
INTRODUCTION
Retinitis pigmentosa (RP) is a hereditary neurodegenerative retinal disease characterized by progressive loss of photoreceptors. The ocular phenotype of this disease include pigmentary retinopathy, arteriolar narrowing, waxy pallor of the optic disc, cystic macular lesions, cataracts and refractive errors [1–5]. Patients usually present with decreased night vision and loss of peripheral vision. The diverse pathogenic genes result in varied clinical manifestations. For example, previous studies showed that the severity of refractive errors in patients with RP was associated with the location of the mutated gene and/or the type of genes involved [5–7].
There are no known risk factors for RP except genetic predisposition. Approximately 70% of patients have a positive family history [8]. In the Netherlands, van den Born et al. estimated that ~63% of patients with RP had a positive family history, with ~30% inherited in an autosomal-recessive pattern, 22% autosomal-dominant, 10% X-linked and the remaining 37% isolated cases [9]. To date, more than 100 gene loci related to RP have been identified [10] and at least 17 genes are believed to be involved in ARRP. Gene mutations have been found in ~50% of ARRP cases [11]. The common known types of mutations causing ARRP in Israeli and Palestinian populations are located in CRB1 [12]. Corton M et al reported that CRB1 mutations frequently caused early-onset retinal dystrophies in Spanish populations [13]. Recently, approximately 194 mutations related to the pathogenesis of ARRP were identified in CRB1 [14].
Previous studies demonstrated that targeted-capture NGS can precisely and rapidly identify genetic defects [15–16]. Several novel mutative genes have been identified via the NGS approach in patients with RP [17]. Booij et al analyzed a group of 35 unrelated patients with autosomal recessive juvenile retinitis pigmentosa via NGS and found that 12 patients had mutations. These mutations occurred in CRB1 (11%), GUCY2D (11%), RPE65 (6%), and RPGRIP1 (6%) [11]. Yang et al reported that mutations in CRB1 were found in four Chinese families as well as in some sporadic subjects with RP, with a 5.8% gene mutation frequency in CRB1 [14].
In this study, a Chinese pedigree with ARRP was studied and the compound heterozygous mutations in CRB1 were identified using NGS. To the best of our knowledge, these mutations in CRB1 have not been reported previously.
RESULTS
Clinical findings
The proband (Figure 1 = patient II:1, Figure 2A) is a 14-year-old male. Onset of disease was at age 2. He presented with diminished night vision and subsequent progressive loss of his peripheral and central vision, as well as reduced color vision and night blindness. On his most recent visit, his best corrected visual acuity (BCVA) was CF/30cm (+7.00DS+2.00DDC×105°) in the right eye and 0.03 (+9.00DS+1.50DDC×77°) in the left eye. The intraocular pressures were normal bilaterally. There was 15° esotropia in the left eye. Bilateral fundus examination showed a waxy, pale-appearing optic disc with markedly attenuated retinal arterioles. There were extensive typical bone spicules and round pigment clumping. OCT showed that marked atrophy changes in the macular region (Figure 3). The proband’s brother (Figure 1= patient II:2, Figure 2B) was a 9-year-old male. Disease onset was also at age 2, presenting with similar symptoms and clinical signs as his proband brother. On his most recent visit, the BCVA was 0.2 (+7.75DS/+1.00DDC×100°) in the right eye and 0.1 (+8.50DS/+1.00DC×80°) in the left eye. Fundus patterns and OCT imaging were similar to his proband brother. The disease of the proband’s sister (Figure 1 = patient II:3, Figure 2C) was a 7-year-old female. Disease also occurred at the age of 2, presenting with poor night and central vision. On her most recent visit, her best-corrected visual acuity was 0.3 in both eyes. Optometry values were as follows: 0.3 (+9.75DS/+2.25DC × 75°) right eye and 0.3(+10.50DS/+ 2.75DC × 95°) left eye. Fundus examination showed normal optic discs and attenuated retinal arterioles in both eyes. There was an area of hypo-pigmentation with localized bone–spicule pigmentation in the macular region. ERG responses were extinguished. Rod ERG b-wave disappeared in each eye. (Figure 4)
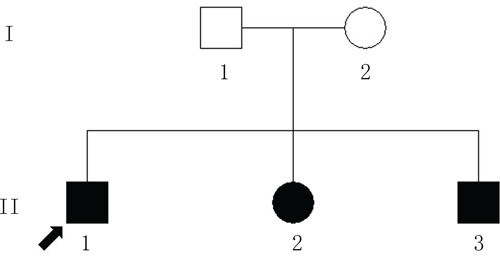
Figure 1: The Chinese pedigree with autosomal-recessive retinitis pigmentosa (ARRP). The circles indicate females; the squares indicate males. The filled shapes indicate the affected individuals with RP. The arrow signifies the proband.
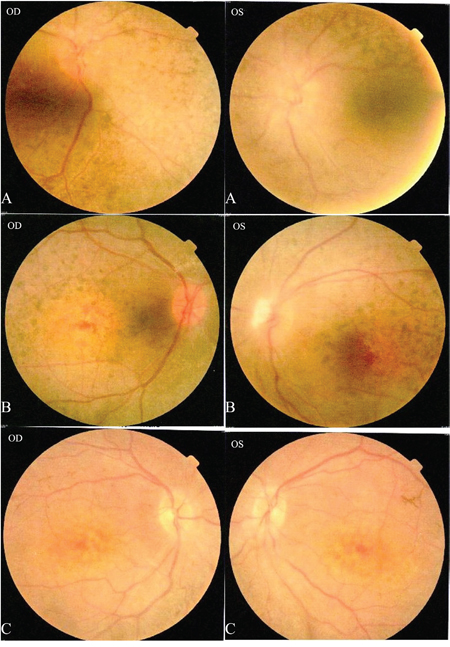
Figure 2: Patients II: A: II:1, B: II:2, C: II:3. Fundus photography in both eyes for patients II:1-3: A. and B. show a waxy pale-appearing optic disc, attenuated retinal arterioles and RPE atrophy. C. shows normal optic discs and attenuated retinal arterioles in each eye. There is an area of hypo-pigmentation with localized bone–spicule pigmentation in the macular region.
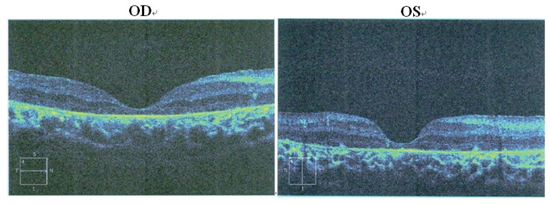
Figure 3: Patient II:1. OCT of the retinal pigment epithelium. OCT shows marked atrophy of the retinal pigment epithelium and loss of photoreceptors. Cystic cavities within the inner and outer nuclear layers are noticed in both eyes. The retinal nerve fiber layer in the macular region is thick, with preservation of the foveal architecture.
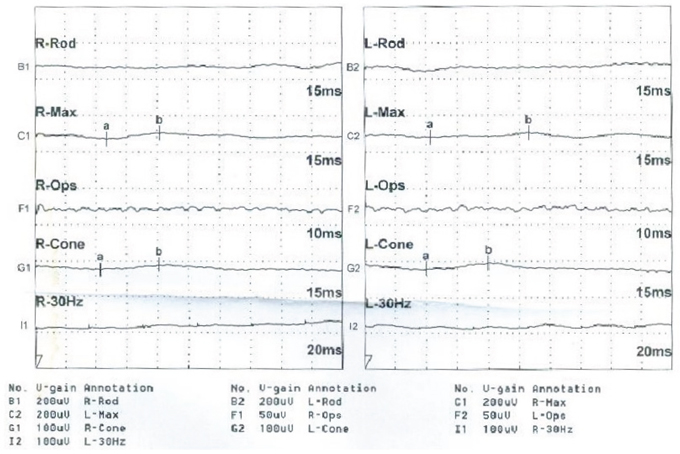
Figure 4: Patient II:3. ERG responses are extinguished, as interpreted by the reduction in amplitude and b-wave disappearance in each eye.
Genetic findings
A total of 57 variants of candidate genes related to ARRP, including RP1, RP2, RPGR, RHO, PRPH2, and CRB1, were detected in the proband. The compound mutations, c.138delA and c.1841G>T, in CRB1 were identified in the proband using gene chip sequencing. This compound heterozygote for c.138delA (p.Asp47IlefsX24) and c.1841G>T (p.Gly614Val) mutations (Figures 5 and 6) were verified in his affected siblings. The unaffected father is a heterozygote carrier of c.1841G>T (p.Gly614Val), and the unaffected mother is a heterozygote carrier of c.138delA (p.Asp47IlefsX24). There were no other candidate gene mutations found in this pedigree.
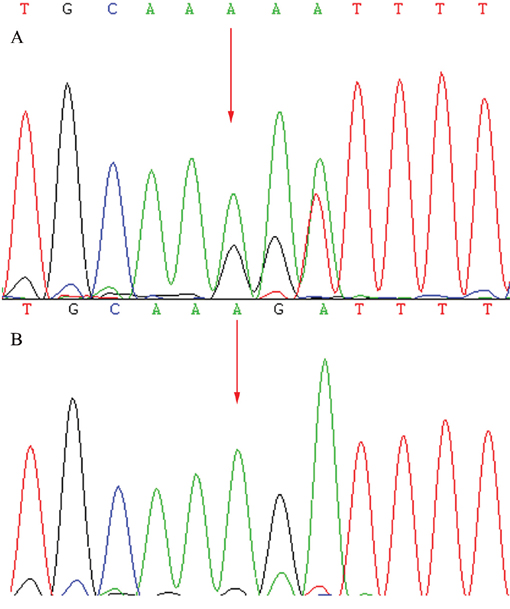
Figure 5: Sequencing results of the CRB1 gene: Sequence analysis showed a heterozygous mutation c.138delA (p.Asp47IlefsX24) in exon 2. (Red arrow indicates the location of the mutation) A. Patients. B. Unaffected mother.
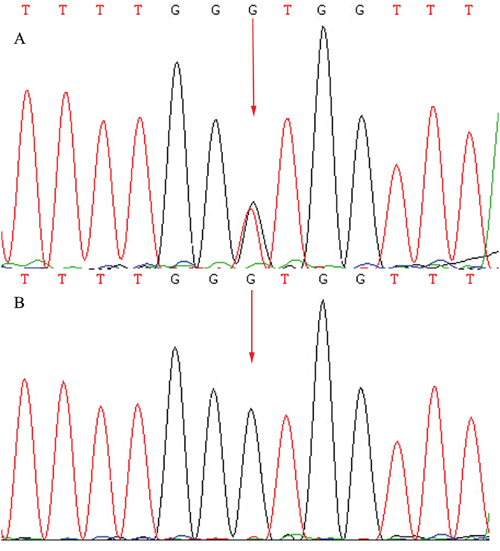
Figure 6: Sequencing results of the CRB1 gene: Sequence analysis showed a heterozygous mutation c.1841G>T (p.Gly614Val) transversion in exon 6. (Red arrow indicates the location of the mutation) A. Patients. B. Unaffected father.
DISCUSSION
CRB1 mutations are associated with a series of autosomal recessive retinal dysthrophies such as Leber congenital amaurosis (LCA), [17–19] early onset RP, [20–21] and preserved para-arteriolar retinal pigment epithelium [18]. RP is one of the most common types of retinal dystrophy in China with a prevalence of nearly 1:4000 [19]. RP may occur as an isolated sporadic disorder, or inherited as an autosomal-dominant, autosomal-recessive, or X-linked pattern. The genes most frequently linked with RP include RHO, USH2A, RPGR, CRB1, GUCY2D, and RPGPIP1 [11]. Previous studies reported CRB1 mutations in ARRP in diverse populations [2, 11, 12, 14]. This study identified a pair of novel compound mutations in CRB1 in a Chinese pedigree with ARRP. All affected patients in the second generation had compound heterozygous mutations for c.138delA (p.Asp47IlefsX24) and c.1841G>T (p.Gly614Val) in CRB1.
CRB1 consists of 12 exons and 11 introns. It encodes two proteins of 1376 and 1406 amino acids and functions as an extra-cellular protein with a signal peptide. This protein contains 19 epidermal growth factor (EGF)-like domains, 3 laminin A globular (AG)-like domains, and C-type lectin (CTL), It plays an important role in the development of the retina [23–25]. Mutations in CRB1 may restrain retinal development and result in the loss of photoreceptor signaling. Yang et al. reported patients with c.3460T>A and c.4207G>C mutations in CRB1 presented with night blindness at the age of 20 and progressive vision lose [14]. In the present study, patients with compound c.138delA and c.1841G>T mutations in CRB1 presented with visual impairment accompanied with high hyperopia at the age of 2. The parents, who carried either c.138delA (p.Asp47IlefsX24) or c.1841G>T (p.Gly614Val)) mutation, were spared from RP, while the three offspring who carried both two mutations from their parents presented with typical RP. The deletion mutation c.138delA results in truncated proteins with the absence of CRB1 products such as transmembrane and cytoplasmic domains. This may stop protein translation. The c.1841G>T mutation caused a replacement of glycine (Gly) with Valine (Val) at the codon 614 which is localized on the 12th EGF-like domain, a highly conserved region. These compound mutations, therefore, seems to be responsible for the pathogenesis of the retinal degeneration seen in this family.
We are aware that all affected subjects in the present study had high hyperopia with spherical equivalent (SE) more than +7.00 diopters (D). Sieving et al analyzed 268 eyes with RP and found a mean SE was at −1.86 diopters [5]. Thomas et al reported that mutations in RP1 in patients with ARRP had SE median at −4.0 diopters, while without RP1 mutation had SE median less than −1.0 diopters [6]. Hanein et al showed that mutations in GUCY2D, RPGRIP1, CRX or CEP290 in patients with Leber congenital amaurosis frequently had high hyperopia [7]. Those studies imply that the types and degrees of refractive errors may be correlated with the types of mutations involved, patients with hyperopic refractive errors may predominantly involve CRB1, GUCY2D, RPGRIP1, CRX or CEP290 mutations in patients with inherited retinal dystrophies.
MATERIALS AND METHODS
Subjects
All three patients in the second generation and two unaffected parents were enrolled in this study. No consanguineous marriage was noticed in the family. This study was approved by the Medical Ethics Committee of the Shenzhen Eye Hospital, Jinan University. Prior to participating in this study, all subjects were given a detailed explanation of the study. Informed consent was obtained from all participants according to the principles of the Declaration of Helsinki.
Clinical examination
All study subjects in this family underwent a complete ophthalmic examination, including visual acuity test with linear Snellen, best corrected visual acuity, intraocular pressure (IOP) measurement, slit-lamp biomicroscopy, and funduscopic examination. Other examinations included fundus photography, visual field testing, full-field electroretinography (ffERG, Diagnosis LLC, Lowell, United States) recorded in accordance with the guidelines of the International Society for Clinical Electrophysiology of Vision (ISCEV), [15] and optical coherence tomography (Spectralis System, Heidelberg Engineering, Heidelberg, Germany, SD-OCT). OCT parameters were used to analyze the retinal pigment epithelium and the retinal nerve fiber layer thickness in the retina.
Criteria for the diagnosis of retinitis pigmentosa
Subjects with typical symptoms, including decreased night vision or night blindness, waxy optic disc pallor, narrowing arterioles, pigment deposits, and ERG amplitude reduction [16].
Mutation screening and sequence analysis
Peripheral venous blood was collected from all study subjects. Genomic DNA was extracted from 200 μL peripheral venous blood using a QIAmp Blood DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer protocols. The integrity of the DNA samples was verified by 1% agarose gel electrophoresis. Fifty-seven candidate genes of non-syndromic inherited RP including the CRB1 gene were chosen as candidate genes to be enriched in the designed sequencing panel using custom-designed NimbleGen SeqCap probe hybridization (Roche NimbleGen, Inc., Madison, WI, USA). Target-Capture sequencing of all coding exons and 20bp of their flanking intronic regions of these genes were performed on the proband to detect the possible disease-causing gene mutations. The indicated DNA samples with equal molar ratios were put into each flow cell along the displayed lanes. Polymerase Chain Reaction (PCR) was observed with HiSeq2000 (Illumina, Inc., San Diego, CA, USA) using the Sequencing-By-Synthesis (SBS) method. The data was analyzed for gene alignment with CASAVA v1.7 (Illumina Inc.) and NextGene (SoftGenetics, State College, PA, USA) software.
Sanger sequencing was used to confirm the indicated mutations and determine whether any of the remaining variants co-segregated with the disease phenotype in this family. PCR amplification was performed. PCR primers were designed by the Primer Premier 5 software. Components of 30 μl of PCR reaction mixture included 15 μl 2× Taq PCR Master Mix (SinoBio, Shanghai, China), 30 ng DNA, 1.0 μM of both forward and reverse primers, and 12 μl ddH2O. The PCR reactions were incubated at 94°C for 3 min, followed by 35 cycles of 94°C for 20 s, annealing for 30 s, and 72°C for 60 s, with final extension at 72°C for 5 min. Purification of PCR products were sequenced using an ABI 377XL automated DNA sequencer (Applied Biosystems, Foster City, CA). The assembly of DNA sequences followed the DNAStar (Madison, WI) software and compared pairwise with online Human Genome databases. All mutations and its variant were interpreted and classified based on the nomenclature recommended by the Human Genomic Variation Society (HGVS).
CONCLUSION
The novel compound heterozygous mutations for c.138delA (p.Asp47IlefsX24) and c.1841G>T (p.Gly614Val) in CRB1 were identified as the pathogenic gene for ARRP in a Chinese pedigree using NGS. To the best of our knowledge, this is the first report that these mutations in CRB1 are responsible for the pathogenesis of ARRP.
ACKNOWLEDGMENTS
We appreciate the family for their understanding and cooperation.
CONFLICTS OF INTEREST
The authors have declared that no competing interests exist.
FUNDING
This work was supported by grants from the National Natural Science Foundation of China (NNSF 81200688, 81570870 and 81500718), Fujian Nature Science Foundation (2016J01544) and the Science and Technology Innovation Committee of Shenzhen (No.JCYJ20140414093858910).
REFERENCES
1. Kanski J. Retinitis Pigmentosa. In: Kanski J, Sehmi K, Bolton A(eds). Clinical Ophthalmology: A Systemic Approach, 6th edn. Philadelphia, PA. Elsevier. 2008; Ch.18.
2. Cordovez JA, Traboulsi EI, Capasso JE, Sadagopan KA, Ganesh A, Rychwalski PJ, Neely KA, Brodie SE, Levin AV. Retinal Dystrophy with Intraretinal Cystoid Spaces Associated with Mutations in the Crumbs Homologue (CRB1) Gene. Ophthalmic genetics. 2015; 36:257-264.
3. Cox SN, HAY E, Bird AC. Treatment of chronic macular edema with acetazolamide. Arch Ophthalmol. 1988;106:1190-1195.
4. Grover S, Apushkin MA, Fishman GA. Topical Dorzolamide for the treatment of cystoid macular edema in patients with retinitis pigmentosa. Am J Ophthalmol. 2006;141:850-858.
5. Sieving PA, Fishman GA. Refractive error of retinitis pigmentosa patients. Br J Ophthalmol Biol. 2006;572:15–20.
6. Chassine T, Bocquet B, Daien V, Avila-Fernandez A, Ayuso C, Collin RW, Corton M, Hejtmancik JF, van den Born LI, Klevering BJ, Riazuddin SA, Sendon N, Lacroux A, Meunier I, Hamel CP. Autosomal recessive retinitis pigmentosa with RP1 mutations is associated with myopia. The British journal of ophthalmology. 2015; 99:1360-1365.
7. Hanein S, Perrault I, Gerber S, Tanguy G, Rozet JM, Kaplan J. Leber congenital amaurosis: survey of the genetic heterogeneity, refinement of the clinical definition and phenotype-genotype correlations as a strategy for molecular diagnosis. Clinical and molecular survey in LCA. Advances in experimental medicine and biology. 2006; 572:15-20.
8. Weleber RG, Gregory-Evans K. Retinitis Pigmentosa and Allied Disorders. In: Retina, Ryan SJ(Ed), Elsevier Mosby. 2006;p.395.
9. Van den Born LI, Bergen AA, Bleeker-Wagemakers EM. A retrospective study of registered retinitis pigmenrosapatients in The Netherlands. Ophthalmic Paediatr Genet. 1992;13:227-236.
10. Berson EL. Retinitis pigmentosa and Allied disease. In: Albert D, Miller J, Azaf D, Blodi B, (eds). Albert and Jakobiec, 3rd edn. Philadelphia, PA. Elsevier. 2008;Ch.177.
11. Booij JC, Florijn RJ, ten Brink JB, Loves W, Meire F, van Schooneveld MJ, de Jong PT, Bergen AA. Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and RPGRIP1 genes in patients with juvenile retinitis pigmentosa. Journal of medical genetics. 2005; 42:e67.
12. Beryozkin A, Zelinger L, Bandah-Rozenfeld D, Harel D, Strom TA, Merin S, Chowers I, Banin E, Sharon D. Mutation in CRB1 are a relative common cause of autosomal recessive early-onset retinal degeneration in the Israeli and Palestinian populations. Invest Ophthalmol Vis Sci. 2013;54:2068-75.
13. Corton M, Tatu SD, Avila-Fernandez A, Vallespin E, Tapias I, Cantalapiedra D, Blanco-Kelly F, Riveiro-Alvarez R, Bernal S, Garcia-Sandoval B, Baiget M, Ayuso C. High frequency of CRB1 mutations as cause of Early-Onset Retinal Dystrophies in the Spanish population. Orphanet journal of rare diseases. 2013; 8:20.
14. Yang L, Wu L, Yin X, Chen N, Li G, Ma Z. Novel mutations of CRB1 in Chinese families presenting with retinal dystrophies. Molecular Vision. 2014;20:359-367.
15. Hartong D T, Berson E L, Dryja T P. Retinitis pigmentosa. The Lancet. 2006; 368:1795-1809.
16. Wang Y, Guo L, Cai S, Dai M, Yang Q, Yu W, Yan N, Zhou X, Fu J, Guo X, Han P, Wang J, Liu X. Exome sequencing identifies compound heterozygous mutations in CYP4V2 in a pedigree with atypical retinitis pigmentosa. PLOS ONE. 2012; 7:e33673.
17. Lotery AJ, Jacobson SG, Fishman GA, Weleber RG, Fulton AB, Namperumalsamy P, Heon E, Levin AV, Grover S, Rosenow JR, Kopp KK, Sheffield VC, Stone EM. Mutations in the CRB1 gene cause Leber congenital amaurosis. Archives of ophthalmology. 2001; 119:415-420.
18. den Hollander AI, Davis J, van der Velde-Visser SD, Zonneveld MN, Pierrottet CO, Koenekoop RK, Kellner U, van den Born LI, Heckenlively JR, Hoyng CB, Handford PA, Roepman R, Cremers FP. CRB1 mutation spectrum in inherited retinal dystrophies. Human mutation. 2004; 24:355-369.
19. Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D, Calvas P, Dollfus H, Hamel C, Lopponen T, Munier F, Santos L, Shalev S, Zafeiriou D, Dufier JL, Munnich A, Rozet JM, Kaplan J. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Human mutation. 2004; 23:306-317.
20. den Hollander AI, Heckenlively JR, van den Born LI, de Kok YJ, van der Velde-Visser SD, Kellner U, Jurklies B, van Schooneveld MJ, Blankenagel A, Rohrschneider K, Wissinger B, Cruysberg JR, Deutman AF, Brunner HG, Apfelstedt-Sylla E, Hoyng CB, Cremers FP. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. American journal of human genetics. 2001; 69:198-203.
21. Khan JA, Ide CH, Strickland MP. Coats’-type retinitis pigmentosa. Surv Ophthalmol. 1988;32:317-332.
22. Bujakowska K, Audo I, Mohand-Said S, Lancelot ME, Antonio A, Germain A, Leveillard T, Letexier M, Saraiva JP, Lonjou C, Carpentier W, Sahel JA, Bhattacharya SS, Zeitz C. CRB1 mutations in inherited retinal dystrophies. Human mutation. 2012; 33:306-315.
23. Tepass U. Crumbs, a component of the apical membrane, is required for zonula adherens formation in primary epithelia of Drosophila. Dev Biol. 1996;177:217-225.
24. Tepass U. Adherens junctions: new insight into assembly, modulation and function.Bioessays. 2002;24:690-695.
25. Pellikka M, Tanentzapf G, Pinto M, Smith C, McGlade CJ, Ready DF, Tepass U. Crumbs, the Drosophila homologue of human CRB1/RP12, is essential for photoreceptor morphogenesis. Nature. 2002; 416:143-149.