF/F mice with Vasa-Cre transgenic mice, in which Cre begins to be expressed in primordial germ cells (PGC), to conditionally delete Rad9a in offspring PGCs (hereafter referred to as Rad9a cKO mice) (Figure 1A) [6, 24]. Western blotting results showed that RAD9A levels were sharply reduced in testis tissues from Rad9a cKO male mice as compared to Rad9aF/F mice. Residual RAD9A observed in knockout mice testes may come from Sertoli cells [23].
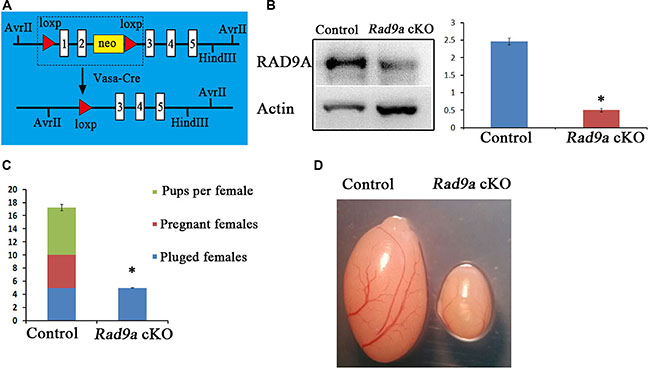
Figure 1: Conditional Rad9a deletion in SSCs via Vasa-Cre. Schematic of deletion of the floxed Rad9a exon1 and exon2 via Vasa-Cre mediated recombination (A) Western blot analysis of RAD9A in P9 testis extracts from both wild type and Rad9a cKO mice (B) Results were normalized to the actin loading control. The experiment was repeated three times. Fertility test (C) Plugged females became pregnant and gave birth to pups after crossing with Rad9aF/F control males, whereas none of the plugged females were pregnant after crossing with Rad9a cKO males. Testes from Rad9aF/F control males and Rad9a cKO males at six months (D) Testis size in Rad9a cKO mice was sharply reduced compared with that of control mice. *P < 0.01.
We assessed the effects of RAD9A deletion in PGCs on the reproductive performance of mutant male mice. None of the plugged female mice became pregnant after crossing with mutant males and no pups were produced (Figure 1C). In comparison, almost all female mice with vaginal plugs that mated with wild type mice became pregnant and produced seven pups each on average. We observed that the testes of Rad9a cKO male mice at six months of age were much smaller than those of Rad9aF/F mice (Figure 1D).
Mutant testes develop disorganized testicular cords
To assess the time course of abnormal testicular development, we compared testis histological morphology from Rad9aF/F and Rad9a cKO male mice at postnatal day nine (P9), three weeks, three months and six months of age. As previously described [25], early spermatocytes began to appear in the testes of wild type mice at P9 (Figure 2Aa). Round spermatids were observed in Rad9aF/F male mouse testes at three weeks (Figure 2Ac). In adulthood (three and six months), spermatogonia, spermatocytes, round spermatids, and condensed mature spermatids were arranged regularly by Sertoli cells in Rad9aF/F male mouse testicular cords (Figure 2Ae and 2Ag). In comparison, Rad9a cKO male mouse testicular cords became vacant as early as P9 (Figure 2Ab). Differences between control and mutant testicular cords became more significant at three weeks of age (Figure 2Ad). Only a single layer of cells remained around the basal membrane in most conditional knockout testicular cords, in contrast to that of control mice. Until three months of age, mutant testicular cords were extremely disorganized (Figure 2Af). Irregularly assembled cell clusters were observed in most cords, and only basal membrane was residual in parts of the abnormal cords. No basal membrane was present in some cases. Phenotypes were more severe in Rad9a cKO male mice testes at six months (Figure 2Ah). Some mutant testicular cords appeared to collapse and some cells penetrated testicular cords into the interstitum of the testes.
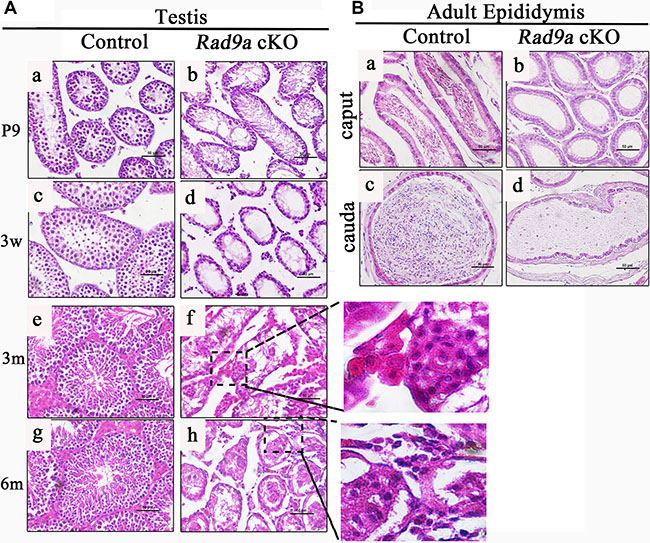
Figure 2: H&E staining of testis and epididymis tissues. Comparison of testicular cords from Rad9aF/F control males and Rad9a cKO males (A) In contrast to control testicular cords (a, c, e, g), Rad9a cKO mouse testes became vacant at as early as nine days of age (B). The difference became more significant at three weeks (d). Testes of Rad9a cKO adult males developed disorganized testicular cords with malformed cell clusters (f). Disorganized testicular cords in mutant testes collapsed (h). Both the caput and cauda epididymides of control adult male mice were full of mature sperm (a, c), but were vacant in adult Rad9a cKO males (b, d) b.
To confirm the testis phenotypes, we examined adult epididymides from both genotypes. Both the caput and cauda of control epididymides were full of mature condensed sperm (Figure 2Ba and 2Bc). However, the caput epididymides of knockout mice were vacant, without any cells (Figure 1Bb). There was also no sperm in mutant cauda epididymides, which were filled with lipid fluid (Figure 2Bd). These data indicated that conditional Rad9a deletion in spermatogonia completely blocked spermatogenisis, led to disorganized cell clusters in the testicular cords, and finally disaggregated the testicular cords.
Malformed cell clusters in mutant testicular cords mainly consisted of Sertoli cells
To investigate the origins of malformed cell clusters observed in mutant testicular cords, we determined cell types using molecular markers. Intermediate filaments in Sertoli cells of postnatal mice are comprised of Vimentin, which is also a Sertoli cell tumor marker [26, 27]. Vimentin accumulated at the boundary between Sertoli and germ cells in control mouse testes at P9. (Figure 3A), and its accumulation was more significant at three weeks (Figure 3E). At adulthood, Vimentin staining presented as a regularly radial pattern in control testicular cords (Figure 3I and 3M). In contrast, Vimentin was evenly dispersed throughout mutant mouse Sertoli cells at P9 (Figure 3B). At three weeks, disorganized Vimentin distributions appeared in knockout mouse testicular cords (Figure 3F, arrows). At three months, Vimentin staining accumulated around malformed cell clusters in the mutant testicular cords (Figure 3J, arrows). In mutant mouse testes at six months, Vimentin immunostaining was observed outside the testicular cords (Figure 3N, arrowheads), as well as around malformed cell clusters (Figure 3N, arrows).
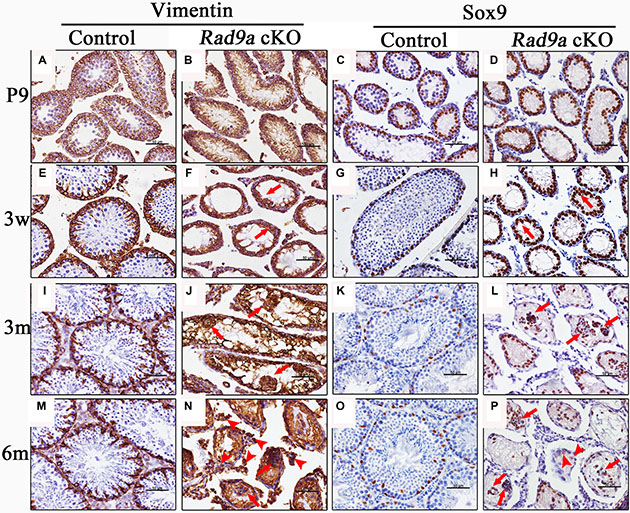
Figure 3: Malformed cell clusters mainly consisted of Sertoli cells. Rad9aF/F control and Rad9a cKO testes at different ages were immunostained with two Sertoli cell markers: Vimentin and Sox9. Nuclei were stained blue with hematoxylin. Vimentin accumulated at the junction between germ cells and Sertoli cells in the control testes at P9. Vimentin presented as a regularly radial pattern from three weeks to adulthood (A, E, I, and M). Vimentin was evenly dispersed throughout Sertoli cells in Rad9a cKO testes at P9, and abnormal Vimentin distribution became increasingly apparent from three weeks to adult hood (B, F, J, and N). In control testes, Sox9-positive cells began to migrate to the basal membrane at P9, and located to the basal membrane (C, G, K, and O). In Rad9a cKO testes, Sox9-positive cells migrated to the lumen from P9, and finally assembled into malformed cell clusters with few Sox9-negative cells (D, H, L, and P).
These findings indicated that malformed cell clusters in mutant testes may consist primarily of Sertoli cells. To confirm this, we compared Sox9 immunostaining, another Sertoli cell marker, in the testes of both genotypes. Most Sertoli cell nuclei localized to the basal membrane as early as P9 in wild type testes (Figure 3C), and staining remained consistent from three weeks to adulthood (Figure 3G, 3K and 3O). However, Sertoli cell nuclei in mutant testes at P9 were poorly orientated to the basal membrane (Figure 3D), and some began to migrate toward the lumen of mutant testicular cords at three weeks (Figure 3H, arrows). Migrating Sertoli cell nuclei assembled to form clusters in adult testicular cords (Figure 3L and 3P, arrows). We observed some cells without Sox9 staining in the malformed cell clusters (Figure 3L and 3P, arrows), as well as cells with weakened Sox9 signal among cells separated from disrupted testicular cords (Figure 3P, arrowheads).
Spermatogonia differentiation is blocked in mutant testes
Vasa (also named MVH) is a germ cell-specific marker in testis, and is expressed throughout the spermatogonium-to-round spermatid development process. We observed moderately Vasa-positive spermatogonia at the basal membrane, as well as early spermatocytes with strong Vasa signal in the lumen of wild type testicular cords at P9 (Figure 4A). Along with spermatogonia and spermatocytes, Vasa-positive round spermatids appeared at three weeks in control testes (Figure 4E), after which Vasa-positive germ cell staining patterns remained consistent (Figure 4I and 4M). In contrast, we observed Vasa-positive spermatogonia only at the basal membrane of mutant testicular cords at P9 (Figure 4B). No Vasa-positive spermatocytes were observed in the lumen of mutant testicular cords, except for moderately Vasa-positive spermatogonia at the basal membrane at three weeks (Figure 4E). We observed only sporadically Vasa-positive germ cells in adult mutant testicular cords (Figure 4J and 4N), some at the basal membrane, and some in the lumen, where they formed disorganized clusters with Sertoli cells (Figure 4J, arrows). We also detected a few Vasa-positive cells among the interstitial cells, indicating leakage of cells from disrupted testicular cords to the interstitium (Figure 4N, arrowheads).
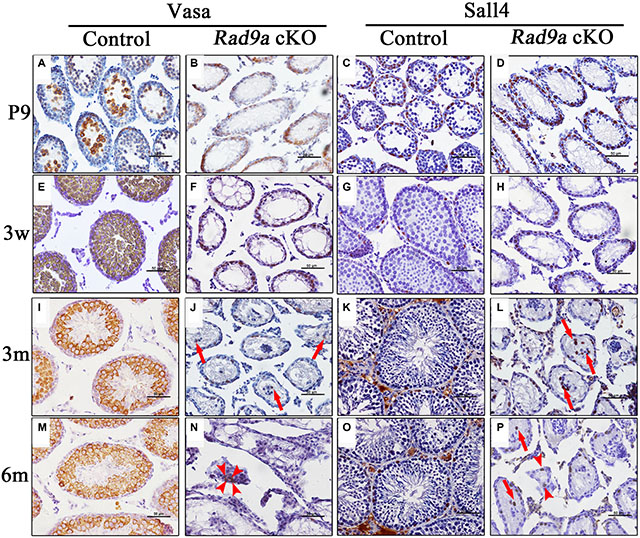
Figure 4: Spermatogonia differentiation was inhibited in mutant testes. Representative seminiferous tubule images at P9 (A–D), three weeks (E–H), three months (I–L). and six months of age (M–P). with staining of MVH (a germ cell marker) and Sall4 (a spermatogonia marker, brown). Nuclei are stained blue with hematoxylin.
These data implied that spermatogonia development might also be disrupted following Rad9a deletion. We traced spermatogonia fates via immunostaining with pluripotency factor Sall4, which is implicated in stem cell maintenance and restricted to gonocytes and undifferentiated spermatogonia [28]. All Sall4-positive cells were restricted at the basal membrane from P9 to adulthood in control testes (Figure 4C, 4G, 4K and 4O). All Sall4-positive cells were also located at the basal membrane in mutant testicular cords at P9 (Figure 4D), but some Sall4-positive spermatogonia in mutant testes began to migrate away from the basal membrane at three weeks (Figure 4H). However, we still observed Sall4-positive spermatogonia at the basal membranes of some mutant testicular cords at adulthood (Figure 4L and 4P). Some Sall4-positive cells had migrated to the lumen of mutant testicular cords, assembling with Sall4-negative cells at adulthood (Figure 4L and 4P, arrows). As indicated by Vasa immunostaining, very few Sall4-positive cells embedded with other cells moved from disorganized testicular cords to the interstitium (Figure 4P, arrowheads). These data indicated that Rad9a deletion blocked spermatogonia differentiation, but spermatogonia nonetheless appeared viable.
Mutant spermatogonia proliferate normally, but are prone to apoptosis
We assessed mutant spermatogonia proliferation rates through immunohistochemical staining with the mitosis marker, phosphorylated histone H3 (pHH3). We observed strong pHH3 signals at P9 in control testes (Figure 5A), with fewer pHH3-positive cells in Rad9a cKO male mouse testicular cords (Figure 5B). Control testicular cords showed increased pHH3-positive germ cells at adulthood (Figure 5E), but we observed only sporadic pHH3-positive cells in mutant testicular cords (Figure 5F). pHH3-positive germ cells in control testis included both spermatogonia and spermatocytes. No significant difference was found in the number of pHH3-positive spermatogonia between control and mutant testicular cords (Figure 5I), indicating that mutant and wild type spermatogonia proliferated at the same rate.
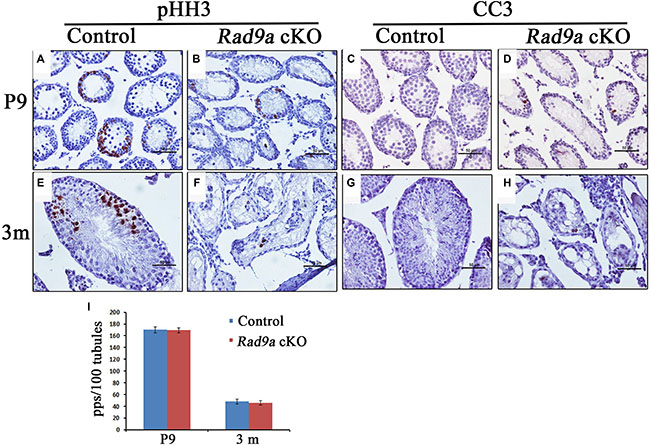
Figure 5: Mutant spermatogonia were prone to apoptosis. More pHH3-positive cells can be seen in Rad9aF/F control testes compared to Rad9a cKO testes at P9 and three months (A, B, E, and F). The number of pHH3-positive spermatogonia was the same for both genotypes (I). Mutant spermatogonia were prone to apoptosis at P9 and three months (C, D, G, and H). pps, pHH3-positive spermatogonia.
RAD9A inhibits apoptosis, and cells in Rad9a null embryos were prone to spontaneous apoptosis [29, 30]. To test whether Rad9a deletion also promotes apoptosis in spermatogonia, we stained tissues for cleaved caspase 3 (CC3). Almost no CC3-positive germ cells were observed in control testicular cords at P9 or adulthood (Figure 5C and 5G). In contrast, we frequently observed CC3-positive germ cells in conditional knockout mouse testicular cords at both P9 and adulthood (Figure 5D and 5H). CC3-positive germ cells included both early spermatocytes and spermatogonia. These results suggest that RAD9A inhibits spermatogonia apoptosis, although spermatogonia devoid of RAD9A proliferate normally.
DISCUSSION
The 9-1-1 complex is a critical participant in several cellular processes, including DNA damage repair, homologous recombination, and cell cycle checkpoint control in both mitotic and meiotic cells [31–33]. However, the function of mammalian RAD9A in spermatogonia differentiation had not yet been elucidated. In this study, we constructed and characterized a mouse model with Rad9a deleted in spermatogonial stem cells (SSCs). Mutant males developed normally with typical reproductive behavior. However, none produced pups, apparently due to reduced testis size. Subsequent histological analyses of conditional knockout mouse testes indicated that mutant testicular cords were vacant as early as P9, leaving only Sertoli cells and spermatogonia at the basal membrane. These results were confirmed by Sertoli and germ cell marker immunostaining. We validated that no mutant spermatogonia entered meiosis. Thus, spermatogonia differentiation was completely blocked in mutant males. When Vasileva, et al. [23] deleted Rad9a in spermatogonia via Stra8-Cre, spermatogenesis was interrupted at the late zygotene or early pachytene stage of meiotic prophase I, and prophase spermatocytes survived normally in mutant testicular cords at adulthood. Differences between these results and those of the present study may result from knockout efficiency variations between the two mouse models. Many RAD9A-positive pachytene spermatocytes are still observed in testis when the Stra8-Cre knockout system is employed. This suggests that spermatocytes and spermatogonia might escape Cre excision [23]. In contrast, Cre excision in our study was much more effective in SSCs. As Rad9a null spermatogonia remained alive and proliferated normally in our study, blocked spermatogonia differentiation may not be attributed to defective DNA damage repair or other known RAD9A functions. Consistent with our results, Rad9a null mouse embryonic stem cells are reportedly viable and proliferate normally, while mouse embryonic fibroblasts with the same genotype cannot be established [6]. These data imply a currently unknown mechanism by which RAD9A protects differentiated cells against apoptosis. More studies are needed to elucidate this mechanism.
We observed malformed testicular cords in mutant testes at adulthood, and cells assembled irregularly to form disorganized cell clusters in mutant testicular cords. Some tubular architecture was also disrupted, and cells broke away into the interstitia of mutant testes. Interestingly, we observed Sox9- and Sall4-positive cells among these interstitium-localized cells. This was a novel phenotype, although it was relatively mild compared to disruption of testicular cords by Sertoli cell-specific factor dysfunction [1–4]. Testicular cord architecture remains intact following Stra8-Cre-mediated Rad9a deletion, although spermatogenesis is interrupted. This difference compared to our system may result from the fact that pachytene stage spermatocytes are present in adult male mutant testes in the Stra8-Cre system. This indicates that an intact blood-testis barrier has been constructed in mutant testicular cords. In contrast, no early spermatocytes were observed in mutant testes at P9 in our study. Our data suggest that spermatogonia differentiation blocked by Rad9a deletion may destroy junctions, such as the blood-testis barrier, and distort testicular construction. More detailed studies are needed to define which types of junctions are destroyed, and to elucidate the mechanisms underlying testicular cord malformation.
In conclusion, our findings revealed that RAD9A plays a key role in spermatogonia differentiation. Rad9a null spermatogonia failed to differentiate into spermatocytes, and testicular cord malformation was observed in mutant mouse testes, which led to complete male infertility.
MATERIALS AND METHODS
Mice
Rad9aflox/flox mice (JAX Lab, referred to as Rad9aF/F) were maintained with a mixed genomic background of 129S4/SvJae and C57/BL6, and Vasa-Cre mice were maintained with a C57/BL6 genomic background [6, 24]. Mice with conditional Rad9a deletion (referred to as Rad9a cKO mice) were generated by crossing Rad9aF/F mice with Vasa-Cre mice. Mice were housed under controlled environmental conditions with free access to water and food, and 12-hour alternating light/dark cycles. Animal care and handling were conducted according to the guidelines of the Animal Research Committee of the Institute of Zoology, Chinese Academy of Sciences, China.
Fertility assay
To assess the fertility of Rad9aF/F and Rad9a cKO males, we selected five healthy two-month-old males for each genotype and mated them to five six-week-old C57/BL6 females. Females with vaginal plugs after crossing were maintained separately. At about day 25 after checking vaginal plugs, females were sacrificed and the number of pregnant females and living pups were recorded.
Histological analysis and immunochemistry
Mice were euthanized via cervical dislocation and testes were dissected and fixed immediately in 4% paraformaldehyde overnight at 4°C. Tissues were dehydrated in gradient ethanol, made transparent in xylen, and embedded in paraffin. Embedded tissues were cut into 5 μm sections, which were deparaffinized and rehydrated for histological analysis and immunochemistry. Hematoxylin and eosin (H&E) staining was performed for histological observation. For immunostaining, after antigen retrieval in 10 mM sodium citrate buffer, the endogenous peroxidase was cleaned up by incubating slides in 3% H2O2. Sections were then blocked with 5% bovine serum albumin (BSA) and incubated with primary antibody at 4°C overnight. After incubation in secondary antibody for 1 h, signals were detected using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA). The following primary antibodies were used in this study: rabbit anti-Sox9 (Abcam, Cambridge, UK; ab3697), rabbit anti-Vasa (Abcam, ab13840), rabbit anti-phospho-HH3 (Abcam, ab5176), mouse anti-Sall4 (Santa Cruz, CA, USA; sc101147), rabbit anti-Vimentin (Zhongshan, Beijing, China; za0511), and rabbit anti-cleaved caspase 3 (CC3) (Cell Signaling Technology, Beverly, MA, USA; 9661).
Western blot analysis
Mice were sacrificed at about nine days of age, and testes were separated and lysed in radio immunoprecipitation assay lysis buffer containing Complete Mini Protease Inhibitor Cocktail Tablets (Roche). Samples were mixed with SDS sample buffer and boiled for 5 min at 100ºC for SDS-PAGE. Western blotting was performed as described previously [34]. Proteins were electrophoresed under reducing conditions in 12% SDS-PAGE gels and transferred to nitrocellulose membranes. Blots were blocked in 5% BSA and incubated overnight at 4°C with primary antibody, followed by incubation with secondary antibody for 1 h at room temperature. Signals were detected using the ECL western blotting detection system. The following primary antibodies were used in this study: mouse anti-RAD9A (Zen Biosciences, Chengdu, China; 200784), and rabbit anti-GAPDH (CST, 5174) according to the manufacturer’s instructions. Secondary antibodies were purchased from ZhongShan Golden Bridge Biotechnology Co., Ltd (Beijing, China).
Statistical analysis
All experiments were repeated at least three times. For histological analysis and immunochemistry, one representative image of the results from several independent experiments was selected for presentation. For comparisons, means and standard deviations were calculated, and the difference between two groups was analyzed using Student’s t-test. Differences were considered statistically significant if P < 0.05.
ACKNOWLEDGMENTS
We are grateful to Ying-Chun Ouyang, Shi-Wen Li, Li-Juan Wang and Hua Qin for their technical assistance. We also thank Zhao-Feng Huang, Bo-Lan Yu and Jing-Pian Peng for their useful suggestions.
CONFLICTS OF INTEREST
None.
GRANT SUPPORT
This study was supported by the National Basic Research Program of China (No. 2012CB944404) and the National Natural Science Foundation of China (No. 31272260, 31371451).
REFERENCES
1. Mruk DD, Cheng CY. Sertoli-Sertoli and Sertoli-germ cell interactions and their significance in germ cell movement in the seminiferous epithelium during spermatogenesis. Endocr Rev. 2004; 25:747–806.
2. Gao F, Maiti S, Alam N, Zhang Z, Deng JM, Behringer RR, Lecureuil C, Guillou F ,Huff V. The Wilms tumor gene, Wt1, is required for Sox9 expression and maintenance of tubular architecture in the developing testis. P Natl Acad Sci USA. 2006; 103:11987–11992.
3. Chen SR, Tang JX, Cheng JM, Li J, Jin C, Li XY, Deng SL, Zhang Y, Wang XX ,Liu YX. Loss of Gata4 in Sertoli cells impairs the spermatogonial stem cell niche and causes germ cell exhaustion by attenuating chemokine signaling. Oncotarget. 2015; 6:37012–37027. doi: 10.18632/oncotarget.6115.
4. Zheng QS, Wang XN, Wen Q, Zhang Y, Chen SR, Zhang J, Li XX, Sha RN, Hu ZY, Gao F, Liu YX. Wt1 deficiency causes undifferentiated spermatogonia accumulation and meiotic progression disruption in neonatal mice. Reproduction. 2014; 147:45–52.
5. Lieberman HB. Rad9, an evolutionarily conserved gene with multiple functions for preserving genomic integrity. Journal of Cellular Biochemistry. 2006; 97:690–697.
6. Hopkins KM, Auerbach W, Wang XY, Hande MP, Hang H, Wolgemuth DJ, Joyner AL, Lieberman HB. Deletion of mouse rad9 causes abnormal cellular responses to DNA damage, genomic instability, and embryonic lethality. Mol Cell Biol. 2004; 24:7235–7248.
7. Furuya K, Poitelea M, Guo L, Caspari T, Carr AM. Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev. 2004; 18:1154–1164.
8. Lee J, Kumagai A, Dunphy WG. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem. 2007; 282:28036–28044.
9. Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis. 2006; 21:3–9.
10. Chang DY, Lu AL. Interaction of checkpoint proteins Hus1/Rad1/Rad9 with DNA base excision repair enzyme MutY homolog in fission yeast, Schizosaccharomyces pombe. J Biol Chem. 2005; 280:408–417.
11. Gembka A, Toueille M, Smirnova E, Poltz R, Ferrari E, Villani G, Hubscher U. The checkpoint clamp, Rad9-Rad1-Hus1 complex, preferentially stimulates the activity of apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta in long patch base excision repair. Nucleic Acids Res. 2007; 35:2596–2608.
12. Guan X, Madabushi A, Chang DY, Fitzgerald ME, Shi G, Drohat AC, Lu AL. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates DNA repair enzyme TDG glycosylase. Nucleic Acids Res. 2007; 35:6207–6218.
13. He W, Zhao Y, Zhang CB, An LL, Hu ZS, Liu YH, Han L, Bi LJ, Xie ZS, Xue P, Yang FQ, Hang HY. Rad9 plays an important role in DNA mismatch repair through physical interaction with MLH1. Nucleic Acids Research. 2008; 36:6406–6417.
14. Pandita RK, Sharma GG, Laszlo A, Hopkins KM, Davey S, Chakhparonian M, Gupta A, Wellinger RJ, Zhang JR, Powell SN, Roti JLR, Lieberman HB, Pandita TK. Mammalian Rad9 plays a role in telomere stability, S- and G-phase-Specific cell survival, and homologous recombinational repair. Molecular and Cellular Biology. 2006; 26:1850–1864.
15. Park MJ, Park JH, Hahm SH, Ko SI, Lee YR, Chung JH, Sohn SY, Cho Y, Kang LW, Han YS. Repair activities of human 8-oxoguanine DNA glycosylase are stimulated by the interaction with human checkpoint sensor Rad9-Rad1-Hus1 complex. DNA repair. 2009; 8:1190–1206.
16. Toueille M, El-Andaloussi N, Frouin I, Freire R, Funk D, Shevelev I, Friedrich-Heineken E, Villani G, Hottiger MO, Hubscher U. The human Rad9/Rad1/Hus1 damage sensor clamp interacts with DNA polymerase beta and increases its DNA substrate utilisation efficiency: implications for DNA repair. Nucleic Acids Res. 2004; 32:3316–3324.
17. Wang W, Brandt P, Rossi ML, Lindsey-Boltz L, Podust V, Fanning E, Sancar A, Bambara RA. The human Rad9-Rad1-Hus1 checkpoint complex stimulates flap endonuclease 1. Proc Natl Acad Sci USA. 2004; 101:16762–16767.
18. Dang T, Bao S, Wang XF. Human Rad9 is required for the activation of S-phase checkpoint and the maintenance of chromosomal stability. Genes Cells. 2005; 10:287–295.
19. Pandita RK, Sharma GG, Laszlo A, Hopkins KM, Davey S, Chakhparonian M, Gupta A, Wellinger RJ, Zhang J, Powell SN, Roti Roti JL, Lieberman HB, Pandita TK. Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol Cell Biol. 2006; 26:1850–1864.
20. Loegering D, Arlander SJ, Hackbarth J, Vroman BT, Roos-Mattjus P, Hopkins KM, Lieberman HB, Karnitz LM, Kaufmann SH. Rad9 protects cells from topoisomerase poison-induced cell death. J Biol Chem. 2004; 279:18641–18647.
21. An LL, Wang YL, Liu YH, Yang XA, Liu CC, Hu ZS, He W, Song WX, Hang HY. Rad9 Is Required for B Cell Proliferation and Immunoglobulin Class Switch Recombination. Journal of Biological Chemistry. 2010; 285:35267–35273.
22. Broustas CG, Zhu AP, Lieberman HB. Rad9 Protein Contributes to Prostate Tumor Progression by Promoting Cell Migration and Anoikis Resistance. Journal of Biological Chemistry. 2012; 287:41324–41333.
23. Vasileva A, Hopkins KM, Wang X, Weisbach MM, Friedman RA, Wolgemuth DJ, Lieberman HB. The DNA damage checkpoint protein RAD9A is essential for male meiosis in the mouse. Journal of cell science. 2013; 126:3927–3938.
24. Gallardo T, Shirley L, John GB, Castrillon DH. Generation of a germ cell-specific mouse transgenic Cre line, Vasa-Cre. Genesis. 2007; 45:413–417.
25. Bellve AR, Cavicchia JC, Millette CF, O’Brien DA, Bhatnagar YM, Dym M. Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. The Journal of cell biology. 1977; 74:68–85.
26. Aumuller G, Schulze C, Viebahn C. Intermediate filaments in Sertoli cells. Microscopy research and technique. 1992; 20:50–72.
27. Wilkerson ML, Lin F, Liu H, Cheng L. The application of immunohistochemical biomarkers in urologic surgical pathology. Archives of pathology & laboratory medicine. 2014; 138:1643–1665.
28. Gassei K, Orwig KE. SALL4 expression in gonocytes and spermatogonial clones of postnatal mouse testes. PLoS One. 2013; 8:e53976.
29. Zhu A, Zhou H, Leloup C, Marino SA, Geard CR, Hei TK, Lieberman HB. Differential impact of mouse Rad9 deletion on ionizing radiation-induced bystander effects. Radiat Res. 2005; 164:655–661.
30. Kobayashi M, Hirano A, Kumano T, Xiang SL, Mihara K, Haseda Y, Matsui O, Shimizu H, Yamamoto K. Critical role for chicken Rad17 and Rad9 in the cellular response to DNA damage and stalled DNA replication. Genes Cells. 2004; 9:291–303.
31. Grushcow JM, Holzen TM, Park KJ, Weinert T, Lichten M, Bishop DK. Saccharomyces cerevisiae checkpoint genes MEC1, RAD17 and RAD24 are required for normal meiotic recombination partner choice. Genetics. 1999; 153:607–620.
32. Hofmann ER, Milstein S, Boulton SJ, Ye M, Hofmann JJ, Stergiou L, Gartner A, Vidal M, Hengartner MO. Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr Biol. 2002; 12:1908–1918.
33. Lieberman HB. Rad9, an evolutionarily conserved gene with multiple functions for preserving genomic integrity. J Cell Biochem. 2006; 97:690–697.
34. Zhang D, Ma W, Li YH, Hou Y, Li SW, Meng XQ, Sun XF, Sun QY, Wang WH. Intra-oocyte localization of MAD2 and its relationship with kinetochores, microtubules, and chromosomes in rat oocytes during meiosis. Biol Reprod. 2004; 71:740–748.