
INTRODUCTION
Diffuse large B-cell lymphoma (DLBCL) comprises about 40% of non-Hodgkin lymphoma and is a heterogeneous disease in terms of the pathological changes, cell of origin (COO), clinical course, and genetic alterations. Clinically, DLBCL is a curable disease and 30–50% of patients can achieve a complete remission after first-line treatment with rituximab (R)–cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP). However, up to 40% of DLBCL patients experience disease relapse or are refractory to the initial treatment and have a poor chance of survival [1].
Many clinical, immunohistochemical, and molecular factors have been proposed for predicting the treatment response and prognosis of patients after standard R-CHOP treatment, but no reliable markers have been identified. The International Prognostic Index (IPI) is regarded as a surrogate marker of the biology of the host and tumor. The IPI is calculated using the patient’s age >60 years, abnormal increase in LDH level, Ann Arbor stage III or IV disease, >1 extranodal site, and European Cooperative Oncology Group performance status ≥2. Although the IPI is an excellent prognostic marker, it fails to predict the prognosis and treatment response in many patients [2].
DLBCL is divided into germinal center B-cell-like (GCB) DLBCL and activated B-cell-like (ABC) DLBCL based on the gene expression profile or immunohistochemistry (IHC) [3, 4]. ABC-type DLBCL is characterized by constitutive activation of the NF-κB signaling pathway and has a poorer prognosis with standard R-CHOP chemotherapy compared with GCB-type DLBCL [5]. This difference is exploited therapeutically, and pathway-targeted therapy has been developed to improve the survival of patients with ABC-type DLBCL. In addition to these biological markers, other factors including expression and rearrangement of the BCL-2 gene, [6] expression and mutation of the p53 gene, [7] expression of B2 microglobulin protein, [8] and proliferation fraction [9] have been used to predict the prognosis of patients with DLBCL.
DLBCL is associated with alterations in many different genes, with a median somatic frequency of 3.3 mutations per Mb [10]. Identification of these genetic aberrations of DLBCL is needed to provide more rational molecularly defined approaches to treatment. Deep sequencing is currently the method of choice for cataloging genomic changes in tumors. To obtain a comprehensive overview of the gene expression patterns and genomic alterations in refractory DLBCL, we produced a multidimensional genomic dataset based on data obtained from whole-exome sequencing (WES), transcriptome sequencing, and copy number variation in six patients with refractory DLBCL and seven patients with DLBCL with a good treatment response.
RESULTS
Single nucleotide variants (SNVs) and insertion/deletions (indels)
We performed WES and RNA-Seq for all 13 samples: seven from treatment-responsive patients (S group) and six from treatment-refractory patients (F group). Eight tumor–normal pairs (S1–4, F1–4) were checked primarily and had high-confidence somatic mutations. The average numbers of pathogenic somatic SNVs and indels in coding regions were 38 (range 19–66) in the S group and 71 in the F group (range 28–120). These coding variants tended to occur more frequently in the F group, although the difference was not significant (P = 0.3429, Figure 1).
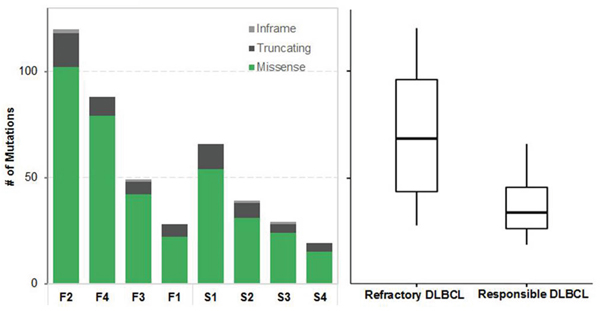
Figure 1: Summary of somatic single nucleotide variants (SNVs). Left, different colors indicate different types of nonsynonymous mutations. F respresents refractory DLBCL and S represents responsible DLBCL. Right, box plot compares number of SNVs of two groups.
Genes mutated more frequently in refractory DLBCL
SNVs of TP53 were the most frequent (50%) and exclusive to refractory DLBCL (Figure 2). All SNVs of TP53 occurred in the DNA-binding domain (Supplementary Figure S1a–S1c). Variant allele frequencies of TP53 were >60% at the DNA level and higher (>90%) when transcribed. RAB11FIP5, PRKCB, PRDM15, FNBP4, AHR, CEP128, BRE, DHX16, MYO6, and NMT1 mutations were identified more than two samples in the F group (Supplement Table S4).
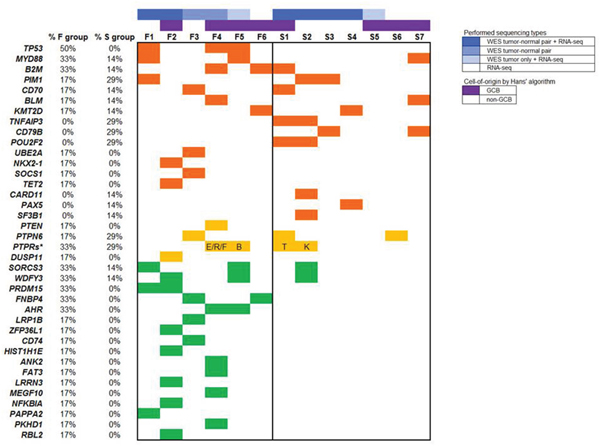
Figure 2: Mutation distribution. Each column represents a DLBCL case. Top panel in different colors represent the sequencing performed for each case and cell-of-origin subtypes. Each row represents a gene which belongs to known DLBCL-associated genes or exclusively/more frequently mutated genes in refractory DLBCL. The asterisk represents a group of protein tyrosine phosphatases receptor type (PTPRs) family genes. Different isoforms are indicated by corresponding alphabet.
MYD88, B2M, SORCS3, and WDFY3 mutations were found in two refractory cases and one responsible sample.(Figure 2). Activating hotspot mutations of MYD88 (p.Leu265Pro) have been reported to be more common in non-GCB type DLBCLs [11]. All four mutations occurred in highly conserved loci in the Toll-IL-1 receptor (TIR) domain of MYD88 (Supplementary Figure S1d). Two of four mutations in these patients occurred on the hotspot; the others involved different loci.
B2M encodes β2 microglobulin, which is a component of the class I major histocompatibility complex and is required for recognition by cytotoxic T cells. Inactivating mutations and deletions in B2M are common and account for up to 12% of DLBCLs [12]. The mutations of B2M in refractory DLBCL patients included a missense mutation (n=1) and frameshift deletion (n=1).
We assessed the mutation distribution of the major pathways associated with the pathogenesis of DLBCL which including BCR signaling, NF-kB, mammalian target of rapamycin (mTOR), phosphoinositide 3-kinase (PI3K)-protein kinase B (Akt), and JAK/STAT pathways. These are closely related with each other. BCR activation is upstream of PI3K/Akt and NF-kB pathways [13]. mTOR pathway is activated by Akt which can activate NF-kB pathway [14]. NF-kB and STAT3 collboratively mediate tumorigenesis [15]. The F group showed more frequent mutations in the PI3K-Akt and mTOR pathways (Figure 3). PTEN and TSC2 both belong to the PI3K–Akt and mTOR pathways and were mutated in F4. These are known tumor-suppressor genes (TSGs). All patients in the F group had ≥1 tumor-suppressor mutations, whereas TSG mutations were identified only in half of the patients in the S group.
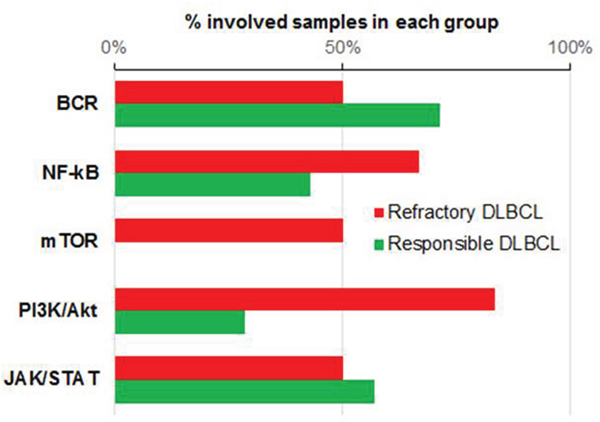
Figure 3: Mutational state of five major pathways. % of involved samples in each pathway indicated by red bar for refractory DLBCL, green for responsible DLBCL. BCR, B-cell receptor signaling pathway.
Common mutations in all DLBCL patients irrespective of treatment response
Of the 485 candidate SNVs and indels (Supplement Table S4), mutation of four genes (TP53, MYD88, PIM1, and B2M) that relate to the major driver genes in DLBCL [16, 17] were shared by three of 13 patients (23.1%). Most somatic SNVs or indels occurred in different positions, except for MYD88, which encodes p.Leu265Pro, and PIM1, which encodes p.Glu226Lys.
In addition, PTPN6, TRIP12, SORCS3, and WDFY3 were mutated in three of the 13 patients (23.1%). PTPN6 and PTEN belong to the Class I classical Cys-based phosphatase family. PTPN6 encodes SHP-1 phosphatase, which attenuates BCR signaling by dephosphorylating the ITAM motifs of the CD79A and CD79B signaling subunits of the BCR [18]. All mutations identified in this study were missense variants (p.Leu63Gln, p.Ser26Asn, p.Lys68Thr, and p.Pro105Leu), and three-quarters of the PTPN6 mutations involved the SH2 domain. One patient (S1) had both a missense mutation and copy-neutral loss of heterozygosity (CN-LOH, Supplementary Figure S3c), which suggested a loss of tumor-suppressor function. Loss-of-function mutations in PTPN6 promote STAT3 deregulation via JAK3 kinase in DLBCL [19].
We checked the mutation of genes belonging to the Class I classical Cys-based phosphatase family and found an additional eight mutations in variable receptor type protein tyrosine phosphatase and one in DUSP11. A total of 12 mutations were found in 53.8% (7/13) of patients. Five of 12 mutations in the phosphatase family involved the phosphatase domain and two occurred in the fibronectin domain, which is located in the extracellular-interacting part. Frequent mutations involving functional domains implies that alterations in phosphatases may play an important role in the pathogenesis of DLBCL.
SORCS3 encodes a type-I receptor transmembrane protein that is a member of the vacuolar protein sorting 10 receptor family. The function of SORCS3 is not known, but a missense mutation was recently reported in 6% of relapsed/refractory DLBCL (rrDLBCL) patients [16, 20]. Thyroid hormone receptor interactor 12 (TRIP12), encoding HECT domain ubiquitin E3 ligase, is vital for the homeostasis of ubiquitin-controlled events after DNA breakage [21]. WDFY encodes a phosphatidylinositol 3-phosphate-binding protein that functions as a master conductor for aggregate clearance by autophagy [22]. Insertion or missense mutations have been reported in up to 21% of rrDLBCL patients [16].
Fusion genes
To identify confident fusion transcripts, we used three different fusion callers. Eight were selected as candidates that were common in >2 fusion callers (Supplementary Table S5, Supplementary Figure S4). Five of eight were identified in two S patients and three in two F patients. We checked for differences in expression across breakpoints in each partner gene to predict functional alterations caused by fusion. Most of the partner genes did not show differences in fusion status; however, REL, BCL11A, and MAPK13 showed distinct read-depth patterns across the breakpoints (Supplementary Figure S5). The fusion transcript of PIM1–MAPK13 comprised each protein kinase domain of PIM1 and MAPK13. Both partner genes of PIM1–MAPK13 were upregulated compared with nonfusion samples, on the assumption that fusion leads to gain of function of the involved genes. In the two patients in the F group with REL–BCL11A fusion, one had both fusion and copy number gain, and the same result was identified for PIM1–MAPK13 fusion (Supplementary Figure S5). The S group exhibited known DLBCL-associated structural variations, such as translocation of BCL6 and IGLL5, and inversion of PRDM1. Inactivation of PRDM1 reported previously in DLBCL patients comprises mainly truncating mutations [23, 24]. We identified PRDM1–ATG5 fusion transcript generated by inversion. The fusion transcript was joined out-of-frame, which may reflect a newly identified mechanism of PRDM1 inactivation. BCL6 and IG are commonly translocated in DLBCL, and we identified fusion transcripts of these genes in two samples from the S group.
Copy number alterations (CNAs)
Segmental CNAs were defined based on a T/N coverage ratio >1.25 or <0.75. Segmental CNAs were found in >20% of genomes in the refractory DLBCL patients F1 and F2 (Figure 4 and Supplementary Figure S2). The minimal common regions (MCRs) of CNAs identified exclusively in refractory DLBCL are listed in Table 1. We checked the expression levels of the genes included in each MCR; the genes with concordant alterations between CNA and expression are listed in the column ‘Genes validated by RNA-seq’ in Table 1.
Table 1: Minimal common regions of copy number alterations exclusive in the refractory DLBCL
Chr |
Start |
End |
Cytoband |
Genes validated by RNA-seq |
CNA type |
F1 |
F2 |
F3 |
F4 |
F5 |
|---|---|---|---|---|---|---|---|---|---|---|
1 |
7792507 |
12726746 |
1p36.22–p36.23 |
CLSTN1, H6PD, MTOR, UBE4B |
Loss |
--- |
-- |
|||
1 |
13940794 |
16776736 |
1p36.13–p36.21 |
DDI2, SPEN |
Loss |
-- |
-- |
|||
1 |
32384570 |
35229325 |
1p34.3–p35.1 |
LCK |
Loss |
--- |
-- |
|||
2 |
58387242 |
68359137 |
2p15–p16.1 |
BCL11A, PAPOLG, REL, SLC1A4, WDPCP |
Gain |
++ |
+++ |
|||
3 |
50598347 |
56592938 |
3p21.2–p14.3 |
CACNA1D |
Gain |
++ |
++ |
|||
6 |
33169521 |
44275966 |
6p21.32–p21.2 |
CNPY3, CUL9, LEMD2, MAPK13, MEA1, NFKBIE, PFDN6, PIM1, POLR1C, TAPBP |
Gain |
++ |
+++ |
|||
7 |
96653580 |
101957868 |
7q22.1–q21.3 |
C7orf61, CLDN15, CUX1, GIGYF1, IFT22, MDSPD3, PILRB, TRRAP |
Gain |
++ |
+++ |
+++ |
||
7 |
102950761 |
112472730 |
7q31.1–q22.2 |
PMPCB, RINT1 |
Gain |
+++ |
+++ |
|||
8 |
8866541 |
11710204 |
8p23.1 |
MTMR9 |
Loss |
-- |
-- |
|||
8 |
121243705 |
142190953 |
8q24.22–q24.3 |
ZFAT |
Gain |
+++ |
++ |
|||
11 |
1256532 |
3390732 |
11p15.5–p15.4 |
Loss |
-- |
-- |
||||
11 |
9761727 |
43364203 |
11p14.1–p12 |
Gain |
++ |
++ |
||||
11 |
92623656 |
115375283 |
11q22.2–q21 |
POU2AF1 |
Gain |
++ |
+++ |
++ |
||
15 |
45335454 |
48059712 |
15q21.1 |
Loss |
--- |
-- |
||||
15 |
56974451 |
63089601 |
15q22.2–q21.3 |
RORA, VPS13C |
Loss |
-- |
-- |
|||
15 |
63092566 |
65257801 |
15q22.31–q22.2 |
SNX1 |
Loss |
-- |
-- |
-- |
+++, tumor/normal (T/N) copy ratio > 1.5; ++, T/N copy ratio > 1.25 and ≤ 1.5; -- T/N copy ratio < 0.75 and ≥ 0.5; ---, T/N copy ratio < 0.5
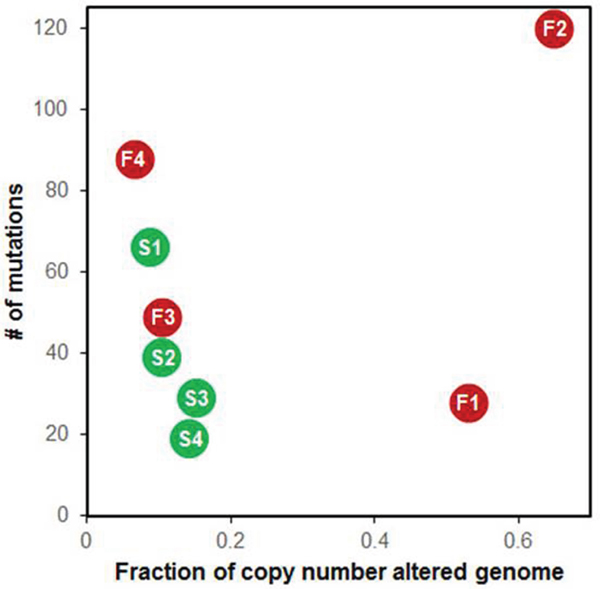
Figure 4: Relationship of somatic single nucleotide variants (SNVs) and copy number alterations. F1-4 in red dots represent refractory DLBCL, S1-4 in green dots represent responsible DLBCL.
CNAs found in refractory DLBCL
Gain of POU2AF1 was found in F1, F4, and F5 (Supplementary Figure S3). POU2AF1 protein, often called Oct-binding factor 1 (OBF-1), Oct coactivator from B cells (OCA-B), or BOB.1, is a transcriptional coactivator that is involved in transcription of immunoglobulin genes [25] and plays a role in B-cell development and Ig expression [26, 27]. Growth-promoting effects of overexpressed POU2AF1 have been demonstrated in human multiple myeloma cells from hematological malignancies [28]. In Hodgkin lymphoma, the percentage of relapsed patients after complete remission was greater in BOB.1-positive patients, and the association was stronger when BOB.1 expression was greater [29].
Gain of BCL11A, REL, XPO1, and FANCL at 2p16.3–p15 was found in two of five refractory DLBCL patients. Fusion or gain of REL, a member of the NF-κB pathway, and BCL11A was reported to be enriched in transformed lymphoma, and this may be a genomic marker for disease progression to clinically more aggressive forms [30, 31]. XPO1 encodes CRM1, an exporter of several tumor suppressor proteins. Cytoplasmic export of tumor-suppressor proteins renders them inactive, which indicates that XPO1 acts as a proto-oncogene. XPO1 mutations were reported to be significantly overrepresented in a relapsed/refractory DLBCL patient cohort and in patients with mediastinal large B-cell lymphoma [20, 32]. Gain of CACNA1D at 3p14.3–p21.1 is recurrent in refractory DLBCL patients. CACNA1D encodes the L-type voltage-gated calcium channel. In the refractory DLBCL patients in our study, CACNA1D was overexpressed compared with the responsive DLBCL patients. Gain of transformation/transcription domain-associated protein (TRRAP) and CUX1 at 7q22.1–q21.3 was present in three patients with refractory DLBCL. As a common component of many HAT complexes, TRRAP is an essential cofactor for both the c-Myc and E1A/E2F oncogenic transcription factor pathways [33, 34]. CUT-like homeobox 1 (CUX1) is a homeobox gene that is implicated in both tumor suppression and progression. Increased CUX1 expression is associated with tumor progression [35]. RNA-seq data showed significantly increased average gene expression for POU2AF1, SLC1A4, REL11, FANCL, CACNA1D, TRRAP, and CUX1 in the F group compared with the S group. Four of five patients in the F group showed a T/N copy ratio ≤0.5 for TP53 (Supplementary Figure S1); this is a CNA that is consistent with one copy deletion.
Gene expression signature in refractory DLBCL patients
We identified the genes expressed differently between the F and S groups. Using the criteria of log2FC ≥1 and FDR <0.05, we identified a total of 1531 differentially expressed genes (DEGs), including 744 upregulated and 787 downregulated genes. Upregulated DEGs were enriched in the canonical pathway gene sets associated with transcription/translation, replication/cell cycle/DNA repair, lipid synthesis, cellular energy metabolism, B cell receptor signaling, and Notch signaling (Supplementary Figure S6 and Table S6). Most significantly enriched gene sets were in the ‘transcription/translation’ group, which includes protein synthesis and degradation, genes encoding S and L ribosomal proteins, RNA polymerase subunits, zinc finger proteins, and exosome components. The ‘replication/cell cycle’ group included E2F1 transcription activator, MCM7 possessing DNA helicase activity, telomerase reverse transcriptase (TERT), and centromere proteins. The second largest group was ‘cellular energy metabolism’, which included members of the NADH: ubiquinone oxidoreductase, ubiquinol–cytochrome C reductase complex as well as adenosine triphosphate (ATP) synthase components.
Downregulated DEGs were enriched in gene sets related to the immune response, especially T-cell receptor signaling, cytokine/chemokine signaling, and the complement cascade. In 2005, a study using whole genome microarray suggested that there are three discrete subsets of DLBCL [36]: ‘oxidative phosphorylation (OxPhos)’, ‘B cell receptor/proliferation (BCR/proliferation)’, and ‘host response (HR)’. We extracted the genes classifying the three groups (‘Consensus Clusters Markers’) to determine which group refractory DLBCL belongs to. The log2FC of these genes differed significantly between the three groups (P = 5.475e–09). The median log2FC was highest in the OxPhos group and lowest in the HR group (Supplementary Figure S7); the refractory DLBCL group had a higher expression of OxPhos genes and lower expression of HR gene compared with the responsive group.
We identified cancer outlier genes (COGs) and checked the overrepresented gene sets in these COGs. The top-ranked gene sets overlapped with many of the upregulated gene sets. Additionally identified genes included the ATP-binding cassette transporter gene family. (Supplementary Table S7) Three of four refractory samples overexpressed at least one ATP-binding cassette transporter, while one of six responsible sample did. (Supplementary Figure S8) Major signaling pathways, such as the mitogen-activated protein kinase (MAPK), insulin, adipocytokine, Toll-like receptor, mTOR, and VEGF pathways, were significantly enriched (FDR <0.05, Supplementary Table S7). MAPK12 and MAPK13, which encode p38 gamma and delta, respectively, are members of the MAPK family and the p38 signaling pathway is involved in resistance to cytotoxic drugs and its inhibition by rituximab sensitizes cells to drug-induced apoptosis [38]. A recent study reported that the p38 expression is increased in CHOP-resistant patients [39]. Insulin, adipocytokine, and Toll-like receptor pathways, which share the PI3K–Akt pathway, were overexpressed in the F group. Taken together with the mutation results, these findings suggest that activation of the PI3K–Akt–mTOR pathway may be important to treatment resistance in DLBCL.
Difference in genetic alterations between cell-of-origin (COO) groups
We checked known DLBCL-associated genetic alterations and compared difference between COO groups. In a total of 13 cases, 8 were GCB and 6 were non-GCB type irrespective of treatment response subtypes. It is well known that genes frequently mutated in non-GCB type are CARD11, MYD88, CDKN2A, CD79A/B TNFAIP3, and PRDM1 while in GCB type, GNA13, EZH2, BCL6 mutations are frequent. TP53, B2M, MEF2B and CREBBP mutations can be are found in both types [39]. In our study, 8 GCB cases showed mutations of MYD88(2/8), truncating mutations of TNFAIP3 (1/8), missense mutations of CD79B (1/8), B2M(3/8), and TP53(2/8) while 6 non-GCB cases had mutations of MYD88(1/6), truncating mutations of TNFAIP3 (1/6), missense mutations of CD79B (1/6), non-frameshift deletion of CARD11(1/6), MEF2B(2/6), and TP53(1/6). GNA13, EZH2 and BCL6 mutations were not found in both types. These results didn’t show distinct mutations profile as previous reports, which might be due to small sample size.
In GCB subtype, copy gain of REL, and BCL11A were found in two cases and copy neutral LOH of PTEN was found in a case. In non-GCB subtype, copy gain of BCL2 was found in four cases, and copy loss of CDKN2A and CDKN2B were found in two different cases. These findings are in line with those of previous report [40]. Of the 11 samples conducted RNA-seq, up-regulted DEGs were enriched in gene sets of extracellular matrix proteins in non-GCB type and gene sets associated with transcription/translation in GCB type.
DISCUSSION
In the present study, we used exome and transcriptome sequencing to identify the molecular background of refractory DLBCL by comparing two groups of DLBCL patients with different treatment responses. Although the sample size was rather small to make conclusion, we have discovered putative sources of resistance to R-CHOP therapy. These include overexpression of ATP-binding cassette transporter genes, mutations or CNAs related to cellular proliferation and apoptosis, activation of the PI3K–AKT–mTOR pathway, and increased mitochondrial oxidative phosphorylation.
ATP-binding cassette transporters are a family of transporter proteins that contribute to drug resistance via ATP-dependent drug efflux pumps. These transporters are expressed on the cell membrane and transport their substrates across the membrane in an ATP-dependent manner. The overexpression of ATP-binding cassette transporters reduces the intracellular concentration of the substrate agents, including vincristine and doxorubicin, commonly used in the treatment of lymphoma patients [41]. In addition, ATP-binding cassette transporters in aggressive lymphoma can modulate exosome release, which leads to exosome-mediated shielding of target cells as a critical determinant of tumor cell susceptibility to antibody therapy [42]. In this study, the ATP-binding cassette transporters, ABCA3, ABCB7, ABCC1, ABCB1, ABCG2, and ABCG1 were overexpressed in the F-group. ABCB1, also called MDR1, is the prototype of ATP-binding cassette transporters and is overexpressed in up to 80% of relapsed lymphomas [43, 44].
Oncogenic signaling pathways can drive metabolic reprogramming, and metabolic adaptation is a mechanism of resistance to targeted therapy [45]. Refractory DLBCL show DEGs that were significantly enriched in genes involved in mitochondrial oxidative phosphorylation, including UQCR11, COX7C, UQCRQ, NDUFB9, NDUFB10, NDUFC1, ATP5D, ATP5O, ATP5G2, and ATP5J2. DLBCL is a heterogeneous disease and can be subdivided into B-cell receptor, OxPhos, and host response tumors based on the transcriptional profile [36]. OxPhos DLBCL cells harbor the signature of genes involved in mitochondrial metabolism. In contrast to the B-cell receptor subtype, OxPhos DLBCLs do not display active or functional BCR signaling and are insensitive to inhibitors of BCR signaling [46]. They are selectively sensitive to pharmacological or genetic inhibition of fatty acid oxidation, which suggests that the metabolic features of this subtype may be exploited therapeutically [47].
Many chemotherapeutic agents exert antitumor effects by inducing apoptosis in tumor cells, and some alterations in apoptosis-signaling pathways are associated with drug resistance. TP53 was the most frequent mutation found in our study and was exclusive to the patients with refractory DLBCL. Half of the F group (3/6) had nsSNVs in the DNA-binding domain of TP53. Each nsSNV was accompanied by a deletion, which resulted in LOH and transcription of altered mRNA. Abnormalities in the tumor-suppressor gene, p53, have also been shown to be associated with drug resistance and short progression-free survival in patients with non-Hodgkin lymphoma [48]. In DLBCL, TP53 mutations have been reported as a marker of poor survival [7, 49, 50]. A recent study of 506 DLBCL patients showed that TP53 mutation was a predictor of survival in R-CHOP-treated patients, whereas TP53 deletion and loss of heterozygosity did not confer worse survival [7]. The TP53 mutation rates in these studies were about 20%, which is similar to the rate observed in our study (23.1%, 3/13). Studies of refractory or recurrent DLBCL using NGS platforms have been published recently, and the reported mutation rates of TP53 were lower than the 50% observed in our study: 32%, [20] 14.9% (32/215), [32] and 21.4% (3/14) [16]).
In this study, activation of the PI3K–Akt–mTOR pathway was predominant in the refractory DLBCL patients. The PI3K–Akt–mTOR signaling pathway plays an important role in controlling proliferation and survival of tumor cells in various types of malignancy, including DLBCL [51]. Activation of the PI3K–Akt–mTOR pathway was reported to be related to poor disease outcome in DLBCL patients treated with CHOP but not in those treated with R-CHOP [52]. In our study, refractory DLBCL patients with an altered mTOR pathway showed a poor response despite R–CHOP treatment. The number of DLBCL patients with refractory disease requires further searching for novel drugs to overcome cell resistance. Agents that directly target the PI3K–Akt–mTOR pathway have potential for the development of future treatments both as single agents and in combination with standard chemotherapeutics [51].
In conclusion, we explored the genetic characteristics of refractory DLBCL and found mutation of genes involved in proliferation and apoptosis, overexpression of drug-resistant genes, metabolic reprogramming with activated mitochondrial oxidative phosphorylation, and abnormal activation of signaling pathways. Although these results must be validated in a larger numbers of samples, our results provide information that may be useful in developing therapeutic strategies for refractory DLBCL.
MATERIALS AND METHODS
Study subjects and sample preparation for next-generation sequencing (NGS)
To identify genomic alterations associated with a therapeutic response in DLBCL patients, we collected fresh-frozen tumor tissues obtained for initial biopsy from 13 patients treated with R-CHOP. Frozen sections were obtained to evaluate the cellularity, and samples with >50% of the tumor cells were included. Patients whose complete remission was maintained for >1 year were classified as the responsive group (S group) and the others were classified as the refractory (F group). Seven of the 13 tumor samples were from the S group (numbered S1–7) and six were from the F group (numbered F1–6). We also obtained blood samples from four patients in each group (NS1–4 and NF1–4), which were paired with the tumor tissues S1–4 and F1–4, respectively. DNA was extracted from all 13 tumors and eight paired blood samples, and RNA was extracted from the 13 tumor samples for deep sequencing. COO subtypes were determined according to Han’s classification by IHC of CD10, Bcl-6, and MUM-1. All samples were negative for EBER in situ hybridization. The clinical information and study platforms applied to each sample are described in Supplementary Table S1. This study was approved by the institutional review board and was in accordance with the Declaration of Helsinki (Approval number: 2015-01-034-001).
Exome and transcriptome sequencing and sequence alignment
We performed WES on 10 tumor samples (S1–5 and F1–5) and eight paired normal blood samples (NS1–4 and NF1–4). To enrich the coding regions, we used SureSelect Human All Exon 50M (Agilent Technologies) for tumors and SureSelect Human All Exon V4 for normal samples. Sequence reads were produced using an Illumina HiSeq2000 instrument with a median on-target depth of 53× for tumor samples (range 47–77×) and 102× (range 96–106×) for normal samples (Supplementary Table S2). We performed the alignment using BWA [53] with the default parameters and hg19 as the reference genome.
RNA-seq was also performed on samples from the 13 tumors (S1–7 and F1–6). Disposable RNA chips (Agilent RNA 6000 Nano LabChip kit) were used to determine the concentration and purity/integrity of RNA samples using an Agilent 2100 bioanalyzer. The sequencing libraries were prepared as previously described [54]. Raw reads from the Illumina HiSeq 2000 were aligned to the human reference genome (hg19) using the STAR [55] 2-pass method. Alignment performance was assessed using RNA-SeqQC (Supplementary Table S3), [56] and two samples (F3-4) with low throughput (# of mapped reads <10 Mb) were excluded from downstream analysis.
Sequence variation analysis
PCR duplicates were removed using Picard (http://broadinstitute.github.io/picard/). A Genome Analysis Tool Kit (GATK) was used for indel realignment and base quality score recalibration. SNVs were identified using MuTect [57]. Somatic indels of eight tumor–normal pairs were called using GATK SomaticIndelDetector (https://www.broadinstitute.org/cancer/cga/indelocator), and indels of unpaired samples were predicted using GATK UnifiedGenotyper and HaplotypeCaller. Variants annotations were obtained using the software tool ANNOVAR [58], which integrated the results into databases on gene, type of variants, and minor allele frequency (MAF) in Exome Aggregation Consortium Version 0.3 (ExAC03, http://exac.broadinstitute.org) and Combined Annotation Dependent Depletion (CADD) score [59].
After listing the nonsynonymous SNVs (nsSNVs), splice site SNVs, and indels of the coding regions from eight paired samples, we applied the criteria and filters to identify candidate mutations as follows: 1) tumor allele frequency ≥10% and normal allele frequency ≤1%; 2) rare mutations based on MAF <0.5% in ExAC03; and 3) deleterious mutations based on a CADD score ≥20. We checked additional mutations of somatic candidate genes in unpaired samples. In samples with both DNA and RNA available, the mutations were checked if they were transcribed or edited in RNA. A full list of mutations is presented in Supplementary Table S4.
Copy number alteration analysis
Somatic copy number alterations (CNAs) were identified in eight tumor–normal pairs using Excavator [60] in somatic mode. For two unpaired samples (S5 and F5), we used Excavator in pooling mode to compare each tumor sample with a mean of the eight normal (NS1–4 and NF1–4) samples. Excavator generates CNA calls based on the ratio of tumor–normal read counts (T/N ratio) and detects segments with a similar copy number. T/N ratios >1.25 and <0.75 were defined as copy gain and loss, respectively. When the T/N ratio was >1.5 or <0.5, the segments were regarded as high-level gain and deep loss, respectively. Minimal common regions (MCRs)[61] were identified using Integrative Genome Browser (IGV) [62]. For MCRs exclusive to F group, we checked the CNAs of individual genes. B allele frequency was calculated from alternate allele frequency of MuTect results and was used to identify CN-LOH.
Fusion gene analysis
To identify fusion transcripts, we used deFuse [63], ChimeraScan [64], and pyPRADA [65]. We first excluded paralog pairs based on Ensembl version 75. Of the remaining results, those supported by ≥2 junction-spanning reads and that were predicted at least two of three tools were selected as fusion candidates.
Differential expression of genes in the two groups
HTSeq [66] was used to count sequence reads for the genes. Differential expression was calculated using DESeq2 [67]. DEGs were defined as genes with a q-value <0.05 and log2(fold change, FC) ≥1. To identify overrepresented gene sets, we used MSigDB (http://software.broadinstitute.org/gsea/msigdb/index.jsp), which integrates gene oncology (GO) terms and KEGG pathways. To identify cancer outlier genes (COGs), we used the criteria suggested previously [68].
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
GRANT SUPPORT
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number : Hl14C3414).
Authors contributions
S.J.K, W.S.K., J.-I.K., and Y.H.K designed and supervised experiments; H.Y.P., S.Y.K, S.L. and H.Y.Y. performed experiments; S.L, H.Y.P, and J.H. analyzed data; H.Y.P. wrote the manuscript.
REFERENCE
1. Swerdlow SH, Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Vardiman, J.W. (2008). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. (Lyon: IARC Press).
2. Panizo C, Rodriguez AJ, Gutierrez G, Diaz FJ, Gonzalez-Barca E, de Ona R, Grande C, Sancho JM, Garcia-Alvarez MF, Sanchez-Gonzalez B, Penalver FJ, Cannata J, Espeso M, Requena MJ, Gardella S, Duran S, et al. Evaluation of clinical and biological prognostic factors in relapsed or refractory diffuse large B-cell lymphoma patients after previous treatment with rituximab and chemotherapy: results of the PRO-R-IPI study. Clinical lymphoma, myeloma & leukemia. 2015; 15:398-403.
3. Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Muller-Hermelink HK, Campo E, Braziel RM, Jaffe ES, Pan Z, Farinha P, Smith LM, Falini B, Banham AH, Rosenwald A, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004; 103:275-282.
4. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Jr., Lu L, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000; 403:503-511.
5. Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015; 125:22-32.
6. Perry AM, Alvarado-Bernal Y, Laurini JA, Smith LM, Slack GW, Tan KL, Sehn LH, Fu K, Aoun P, Greiner TC, Chan WC, Bierman PJ, Bociek RG, Armitage JO, Vose JM, Gascoyne RD, et al. MYC and BCL2 protein expression predicts survival in patients with diffuse large B-cell lymphoma treated with rituximab. British journal of haematology. 2014; 165:382-391.
7. Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Richards KL, Hsi ED, Zhao XF, Choi WW, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2012; 120:3986-3996.
8. Miyashita K, Tomita N, Taguri M, Suzuki T, Ishiyama Y, Ishii Y, Nakajima Y, Numata A, Hattori Y, Yamamoto W, Miyazaki T, Tachibana T, Takasaki H, Matsumoto K, Hashimoto C, Takemura S, et al. Beta-2 microglobulin is a strong prognostic factor in patients with DLBCL receiving R-CHOP therapy. Leukemia research. 2015.
9. Broyde A, Boycov O, Strenov Y, Okon E, Shpilberg O, Bairey O. Role and prognostic significance of the Ki-67 index in non-Hodgkin's lymphoma. American journal of hematology. 2009; 84:338-343.
10. Jardin F. Next generation sequencing and the management of diffuse large B-cell lymphoma: from whole exome analysis to targeted therapy. Discovery medicine. 2014; 18:51-65.
11. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011; 470:115-119.
12. Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, Chan J, Bhagat G, Chadburn A, Gaidano G, Mullighan CG, Rabadan R, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nature genetics. 2011; 43:830-837.
13. Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nature reviews Clinical oncology. 2014; 11:12-23.
14. Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. International journal of cancer Journal international du cancer. 2009; 125:2863-2870.
15. Fan Y, Mao R, Yang J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein & cell. 2013; 4:176-185.
16. Mareschal S, Dubois S, Viailly PJ, Bertrand P, Bohers E, Maingonnat C, Jais JP, Tesson B, Ruminy P, Peyrouze P, Copie-Bergman C, Fest T, Jo Molina T, Haioun C, Salles G, Tilly H, et al. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes, chromosomes & cancer. 2016; 55:251-267.
17. Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, Choi W, Au WY, Srivastava G, Lugar PL, Rizzieri DA, Lagoo AS, Bernal-Mizrachi L, Mann KP, Flowers C, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110:1398-1403.
18. Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harbor perspectives in medicine. 2014; 4.
a19. Demosthenous C, Han JJ, Hu G, Stenson M, Gupta M. Loss of function mutations in PTPN6 promote STAT3 deregulation via JAK3 kinase in diffuse large B-cell lymphoma. Oncotarget. 2015; 6:44703-44713. doi: 10.18632/oncotarget.6300.
20. Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, Grewal J, Yu S, Fornika D, Bushell K, Nielsen TH, Petrogiannis-Haliotis T, Crump M, Tosikyan A, Grande BM, MacDonald D, et al. Genetic Landscapes of Relapsed and Refractory Diffuse Large B-Cell Lymphomas. Clinical cancer research. 2016; 22:2290-2300.
21. Gudjonsson T, Altmeyer M, Savic V, Toledo L, Dinant C, Grofte M, Bartkova J, Poulsen M, Oka Y, Bekker-Jensen S, Mailand N, Neumann B, Heriche JK, Shearer R, Saunders D, Bartek J, et al. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell. 2012; 150:697-709.
22. Deretic V. A master conductor for aggregate clearance by autophagy. Developmental cell. 2010; 18:694-696.
23. Pasqualucci L, Compagno M, Houldsworth J, Monti S, Grunn A, Nandula SV, Aster JC, Murty VV, Shipp MA, Dalla-Favera R. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. The Journal of experimental medicine. 2006; 203:311-317.
24. Tam W, Gomez M, Chadburn A, Lee JW, Chan WC, Knowles DM. Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood. 2006; 107:4090-4100.
25. Luo Y, Roeder RG. Cloning, functional characterization, and mechanism of action of the B-cell-specific transcriptional coactivator OCA-B. Molecular and cellular biology. 1995; 15:4115-4124.
26. Schubart K, Massa S, Schubart D, Corcoran LM, Rolink AG, Matthias P. B cell development and immunoglobulin gene transcription in the absence of Oct-2 and OBF-1. Nature immunology. 2001; 2:69-74.
27. Kim U, Qin XF, Gong S, Stevens S, Luo Y, Nussenzweig M, Roeder RG. The B-cell-specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes. Nature. 1996; 383:542-547.
28. Zhao C, Inoue J, Imoto I, Otsuki T, Iida S, Ueda R, Inazawa J. POU2AF1, an amplification target at 11q23, promotes growth of multiple myeloma cells by directly regulating expression of a B-cell maturation factor, TNFRSF17. Oncogene. 2008; 27:63-75.
29. Valsami S, Pappa V, Rontogianni D, Kontsioti F, Papageorgiou E, Dervenoulas J, Karmiris T, Papageorgiou S, Harhalakis N, Xiros N, Nikiforakis E, Economopoulos T. A clinicopathological study of B-cell differentiation markers and transcription factors in classical Hodgkin's lymphoma: a potential prognostic role of MUM1/IRF4. Haematologica. 2007; 92:1343-1350.
30. Kwiecinska A, Ichimura K, Berglund M, Dinets A, Sulaiman L, Collins VP, Larsson C, Porwit A, Lagercrantz SB. Amplification of 2p as a genomic marker for transformation in lymphoma. Genes, chromosomes & cancer. 2014; 53:750-768.
31. Martinez-Climent JA, Alizadeh AA, Segraves R, Blesa D, Rubio-Moscardo F, Albertson DG, Garcia-Conde J, Dyer MJ, Levy R, Pinkel D, Lossos IS. Transformation of follicular lymphoma to diffuse large cell lymphoma is associated with a heterogeneous set of DNA copy number and gene expression alterations. Blood. 2003; 101:3109-3117.
32. Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P, Maingonnat C, Jais JP, Peyrouze P, Figeac M, Molina TJ, Desmots F, Fest T, Haioun C, Lamy T, Copie-Bergman C, et al. Next-Generation Sequencing in Diffuse Large B-Cell Lymphoma Highlights Molecular Divergence and Therapeutic Opportunities: a LYSA Study. Clinical cancer research. 2016; 22:2919-2928.
33. Murr R, Vaissiere T, Sawan C, Shukla V, Herceg Z. Orchestration of chromatin-based processes: mind the TRRAP. Oncogene. 2007; 26:5358-5372.
34. McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998; 94:363-374.
35. Ramdzan ZM, Nepveu A. CUX1, a haploinsufficient tumour suppressor gene overexpressed in advanced cancers. Nature reviews Cancer. 2014; 14:673-682.
36. Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, Wu B, Pasqualucci L, Neuberg D, Aguiar RC, Dal Cin P, Ladd C, Pinkus GS, Salles G, Harris NL, Dalla-Favera R, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood. 2005; 105:1851-1861.
37. Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Current drug delivery. 2004; 1:27-42.
38. Vega MI, Huerta-Yepaz S, Garban H, Jazirehi A, Emmanouilides C, Bonavida B. Rituximab inhibits p38 MAPK activity in 2F7 B NHL and decreases IL-10 transcription: pivotal role of p38 MAPK in drug resistance. Oncogene. 2004; 23:3530-3540.
39. Vega GG, Aviles-Salas A, Chalapud JR, Martinez-Paniagua M, Pelayo R, Mayani H, Hernandez-Pando R, Martinez-Maza O, Huerta-Yepez S, Bonavida B, Vega MI. P38 MAPK expression and activation predicts failure of response to CHOP in patients with Diffuse Large B-Cell Lymphoma. BMC cancer. 2015; 15:722.
40. Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, Powell J, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:13520-13525.
41. Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nature reviews Cancer. 2010; 10:147-156.
42. Aung T, Chapuy B, Vogel D, Wenzel D, Oppermann M, Lahmann M, Weinhage T, Menck K, Hupfeld T, Koch R, Trumper L, Wulf GG. Exosomal evasion of humoral immunotherapy in aggressive B-cell lymphoma modulated by ATP-binding cassette transporter A3. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:15336-15341.
43. Wilson WH, Bates SE, Fojo A, Bryant G, Zhan Z, Regis J, Wittes RE, Jaffe ES, Steinberg SM, Herdt J, et al. Controlled trial of dexverapamil, a modulator of multidrug resistance, in lymphomas refractory to EPOCH chemotherapy. Journal of clinical oncology. 1995; 13:1995-2004.
44. Liu Q, Ohshima K, Kikuchi M. High expression of MDR-1 gene and P-glycoprotein in initial and re-biopsy specimens of relapsed B-cell lymphoma. Histopathology. 2001; 38:209-216.
45. McQuade JL, Vashisht Gopal Y. Counteracting oxidative phosphorylation-mediated resistance of melanomas to MAPK pathway inhibition. Molecular & cellular oncology. 2015; 2:e991610.
46. Chen L, Monti S, Juszczynski P, Daley J, Chen W, Witzig TE, Habermann TM, Kutok JL, Shipp MA. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood. 2008; 111:2230-2237.
47. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, Polak K, Tondera D, Gounarides J, Yin H, Zhou F, Green MR, Chen L, Monti S, Marto JA, Shipp MA, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer cell. 2012; 22:547-560.
48. Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, Morel P, Fenaux P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994; 84:3148-3157.
49. Leroy K, Haioun C, Lepage E, Le Metayer N, Berger F, Labouyrie E, Meignin V, Petit B, Bastard C, Salles G, Gisselbrecht C, Reyes F, Gaulard P, Groupe d'Etude des Lymphomes de lA. p53 gene mutations are associated with poor survival in low and low-intermediate risk diffuse large B-cell lymphomas. Annals of oncology. 2002; 13:1108-1115.
50. Stefancikova L, Moulis M, Fabian P, Vasova I, Zedek F, Ravcukova B, Muzik J, Kuglik P, Vranova V, Falkova I, Hrabalkova R, Smardova J. Prognostic impact of p53 aberrations for R-CHOP-treated patients with diffuse large B-cell lymphoma. International journal of oncology. 2011; 39:1413-1420.
51. Majchrzak A, Witkowska M, Smolewski P. Inhibition of the PI3K/Akt/mTOR signaling pathway in diffuse large B-cell lymphoma: current knowledge and clinical significance. Molecules. 2014; 19:14304-14315.
52. Xu ZZ, Xia ZG, Wang AH, Wang WF, Liu ZY, Chen LY, Li JM. Activation of the PI3K/AKT/mTOR pathway in diffuse large B cell lymphoma: clinical significance and inhibitory effect of rituximab. Annals of hematology. 2013; 92:1351-1358.
53. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754-1760.
54. Ju YS, Kim JI, Kim S, Hong D, Park H, Shin JY, Lee S, Lee WC, Kim S, Yu SB, Park SS, Seo SH, Yun JY, Kim HJ, Lee DS, Yavartanoo M, et al. Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nature genetics. 2011; 43:745-752.
55. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15-21.
56. DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012; 28:1530-1532.
57. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013; 31:213-219.
58. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010; 38:e164.
59. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics. 2014; 46:310-315.
60. Magi A, Tattini L, Cifola I, D'Aurizio R, Benelli M, Mangano E, Battaglia C, Bonora E, Kurg A, Seri M, Magini P, Giusti B, Romeo G, Pippucci T, De Bellis G, Abbate R, et al. EXCAVATOR: detecting copy number variants from whole-exome sequencing data. Genome biology. 2013; 14:R120.
61. Aguirre AJ, Brennan C, Bailey G, Sinha R, Feng B, Leo C, Zhang Y, Zhang J, Gans JD, Bardeesy N, Cauwels C, Cordon-Cardo C, Redston MS, DePinho RA, Chin L. High-resolution characterization of the pancreatic adenocarcinoma genome. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:9067-9072.
62. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nature biotechnology. 2011; 29:24-26.
63. McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M, Heravi Moussavi A, Senz J, Melnyk N, Pacheco M, Marra MA, Hirst M, Nielsen TO, Sahinalp SC, Huntsman D, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS computational biology. 2011; 7:e1001138.
64. Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011; 27:2903-2904.
65. Torres-Garcia W, Zheng S, Sivachenko A, Vegesna R, Wang Q, Yao R, Berger MF, Weinstein JN, Getz G, Verhaak RG. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics. 2014; 30:2224-2226.
66. Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166-169.
67. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014; 15:550.
68. Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, Lee J, Jung YJ, Kim JO, Shin JY, Yu SB, Kim J, Lee ER, Kang CH, Park IK, Rhee H, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome research. 2012; 22:2109-2119.