
INTRODUCTION
An estimated 1.8 million new lung cancer cases occurred in 2012, accounting for approximately 13% of total cancer diagnoses. In men, lung cancer was the most commonly diagnosed cancer and the leading cause of cancer-related death in 2012. In women, lung cancer was the leading cause of cancer-related death in more developed countries and the second leading cause of cancer-related death in less developed countries [1]. Smoking, particularly of cigarettes, is by far the main contributor to lung cancer, and other risk factors include exposure to radon gas, asbestos, air pollution and some metals, and genetic alterations [2–8]. Similar to many other cancers, lung cancer is initiated by activation of oncogenes or inactivation of tumor suppressor genes [9–11]. The molecular mechanisms involved in lung tumorigenesis remain incompletely understood.
STIM1 (stromal interaction molecule 1) was identified as a novel human gene that maps to a region of chromosome 11p15.5 [12]. STIM1 protein is mainly resident in the endoplasmic reticulum (ER), and activates a set of plasma membrane Ca2+ channels termed store-operated calcium channels (SOCs) when the concentration of free Ca2+ within the ER drops transiently as a result of Ca2+ release from this compartment [13]. It has been reported that STIM1 protein has novel and unexpected physiological and pathophysiological roles in several tissues [14], the clinical phenotype of STIM1-deficient patients was characterized by immunodeficiency together with autoimmune disease, congenital myopathy and ectodermal dysplasia [15].
STIM1 was initially suggested to be a tumor suppressor given that it induced the inhibition of rhabdoid tumor and rhabdomyosarcoma cell lines [16]. Suyama et al. reported that knocking down STIM1 accelerated the cell motility of melanoma cells, and STIM1 was identified as an anti-metastasis gene [17]. However, recent data reveal opposite functions. Wong et al. reported that the presence of STIM1 overexpression in colon adenocarcinomas was associated with cell migration and cell motility properties [18]. Yang et al. reported that STIM1 silencing inhibited the migration and metastasis of breast cancer cells [19]. Chen et al. reported that a poorer clinical outcome was statistically significant in primary tumors with STIM1 upregulation. STIM1 overexpression markedly enhanced cervical tumor cell growth, local spread and angiogenesis and promoted cancer cell migration and invasion [20]. Additional studies showed that knocking down STIM1 resulted in reduced cancer metastasis in colorectal cancer [21], melanoma [22, 23] and hepatocellular carcinoma [24].
However, the potential role of STIM1 in lung tumorigenesis has not yet been clearly understood. One published paper reported that higher levels of STIM1 protein expression were observed in the samples of patients with non-small cell lung cancer (NSCLC) compared with adjacent non-neoplastic lung samples as detected by immunoblotting analysis and immunohistochemistry [25]. To further understand the potential role of STIM1 in lung tumorigenesis, we measured the expression levels of STIM1 in malignant human bronchial epithelial (16HBE) cells transformed by benzo(a)pyrene (BaP) and mice lung tissues treated with BaP. We also investigated the influences of STIM1 silencing on the proliferation and tumor formation of A549 cells in in vitro and in vivo experiments, and the distribution of cell cycle of A549 cells.
RESULTS
Elevated STIM1 expression in human lung tumors
To investigate whether or not STIM1 has a potential role in lung cancer, we examined STIM1 expression by qRT-PCR assay. Thirty human lung cancer tissues and paired adjacent non-neoplastic lung tissues were collected. Our results demonstrated that STIM1 mRNA expression was higher in human lung tumors than that in adjacent non-neoplastic lung tissues (P = 0.001) (Figure 1).
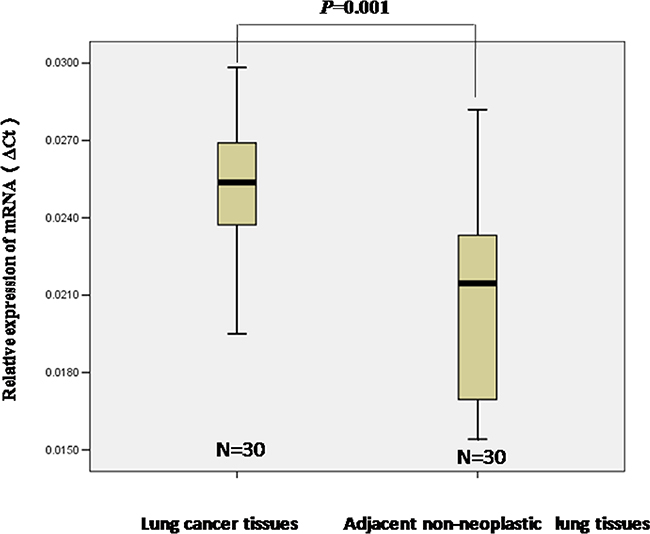
Figure 1: The relative levels of STIM1 mRNA expression was detected by qRT-PCR. The level of STIM1 mRNA expression was higher in lung tumors than that in adjacent non-neoplastic lung tissues. Median values are indicated, and the Mann-Whitney U test was performed.
Elevated STIM1 expression in malignant 16HBE cells transformed by BaP and mice lung tissues treated with BaP
STIM1 mRNA and protein expression profiles were examined by qRT-PCR and western blot assay. Increased STIM1 protein (Figure 2A) and mRNA (Figure 2B) expression was observed in 16HBE-BaP cells compared with 16HBE-control cells. Consistent with the data from cell line, increased expression of STIM1 protein (Figure 2C) and mRNA (Figure 2D) was observed in mice lung tissues treated with BaP as compared with the control group. The immunohistochemistry analysis demonstrated that the staining signal of STIM1 was strong in the nucleus and cytoplasm in some cells in the BaP-treated group but weak in the control group (Figure 2E).
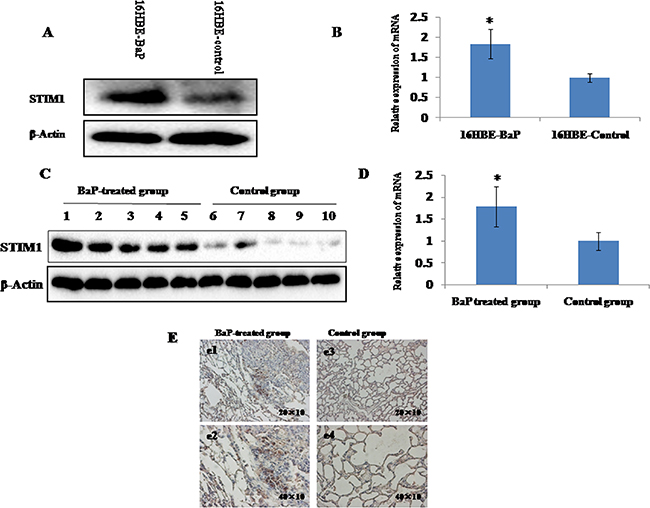
Figure 2: STIM1 expression in malignant 16HBE cells transformed by BaP and BaP-treated mice lung tissues were measured by qRT-PCR and western blot assay. The levels of STIM1 protein A. and mRNA B. were higher in malignant 16HBE cells transformed by BaP than those in control cells. The levels of STIM1 protein C. and mRNA D. were higher in BaP-treated mice lung tissues than those in the control group. The level of STIM1 in BaP-treated mice lung tissues was detected by immunohistochemistry E. Plates e1 (200×) and e2 (400×) represent the lung sections of BaP-treated group, and plates e3 (200×) and e4 (400×) represent the lung sections of the control group. The BaP-treated group exhibited strong cytoplasmic and nuclear STIM1 staining in some cells (e1 and e2). * P < 0.05.
Silencing efficiency of STIM1 gene in A549 cells
Figure 3A showed that the markedly decreased expression of STIM1 protein was observed in A549-shRNA-STIM1 cells as compared to A549-shRNA-control cells. This observation suggested that an A549 cell line stably silencing STIM1 was successfully engineered.
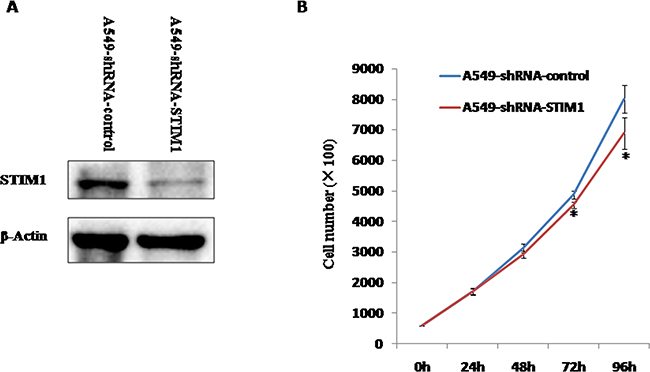
Figure 3: The silencing efficiency of STIM1 in A549 cells and the effects of silencing STIM1 on A549 cell proliferation. A. Western blot analysis showed that shRNA specifically targeting STIM1 markedly decreased the level of STIM1 protein expression in A549 cells compared with control. B. STIM1 silencing inhibited A549 cell proliferation at 72 h and 96 h. *P<0.05.
Silencing STIM1 suppressed the proliferation of A549 cells
We investigated the effects of STIM1 silencing on the proliferation of A549 cells. Our results indicated that the significantly slower growth rate was observed in A549-shRNA-STIM1 cells as compared to A549-shRNA-control cells at 72 h and 96 h (P < 0.05).
Silencing STIM1 attenuated the colony formation in soft agar of A549 cells
After observing that silencing STIM1 inhibited A549 cell proliferation, we further investigated whether or not silencing STIM1 had effects on the colony formation in soft agar of A549 cells in vitro. As shown in Figure 4A and 4B, A549-shRNA-STIM1 cells (18±4) exhibited markedly decreased colony formation compared with A549-shRNA-control cells (35±5) (P < 0.05).
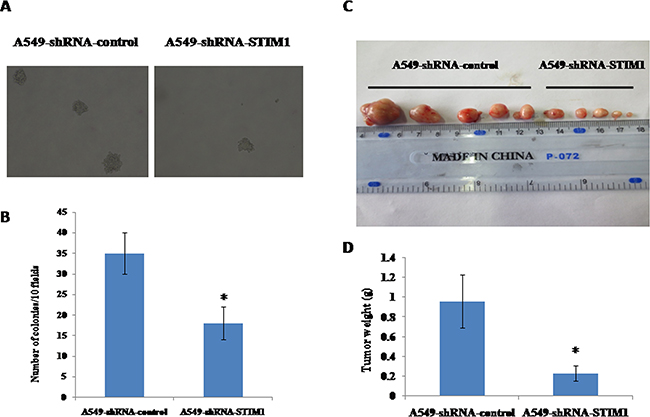
Figure 4: Effects of silencing STIM1 on colony formation and tumor growth in mouse xenograft models. Dramatically decreased colony formation was observed in A549-shRNA-STIM1 cells compared with A549-shRNA-control cells A and B. The mean of tumor weight was significantly reduced in A549-shRNA-STIM1 cells compared with A549-shRNA-control cells C and D. *P<0.05.
Silencing STIM1 suppressed tumor growth of A549 cells in vivo
We observed that silencing STIM1 inhibited colony formation in soft agar of A549 cells in vitro. To test the potential role of silencing STIM1 in the ability of tumor growth of A549 cells in vivo, nude mice xenograft models were established by subcutaneous injection with A549-shRNA-STIM1 cells and A549-shRNA-control cells. As shown in Figure 4C and 4D, on the 16th day, the mean of tumor weight was significantly decreased in the group of mice injected with A549-shRNA-STIM1 cells (0.23±0.08 g) as compared to the group of those injected with A549-shRNA-control cells (0.96±0.27 g) (P<0.05), suggesting that silencing STIM1 markedly suppressed the growth and formation of tumors of A549 cells in in vivo experiments.
Silencing STIM1 arrested cell cycle in G1 phase
We observed that silencing STIM1 inhibited A549 cell proliferation. To further investigate whether or not the cell growth inhibition was induced by cell cycle arrest, we performed the flow cytometry assay to detect the distribution of cell cycle. Our results showed that the percentage of G1 phase cells was significantly higher in A549-shRNA-STIM1 cells (57.9±2.1%) than that in A549-shRNA-control cells (47.7±2.9%) (P<0.05), and the percentage of S phase cells was markedly decreased in A549-shRNA-STIM1 cells (33.4±1.2%) than that in A549-shRNA-control cells (41.9±4.9%) (P<0.05) (Figure 5). In addition, we observed that the proliferation index in A549-shRNA-STIM1 cells (42.0±2.2%) was markedly decreased as compared to that in A549-shRNA-control cells (52.3±2.9%) (P<0.05). These findings suggested that silencing STIM1 might inhibit the growth of A549 cells by arresting the cell cycle in the G1 phase.
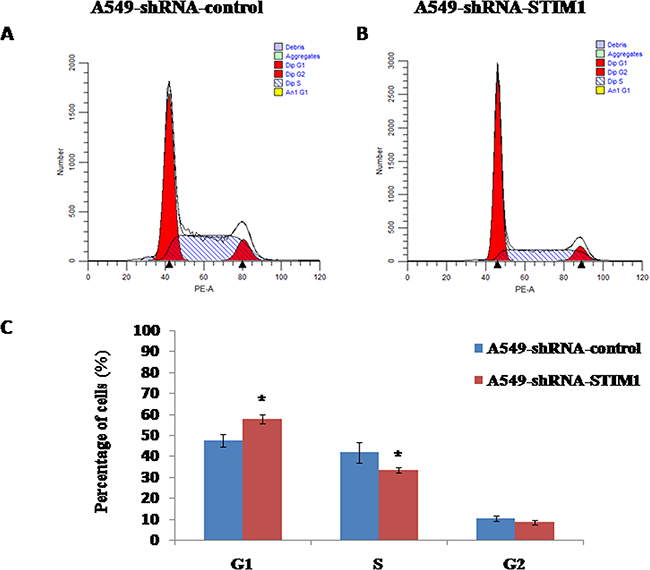
Figure 5: Effects of STIM1 silencing on cell cycle distribution. Representative flow cytometric histograms showed the distribution of cell cycle in A549-shRNA-control cells A. and A549-shRNA-STIM1 cells B. STIM1 silencing increased the percentage of cells in G1 phase and decreased the percentage of cells in S phase C. *P<0.05.
Silencing STIM1 downregulated cyclin D1 expression and upregulated p21 expression
To explore the potential mechanism related to the cell cycle arrest, the expression levels of cell cycle-related proteins were measured by western blot analysis. Our results indicated that silencing STIM1 reduced cyclin D1 protein expression and increased p21 protein expression (Figure 6A). No significant alterations in the expression levels of cyclin B1, cyclin A, CDK4 and phospho-Rb proteins were noted between A549-shRNA-STIM1 cells and A549-shRNA-control cells (Figure 6A).
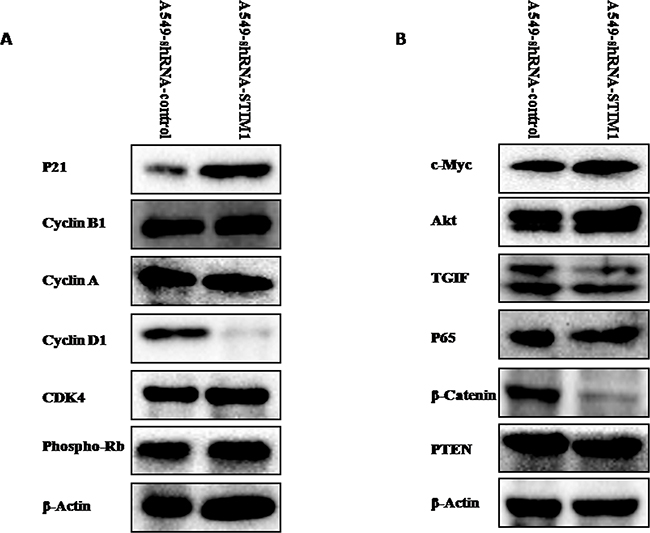
Figure 6: Effects of silencing STIM1 on the expression of several proteins. Western blot analysis showed that silencing STIM1 decreased cyclin D1, TGIF and β-catenin expression and increased p21 expression.
Silencing STIM1 downregulated the expression levels of β-catenin and TGIF proteins
We further tentatively investigated whether or not silencing STIM1 could have effects on the expression levels of Akt, c-Myc, TGIF, β-catenin, p65 and PTEN proteins, since these proteins play important roles in lung tumorigenesis. As shown in Figure 6B, western blot results indicated that the expression levels of β-catenin and TGIF proteins were significantly reduced in A549-shRNA-STIM1 cells as compared to A549-shRNA-control cells. No significant alterations in the expression levels of PTEN, c-Myc, p65 and Akt proteins were noted between A549-shRNA-STIM1 cells and A549-shRNA-control cells.
DISCUSSION
In this present study, higher expression of STIM1 mRNA was observed in lung tumors compared with adjacent non-neoplastic lung tissues, suggesting that STIM1 overexpression may be involved in lung tumorigenesis. A previous study observed higher STIM1 protein expression in lung tumors compared with adjacent non-neoplastic lung tissues detected by immunoblotting analysis and immunohistochemistry [25]. Our results may enhance the confidence of previous findings to a certain extent. To confirm the results observed in human samples, we measured STIM1 mRNA and protein expression in malignant human bronchial epithelial cells transformed by BaP and lung tissues from mice treated with BaP. Our results consistently showed that higher expression of STIM1 mRNA and protein was observed in malignant 16HBE cells transformed by BaP and lung tissues from mice treated with BaP compared with corresponding controls, which supported our findings at the cellular level and in an animal model to a certain extent.
Previous experimental studies showed that suppression of STIM1 inhibited cell proliferation of glioblastoma [26], melanoma [22], hepatocellular carcinoma [27], human epidermoid carcinoma [28] and hypopharyngeal carcinoma [29]. In this current study, we observed that silencing STIM1 inhibited A549 cell proliferation. Combined with previous results, silencing STIM1 inhibited the growth of many types of tumor cells. To further investigate the effects of silencing STIM1 on tumor formation in vitro and in vivo, a soft agar colony formation assay was performed, and a mouse tumor xenograft model was established. Our results demonstrated that suppression of STIM1 reduced the tumor growth and formation of human non-small cell lung cancer cell line of A549 in in vitro experiments and in in vivo experiments, which confirmed our above observations that STIM1 might play an important role in lung tumorigenesis.
One of the major causes of reducing the proliferation of cancer cells is cell cycle arrest. Our data indicated that silencing STIM1 arrested the cell cycle of A549 in G1 phase. The significantly increased percentage of cells in G1 phase was observed in A549-shRNA-STIM1 cells as compared to that in A549-shRNA-control cells. Accompanied with G1 phase arrest, we observed the downregulation of cyclin D1 protein in A549-shRNA-STIM1 cells. Studies showed that increased cyclin D1 expression in cancer cells resulted in an uncontrolled growth advantage [30]. In addition, silencing STIM1 in this study significantly increased p21 protein expression. p21 is one of the major cyclin-dependent kinase inhibitors (CDKI), which exerts negative effects on cell cycle progression and arrests proliferation by binding to active cyclin-CDK (cyclin-dependent kinase) complex and inhibiting their kinase activities [31–34]. Lin et al. observed that STIM1 silencing in U251 cells resulted in a marked decrease in cyclin D1 expression and a marked increase in p21 expression [26]. Chen et al. reported that knocking down STIM1 induced the expression of p21 in cervical cancer SiHa cells [20]. Taken together, the silencing of STIM1 induced G1 phase cell cycle arrest may be mediated via downregulating cyclin D1 and upregulating p21 in A549 cells. However, the potential molecular mechanisms of STIM1 regulating the expression of cyclin D1 and p21 were not investigated in this present study and should be the focus of future studies. A previous study proposed that posttranslational regulation via the 26S-proteasome-dependent pathway might contribute to the mechanisms controlling p21 upregulation in STIM1-knock-down cervical cancer cells [20].
TGIF (TG-interacting factor) was initially reported as a negative regulator of retinoic acid (RA) and the transforming growth factor β (TGF-β) signaling pathway [35, 36]. Zhang et al. reported that TGIF participated in the regulation of Wnt/β-catenin signaling pathway [37]. Our published paper revealed that the increased expression of TGIF was related to lung tumorigenesis [38]. Silencing TGIF suppressed the growth and tumor formation of A549 cells [39]. In this current study, silencing STIM1 suppressed the expression of TGIF and β-catenin proteins. One published paper reported that TGIF could regulate the expression of β-catenin protein in non-small cell lung cancer cells. Overexpression of TGIF increased the expression of β-catenin protein, and knocking down TGIF reduced the expression of β-catenin in A549 cells [40]. Taking these findings together, STIM1 may play a potential role in regulating Wnt/β-catenin signaling in NSCLC cells.
In summary, our data suggest that increased expression of STIM1 has a potential role in lung tumorigenesis. Silencing STIM1 attenuated the tumor growth of A549 cells. In addition, STIM1 suppression induced A549 cell cycle arrest in G1 phase by upregulating p21 and downregulating cyclin D1. This study provides new insights into the molecular oncogenesis of lung cancer and suggests that STIM1 may be a potential molecular target for human lung cancer therapy.
MATERIALS AND METHODS
Cell lines
The A549 cell line was cultured in RPMI-1640 medium according to our published paper [39]. Control shRNA lentiviral particles-A (sc-108080) and STIM1 shRNA (h) lentiviral particles (sc-76589-V) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). We infected A549 cells with lentiviral particles according to the manufacturer’s instructions (Santa Cruz, CA, USA). The infected A549 cells were cultured in RPMI 1640 with 10 μg/ml of puromycin for 3 weeks (Gibco, USA). The stable clone infected with STIM1 shRNA (h) lentiviral particles was termed A549-shRNA-STIM1 and the stable clone infected with control shRNA lentiviral particles was termed A549-shRNA-control. The malignant 16HBE cell line transformed by BaP (16HBE-BaP) and its control cell line (16HBE-control) were maintained in our laboratory [38].
Patient samples
Samples from thirty human lung cancer patients were reported in our previous study [38]. The histological diagnosis of the specific type of lung cancer was determined according to the recommendations of the WHO. The Ethics Committee of the First Affiliated Hospital of Zhengzhou University approved this work, and we carried out the experiments in accordance with the relevant guidelines, including any relevant details. All patients participating in this study provided informed consent.
Cell proliferation analysis
Cells (A549-shRNA-STIM1 and A549-shRNA-control) were seeded in 12-well plates (4×104 cells/well). At 24, 48, 72 and 96 h after seeding, cells were detached and counted in accordance with our previous paper [39].
Colony formation in soft agar assay
The detailed procedures of soft agar colony formation assay were described in our previous publications [38, 39].
Nude mice tumorigenicity
The Ethics Committee of Henan Center for Disease Control and Prevention approved this work. All animal experiments were carried out in accordance with the approved guidelines. Ten female BALB/c nude mice aged four weeks were obtained from Vital River Laboratory Animal Technology Co. Ltd (Beijing, China). Cells (A549-shRNA-STIM1 and A549-shRNA-control) were prepared and injected to each mouse in accordance with our previous papers [38, 39]. The mice were sacrificed at 16 days post-injection.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted using RNAiso Plus (Takara, Japan). cDNA was synthesized using the reverse transcriptase kit (Prime ScriptH RT reagent Kit-Perfect Real Time, Takara, Japan). The cDNA products were analyzed using the SYBR Green PCR Master Mix (Premix Ex TaqTM-Perfect Real Time, Takara, Japan) and 7300 Real-Time PCR machine (Applied Biosystems) according to the manufacturer’s instructions. The primers sequences used were as follows: mouse STIM1, 5’-TGAAGAGTCTACCGAAGCAGA-3’ (sense) and 5’-AGGTGCTATGTTTCACTGTTGG-3’ (antisense); human STIM1, 5’- CCTCGGTACCATCCA TGTTGTAGCA -3’ (sense) and 5’- GCGAAAGC TTACGCTAAAATGGTGTCT-3’ (antisense); and GAPDH, 5’- GACCCCTTCATTGACCTCAAC-3’ (sense) and 5’-CTTCTCCATGGTGGTGAAGA-3’(antisense). Relative STIM1 gene expression was represented as 2-ΔCt (ΔCt=CtSTIM1-CtGAPDH).
Western blot assay
Tissues or cells were lysed in RIPA buffer (Pierce, USA) supplemented with phosphatase inhibitors and protease inhibitors (Pierce, USA), and the protein concentration was quantified by the BCA kit (Pierce, USA). Soluble lysates (30 μg protein) were fractionated by 10% SDS-PAGE gels and transferred to nitrocellulose (NC) membranes (PALL, USA). The membranes were immersed in Tris-buffered saline-Tween 20 (TBST) containing 5% bovine serum albumin (BSA) to block non-specific background staining and then incubated at 4°C overnight with primary antibody. STIM1 (sc-68897), TGIF (sc-9084), Akt (sc-8312), cyclin A (sc-751), cyclin B1 (sc-752), CDK4 (sc-260), cyclin D1 (sc-718), p21 (sc-397), and β-Actin (sc-8432) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). PTEN (#9188S), phospho-Rb (#8516S), p65 (#8242S), β-catenin (#9582S), and c-Myc (#13987S) were obtained from Cell Signaling Technology (Cell Signaling, USA). The membranes were washed with TBST and incubated with goat anti-rabbit or mouse-IgG secondary antibodies (peroxidase conjugated, ZSGB-BIO, Beijing, China) at room temperature for 1 h. The immunoreactive blots were developed [39].
Cell cycle assay
The flow cytometry (BD, Biosciences) was applied to measure the distribution of cell cycle. The detailed procedures of cell cycle analysis were reported in our previous paper [39].
STIM1 immunohistochemistry
BaP-treated mice lung tissues and control lung tissues were maintained in our laboratory [38]. The Ethics Committee of Henan Center for Disease Control and Prevention approved this work. All the animal experiments were performed in accordance with the approved guidelines. Immunohistochemistry analysis was performed according to the manufacturer’s instructions (Beijing ComWin Biotech Co., Ltd; CW0120).
Statistics
The data are presented as the mean ± standard deviation (SD) or median. Student’s t test and Mann-Whitney U test were performed. All the tests were two-sided, P < 0.05 was considered to be significant. Statistical analyses were carried out by using SPSS 13.0 software (SPSS, Chicago, IL).
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (U1404815) and the Henan Collaborative Innovation Center of Molecular Diagnosis and Laboratory Medicine (XTCX-2015-PY7). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no potential conflicts of interest.
Author contributions
Y.W. contributed to method design, implementation, experiments, data analysis, and manuscript writing. H.W. and Y.W. contributed to animal model experiments and soft agar assays. L.L. and Y.W. contributed to western blot analysis. J.L. and Y.W. contributed to immunohistochemistry assays. Y.W. and T.P. contributed to quantitative polymerase chain reaction. Y.W. and H.Y. contributed to cell cycle analysis. H.Y., D. Z. and Y.W. contributed to data analysis.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J and Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65: 87-108.
2. Yoon JY, Lee JD, Joo SW and Kang DR. Indoor radon exposure and lung cancer: a review of ecological studies. Ann Occup Environ Med. 2016; 28: 15.
3. Buonanno G, Giovinco G, Morawska L and Stabile L. Lung cancer risk of airborne particles for Italian population. Environ Res. 2015; 142: 443-451.
4. Schwartz A and Cote M. Epidemiology of Lung Cancer. Adv Exp Med Biol. 2016; 893: 21-41.
5. Larsen JE and Minna JD. Molecular Biology of Lung Cancer: Clinical Implications. Clin Chest Med. 2011; 32: 703-740.
6. Hao C, Wang L, Peng S, Cao M, Li H, Hu J, Huang X, Liu W, Zhang H, Wu S, Pataer A, Heymach JV, Eterovic AK, Zhang Q, Shaw KR, Chen K, Futreal A, Wang M, Hofstetter W, Mehran R, Rice D, Roth JA, Sepesi B, Swisher SG, Vaporciyan A, Walsh GL, Johnson FM and Fang B. Gene mutations in primary tumors and corresponding patient-derived xenografts derived from non-small cell lung cancer. Cancer Lett. 2015; 357: 179-185.
7. Ha SY, Choi SJ, Cho JH, Choi HJ, Lee J, Jung K, Irwin D, Liu X, Lira ME, Mao M, Kim HK, Choi YS, Shim YM, Park WY, Choi YL and Kim J. Lung cancer in never-smoker Asian females is driven by oncogenic mutations, most often involving EGFR. Oncotarget. 2015; 6: 5465-5474. doi: 10.18632/oncotarget.2925.
8. Luanpitpong S, Wang L, Stueckle TA, Tse W, Chen YC and Rojanasakul Y. Caveolin-1 regulates lung cancer stem-like cell induction and p53 inactivation in carbon nanotube-driven tumorigenesis. Oncotarget. 2014; 5: 3541-3554. doi: 10.18632/oncotarget.1956.
9. Cooper WA, Lam DC, O’Toole SA and Minna JD. Molecular biology of lung cancer. J Thorac Dis. 2013; 5 Suppl 5: S479-490.
10. He Y, Zhou Z, Hofstetter WL, Zhou Y, Hu W, Guo C, Wang L, Guo W, Pataer A, Correa AM, Lu Y, Wang J, Diao L, Byers LA, Wistuba, II, Roth JA, Swisher SG, Heymach JV and Fang B. Aberrant expression of proteins involved in signal transduction and DNA repair pathways in lung cancer and their association with clinical parameters. PLoS One. 2012; 7: e31087.
11. De Marco C, Malanga D, Rinaldo N, De Vita F, Scrima M, Lovisa S, Fabris L, Carriero MV, Franco R, Rizzuto A, Baldassarre G and Viglietto G. Mutant AKT1-E17K is oncogenic in lung epithelial cells. Oncotarget. 2015; 6: 39634-39650. doi: 10.18632/oncotarget.4022.
12. Parker NJ, Begley CG, Smith PJ and Fox RM. Molecular cloning of a novel human gene (D11S4896E) at chromosomal region 11p15.5. Genomics. 1996; 37: 253-256.
13. Pozo-Guisado E and Martin-Romero FJ. The regulation of STIM1 by phosphorylation. Commun Integr Biol. 2013; 6: e26283.
14. Soboloff J, Rothberg BS, Madesh M and Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012; 13: 549-565.
15. Berna-Erro A, Woodard GE and Rosado JA. Orais and STIMs: physiological mechanisms and disease. J Cell Mol Med. 2012; 16: 407-424.
16. Sabbioni S, Barbanti-Brodano G, Croce CM and Negrini M. GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development. Cancer Res. 1997; 57: 4493-4497.
17. Suyama E, Wadhwa R, Kaur K, Miyagishi M, Kaul SC, Kawasaki H and Taira K. Identification of metastasis-related genes in a mouse model using a library of randomized ribozymes. J Biol Chem. 2004; 279: 38083-38086.
18. Wong HS and Chang WC. Correlation of clinical features and genetic profiles of stromal interaction molecule 1 (STIM1) in colorectal cancers. Oncotarget. 2015; 6: 42169-42182. doi: 10.18632/oncotarget.5888.
19. Yang S, Zhang JJ and Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009; 15: 124-134.
20. Chen YF, Chiu WT, Chen YT, Lin PY, Huang HJ, Chou CY, Chang HC, Tang MJ and Shen MR. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc Natl Acad Sci U S A. 2011; 108: 15225-15230.
21. Zhang Z, Liu X, Feng B, Liu N, Wu Q, Han Y, Nie Y, Wu K, Shi Y and Fan D. STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer. Oncogene. 2015; 34: 4808-4820.
22. Umemura M, Baljinnyam E, Feske S, De Lorenzo MS, Xie LH, Feng X, Oda K, Makino A, Fujita T, Yokoyama U, Iwatsubo M, Chen S, Goydos JS, Ishikawa Y and Iwatsubo K. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS One. 2014; 9: e89292.
23. Sun J, Lu F, He H, Shen J, Messina J, Mathew R, Wang D, Sarnaik AA, Chang WC, Kim M, Cheng H and Yang S. STIM1- and Orai1-mediated Ca(2+) oscillation orchestrates invadopodium formation and melanoma invasion. J Cell Biol. 2014; 207: 535-548.
24. Yang N, Tang Y, Wang F, Zhang H, Xu D, Shen Y, Sun S and Yang G. Blockade of store-operated Ca(2+) entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013; 330: 163-169.
25. Li W, Zhang M, Xu L, Lin D, Cai S and Zou F. The apoptosis of non-small cell lung cancer induced by cisplatin through modulation of STIM1. Exp Toxicol Pathol. 2013; 65: 1073-1081.
26. Li G, Zhang Z, Wang R, Ma W, Yang Y, Wei J and Wei Y. Suppression of STIM1 inhibits human glioblastoma cell proliferation and induces G0/G1 phase arrest. J Exp Clin Cancer Res. 2013; 32: 20.
27. Wu Z, Qing J, Xia Y, Wang K and Zhang F. Suppression of stromal interaction molecule 1 inhibits SMMC7721 hepatocellular carcinoma cell proliferation by inducing cell cycle arrest. Biotechnol Appl Biochem. 2015; 62: 107-111.
28. Yoshida J, Iwabuchi K, Matsui T, Ishibashi T, Masuoka T and Nishio M. Knockdown of stromal interaction molecule 1 (STIM1) suppresses store-operated calcium entry, cell proliferation and tumorigenicity in human epidermoid carcinoma A431 cells. Biochem Pharmacol. 2012; 84: 1592-1603.
29. Sun Y, Cui X, Wang J, Wu S, Bai Y, Wang Y, Wang B and Fang J. Stromal interaction molecule 1 (STIM1) silencing inhibits tumor growth and promotes cell cycle arrest and apoptosis in hypopharyngeal carcinoma. Med Oncol. 2015; 32: 150.
30. Hall M and Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996; 68: 67-108.
31. Toyoshima H and Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994; 78: 67-74.
32. Grana X and Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene. 1995; 11: 211-219.
33. Kagawa S, Fujiwara T, Hizuta A, Yasuda T, Zhang WW, Roth JA and Tanaka N. p53 expression overcomes p21WAF1/CIP1-mediated G1 arrest and induces apoptosis in human cancer cells. Oncogene. 1997; 15: 1903-1909.
34. Munoz-Alonso MJ, Acosta JC, Richard C, Delgado MD, Sedivy J and Leon J. p21Cip1 and p27Kip1 induce distinct cell cycle effects and differentiation programs in myeloid leukemia cells. J Biol Chem. 2005; 280: 18120-18129.
35. Wotton D, Lo RS, Lee S and Massague J. A Smad transcriptional corepressor. Cell. 1999; 97: 29-39.
36. Bartholin L, Powers SE, Melhuish TA, Lasse S, Weinstein M and Wotton D. TGIF inhibits retinoid signaling. Mol Cell Biol. 2006; 26: 990-1001.
37. Zhang MZ, Ferrigno O, Wang Z, Ohnishi M, Prunier C, Levy L, Razzaque M, Horne WC, Romero D, Tzivion G, Colland F, Baron R and Atfi A. TGIF governs a feed-forward network that empowers Wnt signaling to drive mammary tumorigenesis. Cancer Cell. 2015; 27: 547-560.
38. Wang Y, Wang H, Gao H, Xu B, Zhai W, Li J and Zhang C. Elevated expression of TGIF is involved in lung carcinogenesis. Tumour Biol. 2015; 36: 9223-9231.
39. Wang Y, Pan T, Wang H, Li L, Li J, Zhang C and Yang H. Silencing of TGIF attenuates the tumorigenicity of A549 cells in vitro and in vivo. Tumour Biol. 2016; 37: 12725-12730.
40. Xiang G, Yi Y, Weiwei H and Weiming W. TGIF1 promoted the growth and migration of cancer cells in nonsmall cell lung cancer. Tumour Biol. 2015; 36: 9303-9310.