
INTRODUCTION
Esophageal squamous cell carcinoma (ESCC) is esophageal cancer’s dominate pathologic subtype with remarkable geographic variation in incidence. Eastern Asia demonstrates a comparatively high morbidity in comparison with western nations in the world [1]. For example, over 50% of all ESCC cases reported worldwide occurred in China [2]. Despite tremendous advances in diagnostic techniques and therapeutic methods, the prognosis for patients with ESCC continues to be unfavorable on account of deferred diagnosis and the inadequate efficacy of conventional therapy [3]. Therefore, it is presently pressing to develop novel agents for ESCC patients.
Natural products tend to be a source of inspiration for synthetic chemists attempting to acquire new molecular entities with distinct pharmacological activity [4, 5]. Isodon rubescens (“Donglingcao” in Chinese) is a significant source of a traditional Chinese herbal medicine that has been widely used for esophageal and cardia cancer’s treatment for many years in China [6, 7]. Many ent-kaurane diterpenoids were isolated from this herb, such as Oridonin [8], Jaridonin [9] and Eriocalyxin B [10]. In recent years, a rising attention has been being attracted by ent-kaurane diterpenoids due to their unique and extensive pharmacological activities, especially anticancer activity. In China, the injection of oridonin was used alone or in combination with other chemotherapy drugs for liver cancer’s treatment [11]. Increasing studies have also illustrated that oridonin exerts broad anti-tumor activities through regulating the cell cycle [12, 13], apoptosis [14, 15], and autophagy [16, 17], as well as the inhibition of migration and invasion [18]. Recently, from Isodon rubescens, another new ent-kaurene diterpenoid was isolated by us, named Jaridonin. Importantly, our previous results have shown that Jaridonin was more potent than oridonin in inhibiting proliferation and pro-apoptotic in various human cancer cell lines, such as colon cancer, gastric cancer and esophageal cancer cell lines [9, 19]. We were intrigued by Jaridonin’s anticancer profile to make use of its special scaffold as an introductory template to synthesize novel Jaridonin derivatives to develop effective and safe anticancer agents. Numerous studies have identified sulfur-containing compounds possess multiple biological effects supporting their potential use in multi-targeted cancer prevention and treatment [20]. And recently, expeditious synthetic methods that were based on the Jaridonin scaffold were successfully built by our group to access a series of disulfide bond-substituted derivatives with improved anticancer activity, among them, DS2 (the synthesis and the structure identification of DS2 are shown in Supplementary Figures S1, S2 and S3) suggesting that disulfide bond-substituted modifications seem to be tolerated for producing biologically interesting molecules.
In the present study, we report the effect and potential mechanisms of DS2 in cancer cells. The results indicate that DS2’s activity in ESCC cells involves the Bax regulation, and ROS generation and the activation of the mitochondria-mediated apoptotic pathway. It guarantees further research as an expected therapeutic agent for ESCC’s treatment, and will increase our knowledge about the disulfide bond-substituted derivatives.
RESULTS
The anti-proliferation effects of DS2 in human cancer cells
Chemical structures of DS2 and oridonin, a representative ent-kaurane diterpernoid from Isodon rubescens, are shown as Figure 1A. Figure 1B shows that DS2 inhibits the growth of four human tumor cell lines, including gastric carcinoma cell MGC-803, prostate cancer cell PC-3, as well as ESCC cell lines EC9706 and EC109 in a time- and dose-dependent manner. Compared to control treatment, DS2 treatment at 4 μM concentration for 24 h or 48 h resulted in about 33% or 48%, 30% or 50%, 32% or 70% and 40% or 80% growth inhibition of MGC-803, PC-3, EC9706 and EC109 cells. However, these inhibitions were not observed on proliferation of all four cell lines, with 4 μM oridonin treatment even for 48 h. It appears that DS2 is more potent in inhibiting the growth of human cancer cells than oridonin. Since EC9706 and EC109 cells, in particular, were relatively sensitive to DS2, the subsequent studies were performed on the two cell lines.
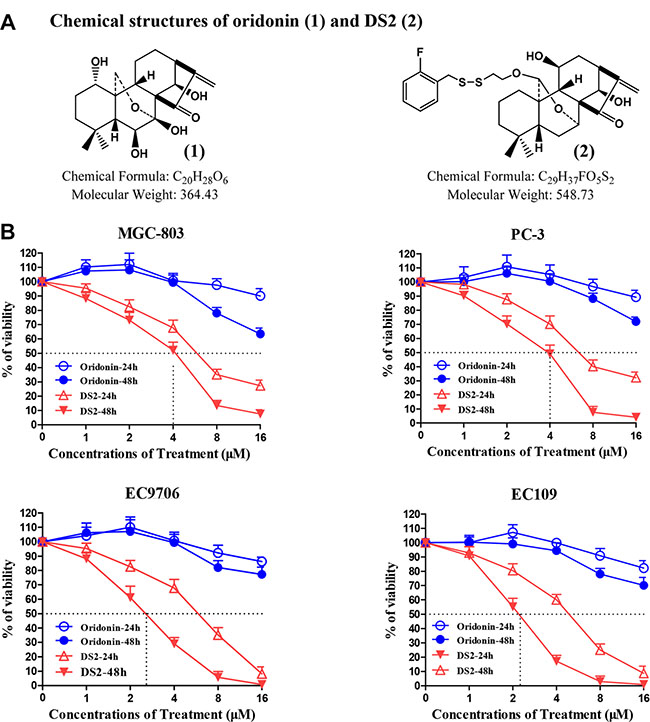
Figure 1: Effects of DS2 on the growth of human cancer cells. (A) Chemical structures of oridonin (1) and DS2 (2). (B) 6 × 103 MGC-803, PC-3, EC9706 and EC109 cells were plated in 96-well culture plates. 24 hours later, the cells were changed with fresh medium and treated with DS2 or oridonin at the indicated doses for 24 hours or 48 hours, 0.1% DMSO was used as controls. Cell viability was measured by MTT assay. Data are mean ± SD (n = 3).
DS2 resulted in G2/M phase cell cycle arrest in ESCC cells
To identify whether DS2-induced anti-proliferation implicates changes in cell-cycle progression, cell cycle phase distribution was examined by us using flow cytometry. As shown in Figure 2, a dramatic increase of G2/M phase was observed in EC9706 and EC109 cells treated by DS2 at 2, 4 and 8 μM for 12 h, and a decrease in G0/G1 and S phase cells was simultaneously observed. (Figure 2A, 2B, 2C and 2D). Moreover, as a cyclin-dependent kinase inhibitor (CDKI), p21 protein levels were observably increased by DS2 in a dose-dependent manner (Figure 2E, 2F). However, all these alterations were not observed in both EC9706 and EC109 cells treated with 8 μM oridonin. These results showed that the antiproliferative activity of DS2 was related to G2/M phase cell cycle arrest.
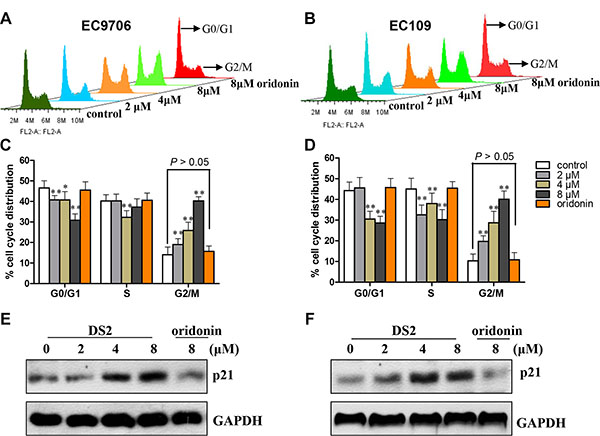
Figure 2: DS2 inhibits cell growth through G2/M phase arrest in ESCCs. (A) and (B) Representative histograms depicting cell cycle distribution in EC9706 and EC109 cell cultures treated with 0.1% DMSO (control) or 2, 4, and 8 μM DS2 or 8 μM oridonin for 12 h. Similar results were observed in three independent experiments. (C) and (D) Data are presented as mean ± SD of triplicate samples. *P < 0.05; **P < 0.01 as compared with control. (E) and (F) After ESCC cells treated with 0.1% DMSO (control) or DS2 or oridonin at the indicated doses for 12 h, the protein levels of cell cycle regulatory molecule p21 were detected by western blot. The results were representatives of three independent experiments. GAPDH was used as loading control.
DS2 induces apoptosis of ESCC cells
To determine whether the proliferation inhibitory effect of DS2 was also due to apoptosis, the EC9706 cells morphology was examined by the fluorescence microscopy stained with Hoechst 33258. As shown in the Figure 3A, cells treated by DS2 present typical apoptotic morphologies, such as cell crimp and rounding, as well as nuclear condensation and generation of apoptotic bodies, compared to controls. Similar phenomena were observed when EC109 cells were treated with DS2 (Figures not shown). The percentage of apoptotic cells with control and different doses of DS2 treatments was further determined by the flow cytometry analysis, following FITC-annexin V/PI staining. 2, 4 and 8 μM DS2 treatments of EC9706 cells for 24 hours resulted in a significant increment of FITC-Annexin V+/PI− (early apoptosis) and FITC-Annexin V+/PI+ (late apoptosis) population, compared to untreated cells (Figure 3B, 3C and 3D). Similar result was obtained when EC109 cell line was treated with DS2 (Figure 4A and 4C). Interestingly, HEECs were significantly more resistant to DS2-induced apoptosis even at 8 μM concentration (Figure 4B and 4D). Furthermore, we also studied the sensitivities of HEECs and HL-7702 cells to DS2 by trypan blue dye exclusion assay, showed relatively insensitivity to DS2 as compared to those ESCC cell lines (Figure 4E and 4F). These results indicated that DS2 selectively caused apoptosis and proliferation inhibition in cancer cell lines without affecting normal cells significantly.
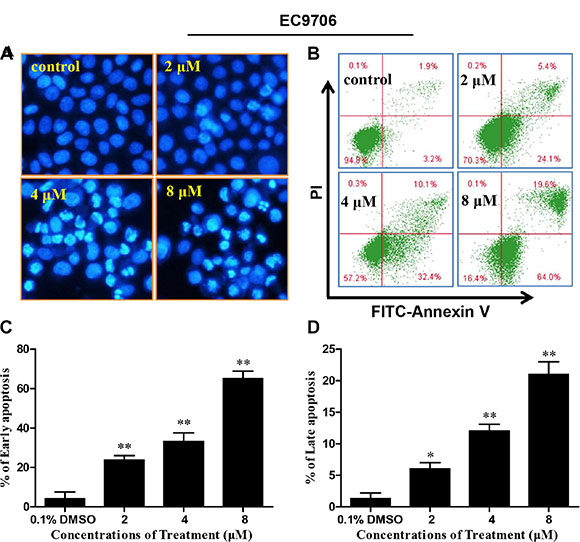
Figure 3: DS2 inhibits viability of EC9706 cells in association with apoptosis induction. (A) Morphological changes in EC9706 cells treated with DS2 as indicated doses. After treatment with DS2 for 24 h, EC9706 cells were stained with Hoechst 33258 and observed under a fluorescence microscope (magnification 200×). (B) A representative picture of flow cytometry, and percentages of cells with apoptosis were given. After EC9706 cells were treated with 0.1% DMSO or DS2 at the indicated doses for 24 h, the cells were stained with FITC-Annexin V/PI and then analyzed by flow cytometry. (C) and (D) Statistical analysis of data obtained in flow cytometer. The experiments were repeated three times and the results were presented as mean ± SD. *P < 0.05 and **P < 0.01 as compared with control.
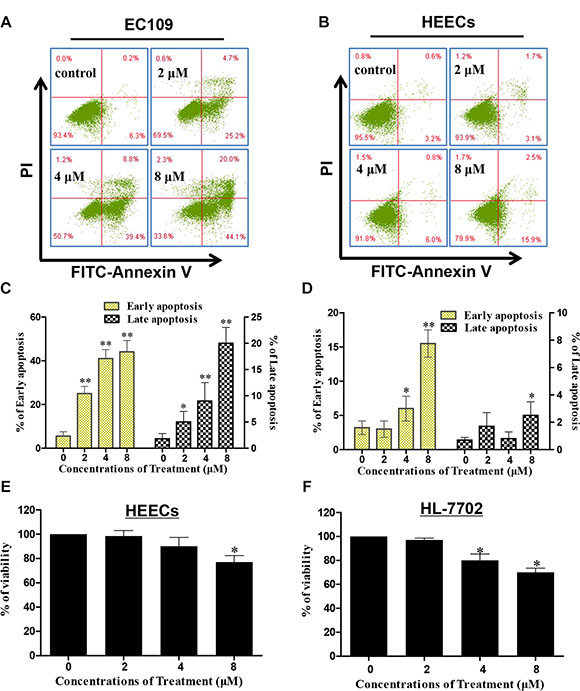
Figure 4: Effect of DS2 treatment for 24 h on EC109 cells, HEECs and normal human liver cells (HL-7702). (A) and (B) After EC109 cell and HEECs were treated with 0.1% DMSO or DS2 at the indicated doses for 24 h, the cells were stained with FITC-Annexin V/PI and then analyzed by flow cytometry. (C) and (D) Percentages of cells with early apoptosis or late apoptosis were summarized with histogram graphs. The experiments were repeated three times and the results were presented as mean ± SD. *P < 0.05 and ** P < 0.01 as compared with control. (E) and (F) Survival rates of HEECs and HL-7702 cells are determined by trypan blue dye exclusion assay. Data are presented as means ± SD of triplicate samples. *P < 0.05;**P < 0.01 as compared with control.
DS2 results in MMP drop, cytochrome c release and cleavage of caspase-3/9 in EC9706 cells
On account of the MMP collapse is a key trigger for the mitochondria dependent apoptotic pathway [21], we next examined whether DS2 affected the MMP by the JC-1 staining. JC-1 is mitochondria selective and forms aggregates in normal mitochondria that leads to an orange fluorescence under exciting at 490 nm. However, in depolarized mitochondrial membranes, JC-1 forms monomeric and emits green fluorescence [22]. After EC9706 cells were treated with 2, 4 and 8 μM DS2 for 24 hours, a clear shift in JC-1 staining of the mitochondria from orange to green fluorescence was observed in a dose-dependent manner (Figure 5A). Figure 5B presents an increase in the percentage of cells in red gate, which represents cells with depolarized mitochondrial membranes. Compared with 8.3% in the control group, the number of cells with loss of MMP increased up to 20.6%, 35.6% and 49.4%, respectively, after the treatment with 2, 4 and 8 μM DS2 for 24 hours. Similar results were also observed in EC109 cells (Supplementary Figure S4). The loss of MMP will result in cytochrome c release from mitochondria to cytosol. Consistent with the above conclusions, a distinct cytochrome c release from mitochondria into cytosol in EC9706 cells in dose-dependent manner was observed (Figure 5C), leading to an increased expression of cleaved caspases 3/9 and a decreased expression of their proenzymes (Figure 5D).
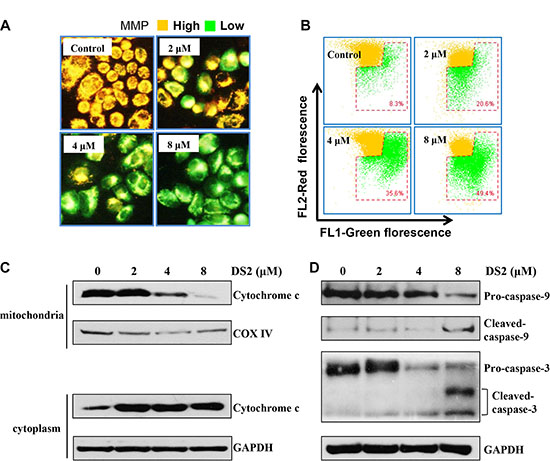
Figure 5: DS2 reduces the MMP and induces release of cytochrome c and the cleavage of caspase-9/3. EC9706 cells were incubated with DS2 at the indicated concentrations for 24 h, stained with JC-1 and then imaged by fluorescent microscope (A) and analyzed by flow cytometry, numbers in the bottom right gate, percentage of cells with low MMP (B). (C) Mitochondria and cytosolic extracts from indicated treatments for 24 h were prepared as described in Materials and Methods. Western blotting analysis was performed for cytochrome c levels. (D) Protein levels of cleaved caspase-9/-3 were determined by Western blot. GAPDH and COX IV were used as loading control.
DS2 increases protein expressions of Bax in a concentration- and time-dependent manner
Because DS2 treatment disrupted MMP resulting in cytosolic release of cytochrome c in EC9706 cells, we subsequently tested whether the apoptosis induced by DS2 was regulated by Bax, a key proapoptotic molecule in the mitochondria-dependent apoptotic pathway [23]. Treatment of EC9706 and EC109 cells with the indicated concentrations DS2 resulted in a significant up-regulation of Bax protein in a concentration-dependent manner, compared with control (Figure 6A, and 6B). Moreover, treatment of EC9706 cells with 4 μM DS2 for 6, 12, and 24 hours caused a noticeable up-regulation of Bax expression by 18-, 20-, and 31-fold, respectively, compared with control (Figure 6C). The similar results were also observed in EC109 cells, and the induction of Bax protein by DS2 was obvious as early as 6 h after treatment and was sustained for the whole experimentation (Figure 6C and 6D). However, as shown in Figure 6, the level of Bcl-2, a key anti-apoptotic protein, did not alter in EC9706 and EC109 cells treated by DS2 in indicated concentrations and times. These results showed that the DS2-induced apoptosis in ESCC cells was probably attributable to the up-regulation of Bax.
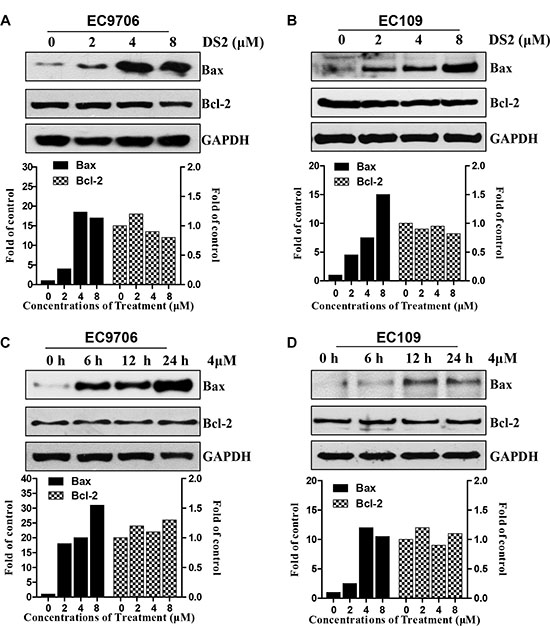
Figure 6: Effect of DS2 treatment on levels of Bax proteins. Cells were treated with DS2 as indicated doses for 24 h or 4 μM DS2 for 6, 12, and 24 h, and then lysed in cell lysis buffer. (A) and (B) Immunoblotting for Bax using lysates from EC9706 or EC109 cells treated with the indicated doses. (C) and (D) Immunoblotting for Bax using lysates from EC9706 or EC109 cells treated with 4 μM DS2 for 6, 12, and 24 h. GAPDH was used as a loading control. Relative levels of Bax protein were quantified by densitometry measurement after adjusting levels of loading controls.
The loss of MMP and apoptosis caused by DS2 are associated with Bax
To further investigate potential molecular mechanisms of DS2-induced apoptosis in EC cells, we hypothesized that Bax might play an important role in the regulation of DS2-induced apoptosis. Then we confirmed this hypothesis by using Bax siRNA. The transfection efficiency was determined by with a control fluorescent-labeled siRNA, and fluorescence was visualized using fluorescence microscope (green, Figure 7A). Furthermore, a representative immunoblot for Bax using lysates from control EC9706 cells and Bax siRNA-transfected cells following 12 h treatment with 4 μM DS2 is shown in Figure 7B. The expression of Bax protein was decrease in Bax siRNA-transfected cells even treatment with DS2. These results proved that the expression of Bax protein was silenced in Bax siRNA-transfected cells. The effect of DS2 treatment (4 μM, 12 h) on the loss of MMP and apoptosis were determined using Bax siRNA-transfected cells, and these results are shown in Figure 7C and 7D. The loss of MMP and apoptosis on treatment with DS2 was statistically significantly reversed in Bax siRNA-transfected cells (Figure 7E, 7F). Together, these results indicated that Bax was involved in DS2-induced cell death.
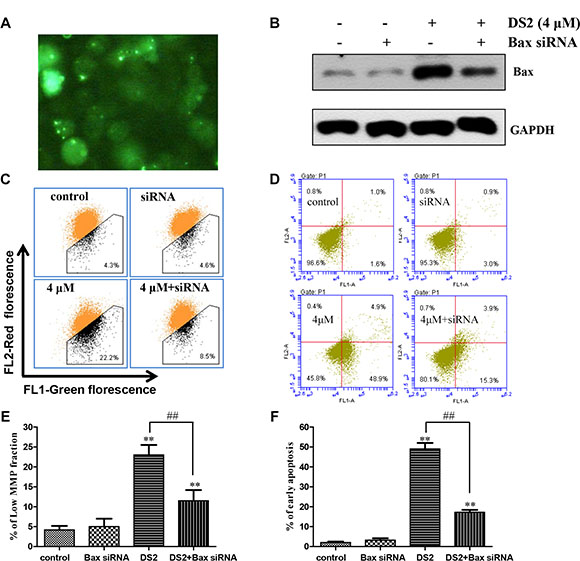
Figure 7: Bax siRNA conferred significant protection against DS2-induced MMP drop and apoptosis. (A) Uptake of fluorescent-labeled siRNA in EC9706 cells. Cells were transfected with control fluorescent-labeled siRNA (green). Shown here are representative areas of cells. (B) EC9706 cells were treated with control, Bax siRNA, and then the cells were treated with 4 μM DS2 for 24 h. Total Bax levels were shown by representative immunoblots. (C) The low MMP fractions in EC9706 cells transiently transfected with a control siRNA or Bax siRNA and treated with either 0.1% DMSO (control) or 4 μM DS2 for 24 h. (D) Cell apoptosis in EC9706 cells transiently transfected with a control siRNA or Bax siRNA and treated with either 0.1% DMSO (control) or 4 μM DS2 for 24 h. (E) and (F) Statistical analysis of data about MMP and apoptosis obtained in flow cytometer. The experiments were repeated three times and the results were presented as mean ± SD. **P < 0.01 as compared with control; ##P < 0.01, significantly different between DS2-treated control siRNA and DS2-treated Bax siRNA groups by paired t-test.
DS2-induced apoptosis and growth inhibition in ESCC cell lines are due to ROS generation
ROS is an upstream initiator for apoptosis and has been proved to be involved in the mitochondrial apoptotic pathway [24]. We have proved previously that Jaridonin-mediated apoptosis in ESCC cell lines correlates with ROS generation [9]. To investigate whether DS2 stimulates ROS generation in EC9706 cells, we measured intracellular ROS by flow cytometry method following staining with DCFH-DA [25]. The DS2-treated EC9706 and EC109 cells exhibited a concentration dependent enhancement in the percentage of DCF-positive cells (Figure 8A and Supplementary Figure S5). Similarly by flow cytometry analysis, 4 μΜ and 8 μΜ DS2 treatment for 8 hours statistically significant increased the mean DCF fluorescence up to about 2.3- and 3.9- fold, respectively, compared control treatment in EC9706 cells (P < 0.01) (Figure 8B). Interestingly, ROS generation was not observed in HEECs treated with similar DS2 for 8h (Figure 8C and 8D).
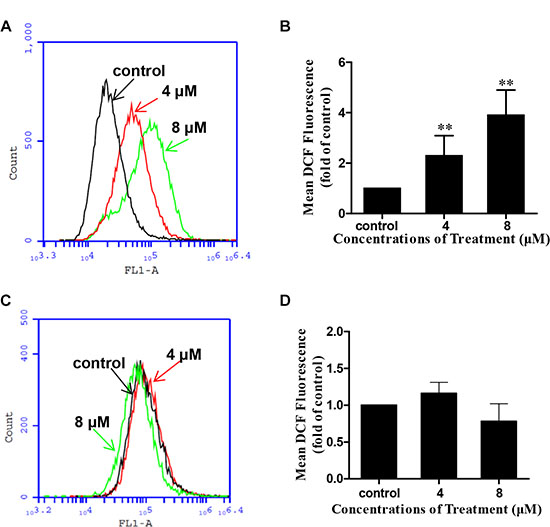
Figure 8: DS2 increased ROS generation in EC9706 cells. EC9706 or HEEC cells were treated with DS2 at indicated doses for 8 h, followed by incubation with 10 μM DCFH-DA for 30 min at 37°C. The intracellular levels of ROS were determined by flow cytometer. (A) Representative percents of DCF positive cells analysed by flow cytometer in EC9706 cultures treated with 0.1% DMSO (control) or the indicated DS2 for 8 h. (B) Columns, mean DCF fluorescence in EC9706 cells of three independent experiments; bars, SD. **P < 0.01, DS2 versus control. (C) Representative percents of DCF positive cells analysed by flow cytometer in HEECs following 8 h exposure to DMSO or the indicated concentrations of DS2. (D) Statistical analysis of data about HEECs. The experiments were repeated three times and the results were presented as mean ± SD.
To further confirm this finding, we used 5 mM ROS scavenger NAC to pre-treat EC9706 and EC109 cells for 2 hours, followed by DS2 treatment. Figures 9A and 9B show that 8 μΜ DS2 induced ROS generation was completely attenuated in EC9706 cell. Moreover, NAC pretreatment conferred near-complete protection against DS2 induced drop of the MMP (Figure 9C and Supplementary Figure S6A) and apoptosis (Figure 9D and Supplementary Figure S6B), as well as up-regulation of Bax (Figure 9E and 9F). To strengthen the involvement of ROS in DS2-induced apoptosis, the effect on cell survival of two antioxidants NAC and GSH was investigated. Consistently, pretreatment with 5 mM L-NAC and 3 mM GSH significantly attenuated the inhibitory effect of DS2 on viabilities of EC9706 and EC109 cells (Figure 9G and 9H). Taken together, our data suggest that ROS production is required, at least in part, for DS2 inducing the mitochondria-dependent apoptotic pathway and inhibiting the growth of ECSS cells.
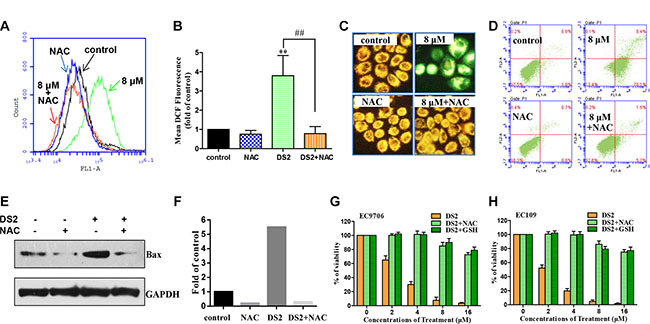
Figure 9: Effect of antioxidants on DS2 caused ROS generation, MMP drop, apoptosis, and Bax expression, as well as growth inhibition in ESCC cell lines. (A) Flow cytometer analysis percentage of DCF-positive cells in EC9706 cells treated for 12 h with 0.1% DMSO (control) or 8 μM DS2 in the absence or presence of 5 mM NAC (2 h pretreatment). (B) Columns, mean DCF fluorescence of three independent experiments; bars, SD. **P < 0.01, DS2 versus control; ##P < 0.01, DS2 versus DS2+NAC. (C) EC9706 Cells were pretreated with or without 5 mM NAC , incubated with 8 μM DS2 for 24 h, stained with JC-1 and then imaged by fluorescent microscope. (D) Cell apoptosis in EC9706 cells following 24 h treatment with 0.1% DMSO (control) or 8 μM in the absence or presence of 5 mM NAC (2 h pretreatment). (E) Western blot analysis of Bax in EC9706 cells pretreated with or without 5 mM NAC for 2 h, and then incubated with 8 μM DS2 for 24 h. (F) Relative levels of Bax protein were quantified by densitometry measurement after adjusting levels of loading controls GAPDH. (G) and (H) EC9706 or EC109 Cells were pretreated with or without 5 mM NAC or 3 mM GSH for 2 h, and then incubated with the indicated doses of DS2 for 48 h. Cell viabilities were evaluated by MTT assay. Data were expressed as means ± SD. Each data point was an average of three independent experiments.
DISCUSSION
Previous studies have indicated that natural ent-kaurane diterpenoids [26, 27] and its derivatives exhibit considerable anti-tumor activity [28, 29]. Especially, HAO472, an oridonin analog, was recently advanced into a phase I clinical trial (CTR20150246) in China by Hengrui Medicine Co. Ltd, for the treatment of acute myelogenous leukemia [30], which further confirms that ent-kaurane diterpenoids and their derivatives are the potential therapeutic agents in human cancers. In this study, we investigated DS2, a novel diterpenoid analog, the potential anti-proliferation activity using a few of human cancer cell lines, and found that DS2 displayed more potent anti-proliferation properties in a tumor-selective manner than oridonin. Compared with other cancer cells, ESCC cell lines EC9706 and EC109 were more sensitive to DS2 cytotoxicity, and this compound dramatically induced ESCC cell lines cycle arrest and apoptosis. Interestingly, these effects mediated by DS2 is not distinctly observed in normal tissue cells (eg. HEECs and HL-7702). Therefore, it is possible that this compound has a clinical translational potential for ESCC patients. Further exploration of the potential mechanisms of DS2-inducing apoptosis is indispensable.
To this end, we have traced from MMP loss and cytochrome c release to cleavage of caspase-9/3. Finally, we found that Bax proteins played a significant role in the DS2-induced apoptosis. Bax belongs to a major pro-apoptosis member of Bcl-2 family proteins, and is a central cell death regulator inducing mitochondrial membrane permeabilization and apoptosis-promoting molecules release from mitochondria to cytosol [31]. Many clinical anticancer drugs are known to induce Bax activation to facilitate apoptosis [32, 33], and increasing new molecular entities are synthesized and evaluated in direct activation of Bax [23, 34]. Intriguingly, accumulating evidence demonstrates that Bax can serve as a promising direct target for drug discovery [23]. Because DS2 treatment disrupted MMP leading to cytosolic release of cytochrome c in EC9706 cells, we then tested whether DS2-induced apoptosis was related to Bcl-2 family proteins. The present results indicated that the Bax protein levels were significantly increased by DS2 in a concentration- and time-dependent manner, and the DS2-mediated induction of Bax protein was obvious as early as 6 h after treatment and was sustained for the whole experimentation. But the change of Bcl-2 levels was not observed in EC9706 and EC109 cells. More interestingly, Bax knockdown in EC9706 cells could afford statistically significant protection against DS2-induced drop of MMP and apoptosis. These results pointed out that the DS2-induced apoptosis in ESCC cells was probably attributable to the up-regulation of Bax. It is important to point out, however, that Bax cannot fully explain the cytotoxicity caused by DS2 because the knockdown of Bax confers only partial protection against DS2-caused MMP collapse and apoptosis. Other regulators of mitochondria-mediated apoptosis, including promoters (Bak for example) or inhibitors (Bcl-xL for example), might be involved in DS2-induced apoptosis. Moreover, in ESCC cells, the parent compound, Jaridonin dramatically increases expression of p53, which is recognized as the upstream regulator of Bax [9]. Whether is p53 required for DS2, a Jaridonin analog induced cancer cells death? Therefore, further investigations are necessary to systematically identify these possibilities.
Cancer is frequently thought of as a cell cycle disease, cell cycle blockade is therefore considered as a valid strategy for wiping out cancer cells [35, 36]. In the present study, we also proved that DS2 worked on cell-cycle progression, and was more potential than oridonin. Previous studies have demonstrated that oridonin and its derivatives induced the expression of p21 and p21 dependent G2/M phase cell cycle arrest [37, 38]. Consistent with the previous reports, DS2 increased the levels of p21 protein in both the EC9706 and EC109 cells in dose-dependent manners. As a major endogenous CDKI, p21 has broad specificity for cyclin-dependent kinase (CDKs), and primarily prevents cell cycle progression. Besides, it also plays an important role in regulating other cellular processes, including apoptosis, senescence and differentiation [39]. It was reasonable to think that p21 was involved in DS2-induced G2/M phase arrest. Although the exact mechanisms remained to be elucidated, it was verified that DS2 inhibited proliferation via G2/M phase cell cycle arrest.
Elevated ROS levels and oxidative stress have been connected with apoptosis that was induced by a number of anticancer agents [40, 41]. Consistent with these reports, we demonstrated, for the first time, that the DS2-induced cell death in ESCC cells correlated with ROS generation. The present study indicates DS2 results in ROS generation in ESCC cells, and the DS2-induced drop of MMP, apoptosis, and increase of Bax are attenuated by NAC. Moreover, the growth inhibition caused by DS2 is also completely reversed on cotreatment with ROS scavenger, NAC and GSH in EC9706 and EC109 cells. It is possible that the level of ROS after treatment of cancer cells with DS2 is sufficient to provoke increase of Bax and apoptosis.
In conclusion, we have identified a novel apoptosis inducer and anticancer agent, DS2, a synthetic analog of natural ent-kaurane diterpene. The mechanisms of DS2–inducing apoptosis are, at least partly, through both ROS generation and the Bax-dependent mitochondria mediated pathway. Our findings should encourage not only the potential of DS2 as a candidate for the treatment of human ESCC but also the development of more hopeful diterpene derivatives for preventing and treating human cancers.
MATERIALS AND METHODS
Reagents and antibody
The primary antibodies for p21, cytochrome c, caspase-9, caspase-3, Bcl-2 and Bax were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). siRNA targeted against Bax and fluorescein conjugate siRNA were also from Santa Cruz Biotechnology. The siRNA transfection reagent Lipofectamine 2000 was from Invitrogen (Carlsbad, CA, USA). The antibody against GAPDH was from Good HERE Biotech Inc. (Hangzhou, China), and anti-COX IV antibody was from Abbkine Inc. (CA, USA). The horseradish peroxidase-conjugated secondary antibodies were obtained from Zhongshan Golden Bridge Biotech Inc. (Beijing, China). FITC-Annexin V/ PI apoptosis assays kit was from Biovision, Inc. (Palo Alto, CA). The ROS detection kit, GSH, NAC and JC-1 probe were all purchased from Beyotime Institute of Biotechnology (Jiangsu, China). Enhanced chemiluminescence (ECL) detection reagents were from Pierce Biotechnology, Inc. (Rockford, IL). Hoechst 33258, propidium iodide (PI) and 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) were from Sigma (St. Louis, USA).
Cell culture conditions and compounds
Human esophageal cancer cell lines EC9706 and EC109, human gastric cancer cell line MGC-803, human prostate carcinoma cell line PC-3, and primary normal human liver cells (HL-7702) were purchased from China Center for Type Culture Collection (CCTCC, Shanghai, China). EC9706 cell line has been proven to be esophageal carcinoma of the fungating type, which is poorly-differentiated squamous cell carcinoma, and EC109 cell line is well-differentiated [19]. HEECs were obtained from Wuhan PriCells Biomedical Technology Co., Ltd. (Wuhan, China). All the cell lines were cultured in RPMI 1640 medium, containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were incubated at 37°C in a humidified atmosphere, containing 5% CO2. Pure oridonin was isolated from Isodon rubescens, and DS2 was synthesized in our laboratory. The chemical structures are shown in Figure 1A and were confirmed by NMR and IR data. Purities were determined by HPLC and were all above 98%. DS2 and oridonin were dissolved in dimethyl sulfoxide (DMSO), aliquoted, and stored at −80°C. The concentration of DMSO in culture medium was kept below 0.1% (v/v), a concentration known not to affect cell proliferation.
Cell viability assay
MGC-803, PC-3, EC9706, and EC109 cells were plated at a density of 6 × 103/well in 96-well plates in complete culture medium. After 24 hours, the cells were refreshed with fresh medium, untreated or treated as indicated in the figure legends. 24 h or 48 h later, MTT was added at a final concentration of 1 mg/mL and incubated at 37°C for 4 hours. The absorbance was determined at 490 nm. The effect of DS2 on HEECs and HL-7702 viability was determined by trypan blue dye exclusion assays as described previously [19].
Cell cycle analysis
Cells were seeded in six-well plates and treated with DS2 at the indicated concentrations for 12 h, trypsinized, washed in PBS, and fixed in ice cold 70% ethanol overnight. The fixed cells were washed twice with PBS, and then resuspended in mixture solution (0.5% Triton X-100, 0.5 mg/ml RNase and 100 μg/ml PI in PBS), incubated at 37°C for 30 minutes. DNA contents/cells were analyzed in Accuri C6 flow cytometer (Becton, Dickinson & Co.; Franklin Lakes, NJ), with a total of 10,000 cells tested. The histograms of DNA distribution were modeled as a sum of G0/G1, S and G2/M phase, using FlowJo software.
Hoechst 33258 staining and apoptosis analysis by flow cytometry
As we have described previously [9], the DS2-induced apoptosis was examined and identified according to the condensation and fragmentation of their nuclei by fluorescence microscopy after Hoechst 33258 staining. FITC- annexin V and PI was used to stain and identify subpopulations of cells with membrane changes (early stage apoptosis) and the associated loss of membrane integrity (necrotic or late apoptotic cells), respectively.
Detection of MMP
MMP was measured using a potential-sensitive dye JC-1 [42]. Briefly, cells were exposed to DS2 alone at the indicated concentrations or DS2 in combination with NAC (5 mM) for 24 h. At the end of the incubation period, the cells were collected and washed twice with cold PBS, and then incubated with medium containing 10 μg/mL JC-1 for 30 min at 37°C. The MMP was measured under an excitation at 488 nm using a fluorescence microscopy, or analyzed by flow cytometry.
Determination of cytochrome c release from the mitochondria
EC9706 cells were treated with 2, 4 and 8 μM DS2 for 24 hours. The mitochondria and cytosol fractions were prepared as described before [9]. Briefly, the homogenate was subjected to centrifuging at 600 g for 10 minutes to remove nuclei and unbroken cells. Then the supernatant was collected and centrifuged again at 12,000 g for 30 minutes at 4°C to obtain the cytosol (supernatant) and the mitochondria (pellet) fraction. Samples of the cytosol and the mitochondria were dissolved in lyses buffer were subjected to western blotting.
Western blotting analysis
After treatment under each experimental condition, cells were lysed as described previously. Clarified protein lysates (30–80 μg) were electrophoretically resolved by 10% SDS-polyacrylamide gel, transferred to nitrocellulose membranes, and probed with primary antibodies. Immunoblotting was performed as described by us previously [9].
Measurement of ROS generation
Intracellular ROS generation was determined using the redox-sensitive probes 2′, 7′-dichlorodihydrofluorescein diacetate (DCFH-DA) [43]. Control and treated cells as indicated in the figure legends were washed twice with PBS, and then incubated with serum free medium containing 10 μM DCFH-DA for 30 minutes at 37°C. Cells were washed three times with serum free RPMI1640 medium to remove extracellular fluorescent dye. The fluorescence signal was determined by a flow cytometer.
siRNA transfection
EC9706 were transfected with Bax siRNA, control siRNA, fluorescein conjugate control siRNA as per the manufacturer’s instruction with minor modifications. For transfection, cells were plated in 6-well plates and transfected at 30% confluence with siRNA duplexes (final concentration 70 nM) using Oligofectamine according to the manufacturer’s recommendations. After 48 h, cells were treated with 4 μM DS2 or 0.1% DMSO for 12 h. Both floating and adherent cells were collected, washed with PBS, and processed for MMP and apoptosis analysis as described above.
Statistics
Data are expressed as mean ± standard deviation (SD). The significance of two groups was determined with a two-tailed student’s t-test. Analysis of multiple groups was performed by analysis of variance (ANOVA). The differences were considered significant at P < 0.05.
ACKNOWLEDGMENTS AND FUNDING
This work was financially supported by the National Natural Science Foundation of China (no. 81502952) and the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (no. 2009ZX09102-154).
CONFLICTS OF INTEREST
The authors confirm that this article content has no conflicts of interest.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69–90.
2. Zhao CL, Yu WJ, Gao ZQ, Li WT, Gao W, Yang WW, Feng WG, Ju JY. Association of BCSC-1 with human esophageal squamous cell carcinoma. Neoplasma. 2015; 62:765–769.
3. Ning ZH, Zhao W, Li XD, Chen LJ, Xu B, Gu WD, Shao YJ, Xu Y, Huang J, Pei HL, Jiang JT. The status of perineural invasion predicts the outcomes of postoperative radiotherapy in locally advanced esophageal squamous cell carcinoma. Int J Clin Exp Pathol. 2015; 8:6881–6890.
4. Barnes EC, Kumar R, Davis RA. The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat Prod Rep. 2016; 33:372–381.
5. Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015; 14:111–129.
6. Ding CY, Zhang YS, Chen HJ, Yang ZD, Wild C, Chu LL, Liu HL, Shen Q, Zhou J. Novel nitrogen-enriched oridonin analogs with thiazole-fused a-ring: protecting group-free synthesis, enhanced anticancer profile, and improved aqueous solubility. J Med Chem. 2013; 56:5048–5058.
7. Sun HD, Huang SX, Han QB. Diterpenoids from isodon species and their biological activities. Natural Product Reports. 2006; 23:673–698.
8. Zhou GB, Kang H, Wang L, Gao L, Liu P, Xie J, Zhang FX, Weng XQ, Shen ZX, Chen J, Gu LJ, Yan M, Zhang DE, et al. Oridonin, a diterpenoid extracted from medicinal herbs, targets AML1-ETO fusion protein and shows potent antitumor activity with low adverse effects on t (8;21) leukemia in vitro and in vivo. Blood. 2007; 109:3441–3450.
9. Ma YC, Ke Y, Zi XL, Zhao W, Shi XJ, Liu HM. Jaridonin, a novel eiterpenoid from isodon rubescens, induces reactive oxygen species-mediated apoptosis in esophageal cancer cells. Current Cancer Drug Targets. 2013; 13:611–624.
10. Wang L, Zhao WL, Yan JS, Liu P, Sun HP, Zhou GB, Weng ZY, Wu WL, Weng XQ, Sun XJ, Chen Z, Sun HD, Chen SJ. Eriocalyxin B induces apoptosis of t (8;21) leukemia cells through NF-jB and MAPK signaling pathways and triggers degradation of AML1-ETO oncoprotein in a caspase-3-dependent manner. Cell Death and Differentiation. 2007; 14:306–317.
11. Ding CY, Zhang YS, Chen HJ, Yang ZD, Wild C, Ye N, Ester CD, Xiong AL, White MA, Shen Q, Zhou J. Oridonin ring a-based diverse constructions of enone functionality: identification of novel dienone analogues effective for highly aggressive breast cancer by inducing apoptosis. J Med Chem. 2013; 56:8814–8825.
12. Cheng Y, Qiu F, Ye YC, Tashiro S, Onodera SS, Ikejima T. Oridonin induces G2/M arrest and apoptosis via activating ERK-p53 apoptotic pathway and inhibiting PTK-Ras-Raf-JNK survival pathway in murine fibrosarcoma L929 cells. Arch Biochem Biophys. 2009; 490:70–75.
13. Qi XL, Zhang DR, Xu X, Feng FF, Ren GJ, Chu QQ, Zhang Q, Tian KL. Oridonin nanosuspension was more effective than free oridonin on G2/M cell cycle arrest and apoptosis in the human pancreatic cancer PANC-1 cell line. Int J Nanomedicine. 2012; 7:1793–1804.
14. Bao RF, Shu YJ, Wu XS, Weng H, Ding Q, Cao Y, Li ML, Mu JS, Wu WG, Ding QC, Tan ZJ, Liu TY, Jiang L, et al. Oridonin induces apoptosis and cell cycle arrest of gallbladder cancer cells via the mitochondrial pathway. BMC Cancer. 2014; 14:217.
15. Shi M, Lu XJ, Zhang J, Diao H, Li G, Xu L, Wang T, Wei J, Meng W, Ma JL, Yu H, Wang YG. Oridonin, a novel lysine acetyltransferases inhibitor, inhibits proliferation and induces apoptosis in gastric cancer cells through p53- and caspase-3-mediated mechanisms. Oncotarget. 2016; 7:22623–22631. doi: 10.18632/oncotarget.8033.
16. Cheng Y, Qiu F, Ikejima T. Molecular mechanisms of oridonin-induced apoptosis and autophagy in murine fibrosarcoma L929 cells. Autophagy. 2009; 5:430–431.
17. Li Y, Wang Y, Wang SH, Gao YJ, Zhang XF, Lu CH. Oridonin phosphate-induced autophagy effectively enhances cell apoptosis of human breast cancer cells. Med Oncol. 2015; 32:365.
18. Wang S, Zhong Z, Wan J, Tan W, Wu G, Chen M, Wang Y. Oridonin induces apoptosis, inhibits migration and invasion on highly-metastatic human breast cancer cells. Am J Chin Med. 2013; 41:177–196.
19. Ma YC, Su N, Shi XJ, Zhao W, Ke Y, Zi X, Zhao NM, Qin YH, Zhao HW, Liu HM. Jaridonin-induced G2/M phase arrest in human esophageal cancer cells is caused by reactive oxygen species-dependent Cdc2-tyr15 phosphorylation via ATM-Chk1/2-Cdc25C pathway. Toxicol Appl Pharm. 2015; 282:227–236.
20. Fimognari C, Lenzi M, Hrelia P. Apoptosis induction by sulfur-containing compounds in malignant and nonmalignant human cells. Environmental and Molecular Mutagenesis. 2009; 50:171–189.
21. Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998; 281:1309–1312.
22. Yan J, Pang YQ, Sheng JF, Wang YL, Chen J, Hu JH, Huang L, Li XS. A novel synthetic compound exerts effective anti-tumour activity in vivo via the inhibition of tubulin polymerisation in A549 cells. Biochemical Pharmacology. 2015; 97:51–61.
23. Liu ZQ, Ding Y, Ye N, Wild C, Chen HY, Zhou J. Direct activation of Bax protein for cancer therapy. Med Res Rev. 2016; 36:313–341.
24. Yang Y, Karakhanova S, Hartwig W, D’Haese JG, Philippov PP, Werner J, Bazhin AV. Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. 2016; 231:2570–2581.
25. Aranda A, Sequedo L, Tolosa L, Quintas G, Burello E, Castell JV, Gombau L. Dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay: a quantitative method for oxidative stress assessment of nanoparticle-treated cells. Toxicology In Vitro. 2013; 27:954–963.
26. Yang J, Wang WG, Wu HY, Du X, Li XN, Li Y, Pu JX, Sun HD. Bioactive Enmein-Type ent-Kaurane Diterpenoids from Isodon phyllostachys. J Nat Prod. 2016; 79:132–140.
27. Owona BA, Schluesener HJ. Molecular insight in the multifunctional effects of oridonin. Drugs R D. 2015; 15:233–244.
28. Wu J, Ding Y, Chen CHZ, Zhou ZM, Ding CY, Chen HY, Zhou J, Chen CS. A new oridonin analog suppresses triple-negative breast cancer cells and tumor growth via the induction of death receptor 5. Cancer Letters. 2016:380:393–402.
29. Chen W, Zhou JC, Wu KJ, Huang J, Ding Y, Yun EJ, Wang B, Ding CY, Hernandez E, Santoyo J, Chen HY, Lin H. Sagalowsky A, et al. Targeting XBP1-mediated β-catenin expression associated with bladder cancer with newly synthetic oridonin analogues. Oncotarget. 2016; 7:56842–56854. doi: 10.18632/oncotarget.10863.
30. Ding Y, Ding C, Ye N, Liu Z, Wold EA, Chen H, Wild C, Shen Q, Zhou J. Discovery and development of natural product oridonin-inspired anticancer agents. Eur J Med Chem. 2016; 122:102–117.
31. Brenner D, Mak TW. Mitochondrial cell death effectors. Curr Opin Cell Biol. 2009; 21:871–877.
32. Unterkircher T, Cristofanon S, Vellanki SH, Nonnenmacher L, Karpel-Massler G, Wirtz CR, Debatin KM, Fulda S. Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBid stability and mitochondrial apoptosis. Clin Cancer Res. 2011; 17:4019–4030.
33. Ling YH, Lin R, Perez-Soler R. Erlotinib induces mitochondrial-mediated apoptosis in human H3255 non-small-cell lung cancer cells with epidermal growth factor receptor L858R mutation through mitochondrial oxidative phosphorylation-dependent activation of BAX and BAK. Mol Pharmacol. 2008; 74:793–806.
34. Souers AJ, Leverson JD, Boghaert ER, SAckler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013; 19:202–208.
35. Park MT, Lee SJ. Cell cycle and cancer. Journal of Biochemistry and Molecular Biology. 2003; 36:60–65.
36. Buolamwini JK. Cell cycle molecular targets in novel anticancer drug discovery. Current Pharmaceutical Design. 2000; 6:379–392.
37. Li X, Li X, J. Wang, Z. Ye, J.C. Li. Oridonin up-regulates expression of P21 and induces autophagy and apoptosis in human prostate cancer cells. Int J Biol Sci. 2012; 8:901–912.
38. Zhang JH, Kang N, Qiu F, Chen S, Tashiro S, Onodera S, Ikejima T. Induction of G/M phase arrest and apoptosis by oridonin in human laryngeal carcinoma cells. J Nat Prod. 2010; 73:1058–1063.
39. Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst). 2016; 42:63–71.
40. Chen WQ, Zou P, Zhao ZW, Weng QY, Chen X, Ying SL, Ye QQ, Wan Z, Ji JS, Liang G. Selective killing of gastric cancer cells by a small molecule via targeting TrxR1 and ROS-mediated ER stress activation. Oncotarget. 2016; 7:16593–16609. doi: 10.18632/oncotarget.7565.
41. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013; 12:931–947.
42. Xu J, Timares L, Heilpern C, Weng Z, Li C, Xu H, Pressey JG, Elmets CA, Kopelovich L, Athar M. Targeting wild-type and mutant p53 with small molecule CP-31398 blocks the growth of rhabdomyosarcoma by inducing reactive oxygen species-dependent apoptosis. Cancer Res. 2010; 70:6566–6576.
43. Sankpal UT, Nagaraju GP, Gottipolu SR, Hurtado M, Jordan CG, Simecka JW, Shoji M, EI-Rayes B, Basha R. Combination of Tolfenamic acid and curcumin induces colon cancer cell growth inhibition through modulating specific transcription factors and reactive oxygen species. Oncotarget. 2016; 7:3186–3200. doi: 10.18632/oncotarget.6553.