
Background
Sarcomas are a broad family of cancers that arise from cells of mesenchymal origin in virtually every tissue of the body, and they can differentiate along a number of tissue lineages, such as adipose, muscle, fibrous, cartilage, or bone. As such, the pathology of these neoplasms is extremely diverse, with over seventy described subtypes [1]. Historically categorized as either bone or soft tissue, sarcomas are now molecularly classified into two groups: genetically complex, with a high mutational burden and a complex karyotype, or genetically simple, bearing a single disease-specific translocation, mutation, or amplification within a comparatively quiescent genomic background [2].
This histological and molecular heterogeneity makes sarcomas particularly difficult to diagnose, leading to debate surrounding the sufficiency of histological diagnosis versus the need for ancillary molecular diagnostics. Treatment has proven equally challenging, and research findings in one subtype often do not translate to others. These limitations are magnified within the context that sarcomas are among the rarest of cancer diagnoses, making research and trials more difficult. In the US, sarcomas represent 1% of new cancer diagnoses and of cancer-related deaths [3], though they are more prevalent in childhood and adolescence, where they account for 19-21% of cancer-related deaths [4]. Therefore, though the complexity of sarcomas is comparable to that of any of the more common and heavily researched malignancies, there are comparatively few novel therapeutic approaches in advanced development.
Sarcomas, as a group, are resistant to conventional cytotoxic chemotherapy, save for some successes with anthracycline-based therapy for rhabdomyosarcoma, Ewing sarcoma, and osteosarcoma [5]. Late recurrence and metastasis still occur in some subtypes, so when surgery and radiation fail, there are few - if any - effective systemic options available. Clinical trials that include sarcomas are rare and frequently confounded by lumping together results from biologically disparate subtypes, as continues to occur with molecularly divergent subcategories of liposarcoma. Given these accrual and design challenges, it can be difficult to gather convincing high-level evidence to guide the management of sarcomas.
Nonetheless, the past year has seen advances in genomics-based sarcoma science and the publication in major journals of significant positive results from clinical trials. In this review, we aim to summarize recent developments in both diagnostics and treatment, including translational science and clinical trials in chemotherapy, targeted therapy, epigenetic therapy, and the burgeoning field of immune therapy. The scope of this review includes works published from late 2014 to early 2016.
Sarcoma Diagnostics
Genomic landscapes in sarcoma
Multi-platform “omics” approaches were undertaken to elucidate comprehensive mutational landscapes for liposarcomas, epithelioid sarcoma, and rhabdomyosarcomas.
Kanojia et al [6] used a combination of single nucleotide polymorphism (SNP) arrays and whole- and targeted-exome sequencing to characterize the genomic landscape of 86 liposarcomas of all major subtypes. In addition to the expected amplifications in MDM2 and other known 12q amplicon genes CDK4 and HMGA2, they identified a number of novel gene amplifications: UAP1, MIR557, LAMA4, CPM, IGF2, ERBB3, and IGF1R. Of particular interest, CPM (carboxypeptidase M) - located at the edge of the 12q amplicon, outside of what was thought to be the key region defined by CDK4 and MDM2 - was amplified in 39 of 50 well- and de-differentiated liposarcomas. Knockdown of CPM reduced cell line and xenograft growth, migration, and invasion, and reduced expression of phosphorylated EGFR, Akt, and ERK, suggesting that CPM is involved in epidermal growth factor signalling, a targetable pathway that might play an unanticipated role in liposarcomagenesis. This genomic survey also found recurrent mutations in genes associated with cell adhesion, cytoskeletal organization, base excision repair, homologous recombination repair, nucleotide excision repair, and DNA replication: PLEC, MXRA5, FAT3, NF1, MDC1, TP53, and CHEK2. The NF1 (neurofibromin-1) gene was of particular interest, altered in 13 of 50 well- and de-differentiated liposarcomas. Knockdown of NF1 increased cell line proliferation and xenograft growth, suggesting a potential tumor suppressor role for this gene in keeping with its function as a regulator of Ras signalling.
Jamshidi et al [7] published the first next-generation sequencing study of epithelioid sarcoma, a rare but clinically-devastating sarcoma that typically presents in the distal extremities of young adults and does not respond to available systemic therapy. Whole genome and transcriptome sequencing on seven tumor specimens and three cell lines confirmed SMARCB1 loss by variable mechanisms, but revealed a complex genome with an unexpectedly high mutational rate for a tumor of younger patients. This high mutational burden is in direct contrast with the genomic profile of rhabdoid tumor, a pediatric neoplasm also driven by the loss of SMARCB1; the mutation rate of epithelioid sarcoma is three orders of magnitude greater than that of rhabdoid tumor.
Shern et al [8] characterized 147 tumor-normal pairs in rhabdomyosarcomas using a combination of whole- genome, exome, and transcriptome sequencing. The overall burden of somatic mutations was relatively low, but several genes were recurrently altered, including previously reported mutations in NRAS, KRAS, HRAS, FGFR4, PIK3CA, and CTNNB1, and novel mutations in FBXW7 and BCOR. Importantly, authors noted that the receptor tyrosine kinase/RAS/PIK3CA-associated networks were altered in 93% of cases, giving therapeutic implications for this disease. Tumors segregated predictably into subtypes with and without a PAX3/PAX7 gene fusion with FOXO1 or alternative partners, including a novel fusion of PAX3 with the C-terminus of INO80D. They asserted that the presence or absence of a PAX fusion more accurately describes the genomic landscape and biology of rhabdomyosarcoma than the traditional alveolar versus embryonal histology-based subtyping, and that it is a better predictor of clinical behaviour [9] and prognosis [10,11]. This brings up the familiar question as to whether the traditional morphologic diagnosis of sarcomas is adequate, or whether molecular techniques ought to be mandatory for sarcoma diagnoses.
Molecular diagnostics
To interrogate the need for and value of ancillary molecular diagnostics for sarcomas, Italiano et al [12] designed GENSARC, a prospective study across 32 centres in France, that compared the diagnostic accuracy of histological assessment by a sarcoma subspecialty pathologist with and without molecular genetic testing. Sarcomas were accrued whenever one of six subtypes presented, based on histological diagnosis (categorized as certain, probable, or possible diagnosis): dermatofibrosarcoma protuberans (COL1A1-PDGFB translocation, leading to PDGβ overexpression), dedifferentiated liposarcoma (MDM2 amplifications), Ewing sarcoma (EWSR1 translocations), synovial sarcoma (SS18 translocations), alveolar rhabdomyosarcoma (PAX3/7 translocations), and myxoid liposarcoma (FUS-DDIT3 translocations). Expert pathologists committed to a diagnosis, level of certainty, and differential diagnosis prior to completion of molecular tests. Comparative molecular testing included fluorescence in-situ hybridisation (FISH) and/or quantitative or reverse transcriptase polymerase chain reaction. Of 384 cases, molecular testing resulted in a change in diagnosis in 53 (14%) cases, leading to a change in management or prognosis in 45 (12%) cases. Based on these findings, the authors recommend mandatory molecular testing - even when the histological diagnosis was made by a sarcoma subspecialty pathologist - for accurate diagnosis and appropriate clinical management of sarcoma. However, it is important to note that almost no cases where the pathologist was “certain” had their diagnosis change as a result of molecular testing. Cases that were “probable” or “possible” had frequent diagnostic changes, but these are cases where pathologist will almost always order molecular ancillary testing if available, or refer out for testing if not.
Regardless of the necessity for molecular testing to support certain diagnoses, detailed genetic analysis of a patient’s tumor tissue can have important implications for targeted therapy. Studies at many institutions are applying next-generation sequencing strategies in an attempt to identify “actionable” mutations with some clinical significance to cancer, sarcoma centres included. Peds-MiOncoSeq [13] employed exome and transcriptome sequencing in 91 pediatric and young adult cancers, including 25 sarcomas. “Potentially actionable findings” were identified in 42 (46%) cases, leading to an actual change in treatment for 14 (15%) patients. This included one rhabdomyosarcoma patient, whose diagnosis and treatment plan changed following sequencing results, and who remained in remission 6 months after the change in management. This study lacked any control group, so it is not possible to assess whether outcomes improved compared to standard care. A similar study, iCat [14], was an “individualized Cancer therapy” effort in advanced pediatric solid tumors. Profiling included targeted DNA sequencing and copy number assessment. Of the 89 patient tumors profiled - including 12 Ewing sarcomas, 11 osteosarcomas, 11 rhabdomyosarcomas, and 27 other soft tissue sarcomas - 43% had clinical implications: an actionable mutation leading to an FDA-cleared or open clinical trial targeted therapy, a translocation that changed diagnosis, or the identification of an underlying cancer predisposition syndrome. Three patients received matched therapy. Chang et al [15] combined whole exome and transcriptome sequencing with SNP arrays in 59 relapsed and refractory pediatric and young adult patients, including 29 sarcomas. Thirty (51%) had clinically actionable alterations. Actionability included somatic mutations targetable by available therapies (41%), change in diagnosis (12%), or reportable germline mutation for which patients received family genetic counselling (12%).
Outside of the clinic, several more studies searched for actionable/targetable and recurrent/driver mutations across sarcomas. Andersson et al [16] conducted a next-generation sequencing panel of 207 hotspots in 50 cancer-associated genes, in Ewing sarcomas (n = 22), synovial sarcomas (n = 14), gastrointestinal stromal tumors (GIST; n = 9), myxoid liposarcomas (n = 7), and Ewing-like small round cell tumors (n = 3). They identified mutations in 8 driver genes in Ewing sarcoma (NRAS, MET, HRAS), Ewing-like small round cell tumors (BRAF, SMARCB1), GIST (KIT, PDGFRA), and synovial sarcoma (CTNNB1). The BASIC3 study by Parsons et al [17] explored the diagnostic yield of whole-exome sequencing in 121 pediatric solid tumors, including 9 rhabdomyosarcomas, 6 Ewing sarcomas, 4 osteosarcomas, and 7 other soft tissue sarcomas. Nearly 40% yielded actionable mutations, in the form of somatic mutations of established clinical utility (3%) or potential clinical utility (24%), or in diagnostic germline findings related to patient phenotype (10%). CTNNB1 was the most frequently mutated somatic gene, plus KIT, TSC2, and MAPK pathway genes (BRAF, KRAS, NRAS). Movva et al [18] had access to 2539 sarcoma specimens, encompassing 61 bone and soft tissue sarcoma subtypes. Up to 2434 samples were profiled by immunohistochemistry, 1048 by FISH, 591 by next-generation sequencing, and 1250 by Sanger sequencing. By immunohistochemistry, they noted overexpression of TOPO2A in 52.8% of cases, SPARC in 35.9%, and PDGFRA in 22.1%. Low expression of MGMT was noted in 65.3% of cases, and loss of PTEN was seen in 38.6%. By DNA sequencing, the most commonly mutated genes were TP53 (26.3%) and BRCA2 (17.6%). Dual TOPO2A overexpression by immunohistochemistry and TP53 DNA mutation was observed in 85.8% of samples. As study cohorts were not consistently defined, and methodologies were not consistently applied, the results from this study are best considered hypothesis-generating, in need of further validation.
Returning to the question of the necessity of comprehensive molecular analysis for the diagnosis and management of sarcomas, we conclude that the evidence at this point is not strong enough to support mandatory use of these expensive and work-intensive diagnostic tools. As of yet, there has been no significant clinical impact reported in terms of treatment response or patient survival. While comprehensive genomics may certainly prove useful in complex cases, it may be most practical for the use of these tools to remain at the discretion of the sarcoma subspecialty pathologist. In the setting of routine clinical practice, traditional morphological assessment (based on H&E slides, supplemented by immunohistochemistry) is likely sufficient for most sarcoma diagnoses.
Fusion identification
Next-generation sequencing of RNA opens the door to identify both known and novel fusion transcripts. Hofvander et al [19] applied massively parallel paired-end mRNA sequencing to 8 sarcomas, including 2 osteosarcomas, 2 myxofibrosarcomas, 1 low-grade fibromyxoid sarcoma, 1 fibrosarcoma, 1 undifferentiated pleomorphic sarcoma, 1 glomus tumor, and 1 myxoid liposarcoma. By this method, they identified two known fusions (FUS-CREB3L2 and HAS2-PLAG1) and three novel fusions (KIAA2026-NUDT11, CCBL1-ARL1, and AFF3-PHF1), highlighting this technique as an effective strategy for fusion detection. The myxoid liposarcoma was reclassified as a lipoblastoma due to absence of the pathognomonic myxoid liposarcoma FUS-DDIT3 fusion and presence of a HAS2-PLAG1 fusion. Authors note that sequencing data was analysed by three different algorithms, but only two of the fusions were reported by more than one software program. They therefore recommend use of more than one algorithm to analyze sequencing output.
This type of approach may be useful to assist in identifying molecular events that can be useful for the differential diagnosis of some challenging sarcoma categories, such as vascular tumors of bone. Epithelioid hemangioma of bone can be difficult to differentiate from other vascular neoplasms due to its highly variable histological presentation. Ijzendoorn et al [20] identified a balanced t(3;14) translocation in an index case, and transcriptome sequencing revealed that the translocation involved a break in exon 4 of the FOS proto-oncogene, leading to introduction of a stop codon and loss of its transactivation domain. Fusions generating FOS truncations were observed in 5 of 7 samples of epithelioid hemangioma (with variable fusion partners). This finding proffers a new mechanism of tumorigenesis and a potentially useful diagnostic tool and treatment target for epithelioid hemangioma of bone.
New sarcoma classification
RNA sequencing has also proven useful in defining new entities among otherwise unclassifiable sarcomas. Le Loarer et al [21] conducted RNA sequencing on 32 round-cell sarcomas which did not fit into known specific diagnostic categories, and they identified 4 index cases bearing mutations in SMARCA4. Postulating that the partially rhabdoid morphology seen in these cases reflects underlying BAF complex inactivation, they performed a targeted sequencing screen on 18 unclassified sarcomas with partial rhabdoid phenotypes, noting SMARCA4 mutations exclusively in the 6 thoracic tumors of the cohort. After finding 9 additional cases (based on inferred characteristics of the initial cohort), they identified a total of 19 samples with SMARCA4 mutations. By whole transcriptome sequencing, these samples clustered apart from the other unclassified sarcoma samples, defining a new entity of aggressive, poor prognosis thoracic primary sarcomas of young adults: “SMARCA4-deficient thoracic sarcomas.” Comparison to the profiles other SMARCA4-deficient malignancies showed that these are distinct from lung carcinomas but related to malignant rhabdoid tumors and to small-cell carcinoma of the ovary, hypercalcemic type.
Chemotherapy
The treatment paradigm of soft tissue sarcoma with chemotherapy
While basic and translational research delve deeper into tumor biology to identify new strategies to target these multifaceted malignancies, conventional chemotherapy remains a mainstay in the treatment of a number of sarcomas. Indeed, this was the subject of many major findings over the past year.
For patients with primary soft tissue sarcomas, surgery with or without radiotherapy can offer a cure, but nearly half of patients recur and eventually die, with an estimated median survival of 12 to 15 months [22]. As a result, treatment of metastatic or unresectable disease with cytotoxic agents is often given for palliative rather than curative purposes [23]. These cytotoxic agents often incorporate anthracycline- or gemcitabine-based regimens as a first line treatment [24-27]. Other agents such as dacarbazine and ifosfamide, only show clinical improvement in overall response rate and progression free survival (PFS), without significant benefit in overall survival [22,28-31]. Moreover, despite superior PFS observed with these conventional cytotoxic therapies, they are fraught with severe toxicities and attendant high costs, a burden for both patients and health care systems. Currently, there is no consensus standard of care for chemotherapy regimens in metastatic sarcoma patients, in part due to treatment strategies that were developed somewhat empirically, rather than through specific, rational targeting of molecular subtype and/or pathogenic mechanism. Consequently, research efforts are underway to address the value of chemotherapy in sarcoma subtypes, as well as to investigate different and newer systemic therapies. Recently published trials have demonstrated promising positive results using newer cytotoxic agents, including eribulin, trabectedin, and aldoxorubicin. Table 1 summarizes the clinical trials in cytotoxic chemotherapy described below.
Table 1: A survey of recently published chemotherapy clinical trials in sarcoma
Regimen |
Trial Analyzed |
Design |
Stage |
Subtype |
N |
Median PFS (months) |
Median OS (months) |
Eribulin vs. Dacarbazine |
Schoffski et al. Lancet. 2016 |
Phase III |
Advanced |
Liposarcoma LMS |
452 |
2.6 in both arms (NS) |
13.5 vs. 11.5 |
Trabectedin vs. Dacarbazine |
Demetri et al. JCO. 2016 |
Phase III |
Advanced |
Liposarcoma LMS |
518 |
4.2 vs. 1.5 |
12.4 vs. 12.9 (NS) |
Trabectedin vs. BSC |
Kawai et al. Lancet Oncol. 2015 |
Phase II |
Advanced |
Translocation-associated sarcoma |
76 |
5.6 vs. 0.9 |
not reached vs. 8 months |
Trabectedin with doxorubicin |
Pavtier et al. Lancet Oncol. 2015 |
Phase II |
Advanced |
Uterine and soft tissue LMS |
109 |
8.2 in uterine LMS vs. 12.9 in soft tissue LMS |
20.2 uterine LMS vs. 34.5 in soft tissue LMS |
Aldoxorubicin vs. Doxorubicin |
Chawla et al. JAMA Oncol. 2015 |
Phase IIb |
Advanced |
STS |
123 |
5.6 vs. 2.7 |
15.8 vs. 14.3 |
Gemcitabine-docetaxel |
Seddon et al. Clin Sarcoma Res. 2015 |
Phase II |
Advanced |
LMS |
44 |
7.1 |
17.9 |
Gemcitabine-docetaxel-bevacizumab |
Dickson et al. Sarcoma. 2015 |
Phase II |
Advanced |
LMS UPS Pleomorphic liposarcoma Angiosarcoma |
35 |
Not reached (3 months PFS=76%) |
Not reached |
Doxorubicin-ifosfamide vs. Gemcitabine-docetaxel |
Davis et al. Eur J Cancer. 2015 |
Phase II |
Early stage |
STS |
80 |
37 vs. Not reached (NS) |
Not reached |
Abbreviations: PFS = progression-free survival, OS = overall survival, LMS = leiomyosarcoma, STS = soft tissue sarcoma, UPS = undifferentiated pleomorphic sarcoma, NS = not significant
Clinical trials
Eribulin
Eribulin is an analogue of the marine-derived compound halichondrin B that acts as an inhibitor of microtubule dynamics through binding to a single site on tubulin, thereby suppressing the stability and growth of microtubules [32,33]. Eribulin has been shown to promote apoptosis, to suppress migration and invasion of cancer cells, and to induce vascular remodeling [33-35].
The role of eribulin was evaluated in a phase III trial that randomized patients with advanced leiomyosarcomas or liposarcomas to receive either eribulin or dacarbazine [36]. Eligible patients had received at least two previous systemic regimens for advanced disease, including anthracyclines. In this trial, patients treated with eribulin demonstrated a significant improvement of two months for the study’s primary endpoint of overall survival (OS), compared to the dacarbazine standard-of-care arm. However, no significant differences were observed in the secondary endpoint of PFS, and patients experienced a higher toxicity rate of 67% grade 3+ adverse events when treated with eribulin, versus 56% in the dacarbazine arm. Importantly, a pre-planned exploratory subgroup analysis showed that the treatment effects of eribulin were limited to patients with liposarcoma (who had a 7-month improvement in OS), whereas no evidence for eribulin efficacy was observed in the leiomyosarcoma subgroup. Based on this analysis, eribulin was approved in early 2016 by the US Food and Drug Administration (FDA) for metastatic liposarcoma patients. It should be noted that the results of the survival analysis in liposarcomas were not broken down into their three molecularly-distinct subtypes (i.e., dedifferentiated, myxoid, and pleomorphic liposarcoma) due to power concerns [36].
Trabectedin
Trabectedin is a marine-derived drug that has several anti-cancer mechanisms of action [37]. It acts mainly through binding to the minor groove of DNA, inhibiting DNA binding proteins - including transcription and DNA repair complexes - ultimately leading to disruption in cell cycle and induction of apoptosis [37,38].
A phase III trial assessed the role of trabectedin versus dacarbazine in a similar cohort to that included in the aforementioned eribulin trial, again including metastatic leiomyosarcoma or liposarcoma patients who had received at least one prior systemic therapy in addition to anthracyclines [39]. This trial showed a significant improvement of 3.7 months in the secondary endpoint of PFS, favoring trabectedin over dacarbazine, while no statistically significant results were observed for the primary endpoint of OS. Trabectedin PFS benefit was observed across all pre-planned subgroup analyses including different histology subtypes. This data led to trabectedin’s approval by the FDA in October 2015 for use in metastatic liposarcoma and leiomyosarcoma, indications that had already been approved in Europe. Interestingly, the clinical outcomes reported in this study contrast somewhat with those observed in the eribulin trial - which accrued patients with similar inclusion criteria and characteristics - again reporting higher rates of toxicity in the trabectedin arm than with the standard therapy of dacarbazine. Overall, the results reported in these clinical trials do not explicitly support a preferred regimen of either eribulin or trabectedin in the metastatic setting of liposarcoma, but the differences observed in clinical outcomes might reflect differences in the mechanisms-of-action of trabectedin versus eribulin that should be further investigated.
The role of trabectedin as a modulator of the transcription of oncogenic fusion proteins was demonstrated in a phase II trial assessing the efficacy and safety of trabectedin as a second line (or later) therapy for patients with advanced translocation-associated sarcomas [40]. Patients included in this trial had mainly myxoid liposarcoma and synovial sarcoma and were randomized to receive either trabectedin or best supportive care. This trial showed a statistically significant 5-month advantage in the primary endpoint of PFS with trabectedin administration [40]. Importantly, in a subsequent evaluation of 30 patients who crossed over to trabectedin after disease progression on best supportive care, trabectedin was still effective in improving PFS [41].
Beyond the established activity of trabectedin as second-line therapy for sarcomas, its role in the first-line setting was examined in a phase II single-arm clinical trial using a combination of trabectedin and doxorubicin [42]. Among 108 assessable patients with leiomyosarcoma of the uterus or soft tissue, nearly 90% achieved disease control [42]. While this trial showed promising results regarding the combination of trabectedin and doxorubicin in the first-line setting of leiomyosarcoma, a previous study by the European Organization for Research and Treatment of Cancer (EORTC) and Sarcoma Alliance for Research through Collaboration (SARC) groups, comparing trabectedin versus doxorubicin as a first-line therapy in advanced soft tissue sarcoma, did not show any superiority for trabectedin over doxorubicin [43]. Accordingly, the combination of trabectedin and doxorubicin needs to be assessed head-to-head against the effective standard-of-care control arm of doxorubicin monotherapy or doxorubicin-ifosphamide.
Aldoxorubicin
Aldoxorubicin is a novel pro-drug of doxorubicin, where a pH-sensitive linker conjugates doxorubicin to albumin. The exposure of the albumin-drug conjugate to the acidic tumor microenvironment releases doxorubicin, which preferentially localizes in tumor cells that are pinocytotically-active [44,45]. Drug uptake within tumor tissues is further enhanced through defective lymphatic drainage and high permeability, promoting macromolecule retention in tumor tissues [46]. The net result is a capacity to deliver a higher dose of doxorubicin into tumor cells than is received by normal cells (e.g., myocardium), improving the therapeutic index.
Superior clinical activity of aldoxorubicin was recently demonstrated in a phase IIb study that showed a significantly greater PFS (the primary endpoint), in patients with advanced soft tissue sarcoma randomized to receive aldoxorubicin or doxorubicin [47]. However, no significant differences were observed in OS. Currently, the safety and efficacy of aldoxorubicin is being assessed in combination with ifosfamide (phase I/II trial, NCT02235701) or in combination with gemcitabine (phase I trial, NCT02235688) in metastatic soft tissue sarcoma patients. In addition, aldoxorubicin is being investigated in a phase III trial versus investigator’s choice of treatment in metastatic soft tissue sarcomas (NCT02049905).
Gemcitabine and docetaxel
Gemcitabine and docetaxel combinations have previously displayed a proven activity, particularly in advanced leiomyosarcoma [48]. These results have recently led to the design of a phase III trial comparing gemcitabine-docetaxel versus the conventional therapy of doxorubicin in the first-line setting of metastatic soft tissue sarcoma (GeDDis) [48]. This trial did not report significant differences in OS or PFS between the two treatment arms (data not yet published). Furthermore, the gemcitabine-docetaxel combination was stopped early due to toxicity, and thus, doxorubicin was recommended to remain as the standard first-line treatment for metastatic soft tissue sarcoma [48].
Beyond its role in the metastatic setting, the gemcitabine-docetaxel combination was recently assessed in early-stage soft tissue sarcomas. A phase II trial compared the combination of gemcitabine-docetaxel against conventional therapy with doxorubicin-ifosfamide in patients with localized, resectable, high-risk sarcoma. While this trial showed that the gemcitabine-docetaxel combination was tolerable, it did not show a significant superiority of gemcitabine-docetaxel over doxorubicin-ifosfamide in hospitalization rate, which was (somewhat unusually) defined as the primary endpoint of this study [49].
Further phase II trials have also investigated whether the activity of gemcitabine and docetaxel is enhanced by the addition of anti-angiogenic agents such as bevacizumab [50], particularly as targeted therapy for vascular sarcomas [51]. While these studies reported only modest response rates, more than half of the patients achieved stable disease. In an attempt to appropriately evaluate these findings in a phase III trial, a double-blind, placebo-controlled, randomized trial of gemcitabine and docetaxel with or without bevacizumab was designed [26]. Eligible patients had a diagnosis of leiomyosarcoma, undifferentiated pleomorphic sarcoma, pleomorphic liposarcoma, or angiosarcoma. However, due to a slow recruitment rate, the trial was changed to a single-arm study evaluating only the addition of bevacizumab to gemcitabine and docetaxel, reporting a PFS of 76% at 3 months [26]. While this PFS rate is impressive and higher than expected from similar regimens without bevacizumab [52], the absence of a control arm - a common problem in sarcoma trials - makes it hard to achieve a high level of evidence supporting the role of bevacizumab in advanced soft tissue sarcomas. These findings highlight the overall need for more collaborative, randomized phase III clinical trials that could confirm benefits suggested by single-arm phase II studies.
Targeted Therapy
Translational science
Basic science studies have identified a number of targetable oncogenic pathways activated in specific sarcomas. Figure 1 depicts the agents and targeted molecules discussed below, including regulators and cross talk between the pathways.
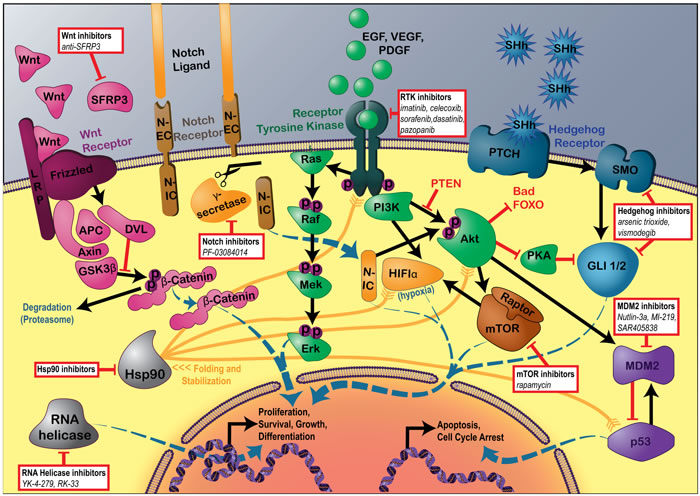
Figure 1: Signaling pathways currently targeted in sarcoma translational research. Details of Wnt, Notch, Receptor Tyrosine Kinase, Hedgehog, MDM2, and mTOR signaling pathways, including common regulators and pathway interactions. Hsp90 protein clients also depicted. Boxes indicate the specific agents described in this review that have seen progress in the past year.
Receptor tyrosine kinases
Evidence behind receptor tyrosine kinase inhibitors in sarcomas is comparatively advanced, with a number of agents in clinical trials. Aside from gastrointestinal stromal tumors, there is little known about biomarkers predictive for response. A phase II study of imatinib in 33 progressive, unresectable desmoid tumors [53] assessed the correlation between CTNNB1 (β-catenin) mutation status and “progression arrest rate” - the proportion of patients showing complete or partial response, or stable disease by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 - 6 months after initiation of imatinib treatment. CTNNB1 mutations are found in 80% of desmoid tumors, either at threonine 41 (T41) or serine 45 (S45), the latter correlating with increased risk of recurrence [54-56]. Progression arrest rate was highest in patients bearing the S45 mutation, at 85%. The next best response was seen in patients with the T41 mutation (70%), followed by those with wild-type CTNNB1 (43%). According to this study, CTNNB1 mutation status may be predictive of response to imatinib; however, given that wild-type CTNNB1 is generally associated with better prognosis [54-56], this study design may have selected for a subpopulation of more clinically aggressive wild-type desmoid tumors. Furthermore, desmoid tumors frequently undergo spontaneous progression arrest, and clinical studies of this vexatious neoplasm really require control arms if improved progression arrest is to be attributed to kinase inhibitors or other systemic interventions [57].
In pre-clinical studies of osteosarcoma, Liu et al [58] show synergy between kinase inhibitor ZD6474 and non-steroidal anti-inflammatory agent celecoxib in cell lines and mouse xenografts. ZD6474, a small-molecule inhibitor of VEGFR-2 and EGFR, inhibited the proliferation of three osteosarcoma cell lines, promoting cell cycle arrest and apoptosis. Based on high COX-2 expression in osteosarcoma cell lines, authors also tested the COX-2 inhibitor celecoxib in vitro and saw a similar anti-proliferative effect. Combination treatment of ZD6474 and celecoxib demonstrated synergistic effects in one cell line and additive effects in the other two. Synergy between the two agents was also observed in cell line xenografts.
mTOR
Malignant peripheral nerve sheath tumors (MPNSTs) have been shown to have receptor tyrosine kinase gene amplifications of PDGFR and cKIT [59]; however, imatinib did not show promise in a phase II trial [60]. mTOR is a key member of the PI3K/AKT axis of RTK signalling, so combining mTOR and RTK inhibition may reduce opportunities for tumor escape and therapeutic resistance. In a study comparing two kinase inhibitors plus or minus mTOR inhibitor rapamycin [61], combination treatment enhanced the anti-proliferative effects against an MPNST cell line from 40-45% per single agent to 70% inhibition of cell proliferation. In a mouse xenograft, addition of rapamycin to kinase inhibitors resulted in enhanced tumor suppression, and following cessation of treatment, reduced tumor regrowth.
Notch
Shang et al [62] observed that nuclear Hes1, a downstream effector of Notch, is highly expressed in desmoid tumor cell lines compared to dermal scar tissue. Gamma-secretase inhibitors inhibit Notch signalling by blocking cleavage of Notch’s intracellular domain, NICD. A recent phase I trial of γ-secretase inhibitor PF-03084014 in desmoid tumors [63] showed promising results, leading to the initiation of an ongoing phase II trial (NCT01981551) [64]. To further evaluate this agent’s activity in desmoid tumors, Shang et al [62] exposed a panel of desmoid tumor cell lines to PF-03084014. Treatment inhibited cell growth and decreased migration and invasion. There was also a drop in NCID and expression of Hes1 in exposed cell lines. Taken together, these studies suggest that Notch signalling is important for desmoid tumor survival and that PF-03084014 is a worthwhile strategy to investigate for the treatment of desmoid tumors.
Wnt
In a rhabdomyosarcoma study aiming to identify targetable proteins associated with alveolar rhabdomyosarcoma fusion oncoprotein [65], microarray expression profiling was performed on PAX3-FOXO1-expressing human skeletal muscle myoblasts. Transcriptome analysis revealed alteration of Wnt pathway gene members, including SFRP3 (secreted frizzled related protein 3). Knockdown of SFRP3 in an alveolar rhabdomyosarcoma cell line led to reduced cell growth and proliferation, cell cycle arrest, and apoptosis. In a conditional alveolar rhabdomyosarcoma murine xenograft system, suppression of SFRP3 reduced tumor growth and increased myogenic differentiation. Combining SFRP3 suppression with chemotherapy agent vincristine was more effective at reducing alveolar rhabdomyosarcoma cell line growth than either treatment alone, and the addition of vincristine ablated tumorigenesis in the conditional murine xenograft.
Hedgehog
Hedgehog pathway inhibitors show promise in osteosarcoma, through targeting of SMO [66] or GLI family [67,68] hedgehog transcription factors. Saitoh et al [69] examined hedgehog inhibitors arsenic trioxide and vismodegib in combination with conventional chemotherapy agents cisplatin, ifosfamide, or doxorubicin in cell-line and xenografts. In vitro and in vivo, osteosarcoma cell proliferation and tumor growth were inhibited by any pairing of a Hedgehog pathway inhibitor with a standard chemotherapy agent. Combinations were designated as synergistic by combination-index analyses.
Hsp90
Hsp90 is a chaperone protein that assists in the folding of many of the signalling molecules mentioned above. In a study of undifferentiated pleomorphic sarcoma [70], Hsp90 was found to be highly expressed in 56.4% of cases and was associated with poor prognosis. Expression correlated with p-Akt, p-mTOR and p-S6RP, potentially implicating Hsp90 in the activation of the mTOR pathway. In vitro, inhibition of Hsp90 decreased cell viability and inactivated the mTOR pathway. In a study by Ernst et al [71] expression of HSP90 was found to be associated with radioresistance by comparing transcriptomes of sarcoma cell lines with radioresistance scores (generated by principal component analysis). Expression of HSP90 strongly correlated with high radioresistance scores, and subsequent HSP90 inhibition sensitized radioresistant sarcoma cell lines to radiation therapy. Hsp90 inhibition was also shown to help overcome kinase inhibitor resistance in myxoid liposarcoma [72], contributing to rapid cell death in cell lines and necrosis in cell line xenografts.
MDM2
MDM2 is a suppressor of genome guardian p53, and its genetic amplification is a hallmark of well- and de-differentiated liposarcoma. Bill et al [73] compared the effectiveness of MDM2 inhibitors Nutlin-3a, MI-219, and novel agent SAR405838. In vitro, all three agents effectively reactivated the p53 pathway, impeding cell proliferation and inducing cell-cycle arrest and apoptosis. The highest potency was seen with SAR405838, and this antitumor effect persisted in treatment of dedifferentiated liposarcoma mouse xenografts. Another study on MDM2 inhibitor Nutlin-3 in DDLPS [74] found that this agent was only effective in cell lines with wild-type p53. Nutlin-3 had little effect on p53-deficient cell lines, presenting p53 mutation as a mechanism of MDM2 inhibitor resistance. In these cell lines, addition of histone deacetylase (HDAC) inhibitor treatment overcame Nutlin-3 resistance, and was associated with PTEN and p21 up-regulation and inactivation of AKT.
RNA helicase
FLI1, the most common translocation partner of EWSR1 in the oncogenic fusion protein driving Ewing sarcoma, requires binding to transcriptional cofactor RNA Helicase A for full activity. YK-4-279, a small molecule that blocks the interaction between FLI1 and RNA helicase A, was tested in a Ewing sarcoma cell line xenograft [75] and induced tumor regression with daily dosing. A related RNA helicase, DDX3, was found to be highly expressed in a number of sarcoma subtypes [76]. Suppression of DDX3 by knockdown or by its small-molecule inhibitor RK-33 was cytotoxic to Ewing sarcoma cell lines, and in a patient-derived Ewing sarcoma xenograft, RK-33 inhibited tumor growth without evident toxicity. Taken together, these studies support the development of RNA helicase inhibition as a new targeted strategy for Ewing sarcoma.
FOXM1
FOXM1 is a transcription factor - sometimes activated as part of the Wnt or Hedgehog pathways - that contributes to proliferation and cell cycle progression. It has been shown to be overexpressed in numerous sarcomas [77,78], which Eisinger-Mathason et al. [77] claim is attributable to deregulation of the Hippo pathway, based on copy number variation analysis of The Cancer Genome Atlas sarcoma database (mostly leiomyosarcoma, dedifferentiated liposarcoma, myxofibrosarcoma, and UPS). In fact, they postulate that overexpression of FOXM1 by Hippo effector protein YAP is necessary for tumorigenesis in some sarcomas, based on reduced proliferation seen in a mouse model of UPS after YAP knockdown. Knockdown or pharmacological (thiostrepton) inhibition of FOXM1 itself also impaired proliferation and reduced tumor size in the UPS mouse model, a finding which was also shown in synovial sarcoma cell lines by Maekawa et al [78]. Together, these findings highlight FOXM1 as a potential therapeutic target for some sarcomas.
Clinical trials
While significant advancements have been reported in clinical trials of cytotoxic chemotherapeutic agents, metastatic soft tissue sarcomas still have poor outcomes and few effective therapeutic options. Table 2 summarizes the clinical trials in targeted therapy described below.
Table 2: A survey of recently published targeted therapy studies in sarcoma
Targeted Therapy |
Study Analyzed |
Design |
Stage |
Subtype |
N |
Median PFS (months) |
Median OS (months) |
Imatinib |
Stacchiotti et al. Clin Cancer Res. 2016 |
retrospective |
Advanced |
DFSP |
10 |
11 |
Not reached |
Imatinib |
Hindi et al. Eur J Cancer. 2015 |
retrospective |
Advanced |
Chordoma |
46 |
9.9 |
30 |
Sorafenib |
Bompas et al. Annals of oncology. 2015 |
Phase II |
Advanced |
Chordoma |
27 |
Not reached (9 months PFS=73%) |
Not reached (9 months OS=87%) |
Dasatinib |
Scheutze et al. Cancer. 2016 |
Phase II |
Advanced |
Ewing LMS Liposarcoma MPNST OSa RMS UPS |
200 |
1.9 |
8.6 |
Pazopanib vs. Placebo |
Kawai et al. Jpn J Clin Oncol. 2016 |
Phase III |
Advanced |
STS |
47 |
1.6 vs. 5.7 |
15.4 vs. 14.9 (NS) |
Pazopanib plus radiotherapy |
Haas et al. Acta Oncol. 2015 |
Phase I |
Preoperative for locally advanced |
STS |
10 |
NA |
NA |
Pazopanib |
Ronellenfitsch et al. BMJ Open. 2016 |
Phase II |
Preoperative for high risk early stage |
STS |
recruiting |
TBD |
TBD |
Abbreviations: DFSP = dermatofibrosarcoma protuberans, LMS = leiomyosarcoma, MPNST = malignant peripheral nerve sheath tumor, OSa = osteosarcoma, RMS = rhabdomyosarcoma, UPS = undifferentiated pleomorphic sarcoma, STS = soft tissue sarcoma, NA = not available, TBD = to be determined, NS = not significant
Imatinib
Imatinib is a tyrosine kinase inhibitor that inhibits several oncogenic pathways. Its impressive activity in sarcomas has been primarily shown in GIST, by interrupting the constitutive activation of KIT-mediated signal transduction characteristic of that sarcoma [79-81]. Imatinib has also demonstrated clinical benefit in the metastatic setting for dermatofibrosarcoma protuberans (DFSP) through inhibiting the activation of platelet derived growth factor receptor beta (PDGFR-β) [82,83]. DFSP tumors are characterized by PDGFB rearrangements (resulting in overexpression of PDGFβ), and 10-20% progress to a more aggressive, higher-grade subtype, designated as fibrosarcomatous transformation (FS-DFSP) [84-86]. Scarce data exists on the role of imatinib specifically in these metastatic FS-DFSP tumors. A recent retrospective study evaluating 10 metastatic FS-DFSP patients treated with imatinib showed that 90% of patients achieved clinical benefit (8 partial responses and 1 stable disease) with a median of 11 months PFS [87]. However, the duration of response observed in this study was lower than that reported ina previous series containing a mix of cases with both low-grade and FS-DFSP [88]. Moreover, this study showed that imatinib failed to eradicate the metastatic disease, as all five patients who had a complete resection of the residual tumor after imatinib still experienced a relapse. These findings suggest that despite a high expression of PDGFβ in DFSP, there might be other putative targets that contribute to disease progression and the development of imatinib resistance in metastatic patients. Interestingly, RNA transcriptional profiling of the study cohort revealed a simultaneous down-regulation of kinase signaling pathways and up-regulation of pathways involved in antigen processing and presentation [87]. These findings support the potential role for incorporating immune system enhancement strategies to achieve higher durable responses in the setting of imatinib-refractory FS-DFSP. Recently, CDK4/CDK6 inhibitors demonstrated pre-clinical activity against imatinib-resistant FS-DFSP [83].
The role of imatinib as a PDGFR-β inhibitor was also tested among metastatic chordoma patients, a disease displaying PDGFR-β protein expression but not amplification or other activating mutation. In a recent retrospective analysis that included 46 PDGFR-β-positive metastatic chordoma cases, median PFS was 9.9 months. Within a median follow up of 24.5 months, 34 of 46 of patients had stable disease by RECIST 1.0, with no partial or complete responses observed [89]. These results are consistent with previous findings reported in a phase II trial in 50 patients with advanced chordoma treated with imatinib (median PFS = 9 months and 70% of patients achieved stable disease) [90]. The very limited response rates observed in these trials, despite reportedly high expression of PDGFR-β, suggest a potential role for targeting related pathways other than PDGFR-β using other therapies. As such, the combination of imatinib and histone deacetylase inhibitor in recurrent chordoma patients is currently under evaluation (NCT01175109).
Sorafenib
Sorafenib, a multi-kinase inhibitor, has been shown to act through different signal transduction pathways, including via inhibition of pro-angiogenic vascular endothelial growth factor receptors (VEGFR and PDGFR-β). In phase II clinical trials, sorafenib has been shown to have activity in metastatic soft tissue sarcoma [91], specifically in leiomyosarcoma [92]. Furthermore, a pre-planned exploratory analysis of a phase II clinical trial indicated that this agent has activity in angiosarcoma [93].
A recently published phase II trial evaluated the role of sorafenib as a PDGFR-β inhibitor in locally advanced and metastatic chordoma patients (n = 27) [94]. In this trial, the 9-month PFS was 73% and the 12-month OS was 86.5%. Compared to previous studies evaluating the role of imatinib in metastatic chordoma, 92% (12/13) of the assessable patients in this trial, who were not pre-selected based on PDGFR-β status, had a stable disease by RECIST criteria.
Dasatinib
Dasatinib is a multi-kinase inhibitor that targets several oncogenes. Its main activity in sarcoma is thought to be through inhibiting the c-SRC kinase pathway. Gene expression profiling has reported the c-SRC pathway to be highly expressed in leiomyosarcoma [95] and chondrosarcoma [96]. Pre-clinical activity has been demonstrated in different cell lines, including rhabdomyosarcoma, osteosarcoma [97], Ewing sarcoma [97,98], and synovial sarcoma [99]. Despite this pre-clinical evidence, a large phase II clinical trial conducted by the SARC group, which included 200 patients from seven different cohorts of advanced sarcoma, showed low clinical benefit from dasatinib [100]. The study was terminated early for futility in five cohorts; only the cohorts of leiomyosarcoma and undifferentiated pleomorphic sarcoma fully accrued. However, dasatinib still did not show clinically-significant activity in these two cohorts, with only two objective responses observed in undifferentiated pleomorphic sarcoma patients [100]. Currently, the activity of dasatinib in combination with the CTLA4 inhibitor ipilimumab is being assessed in a phase I trial in unresectable or advanced soft tissue sarcoma (NCT01643278).
Pazopanib
Pazopanib is a multi-kinase inhibitor that has been shown to have activity in metastatic soft tissue sarcoma, primarily through the phase III PALETTE trial published in 2012, which randomized metastatic soft tissue sarcoma patients to receive either pazopanib or placebo. This trial demonstrated a significantly prolonged PFS by 3 months in pazopanib arm, although no significant differences were observed in OS [101]. In a recent analysis on the Japanese subpopulation in PALETTE, pazopanib demonstrated results consistent with that observed in the PALETTE trial global population [102]. It should be noted that a pre-planned analysis on the original PALETTE trial did not reveal a superior benefit of pazopanib in specific sarcoma subtypes [101]. Moreover, a retrospective analysis limited to the uterine sarcoma cases from the PALETTE trial did not show significant activity of pazopanib against uterine sarcoma when compared to other subtypes [103].
Pazopanib has also been reported to be active against desmoid-type fibromatosis [104], which is being compared to chemotherapy in a phase II clinical trial (NCT01876082).
The role of pazopanib in the neoadjuvant setting was also investigated in several studies, based on recent evidence suggesting a synergistic effect when combining radiotherapy with angiogenesis-targeted therapies that act through inhibiting the supplying vasculature for sarcoma cells [105]. A recent phase I trial assessed the neoadjuvant combination of pazopanib and radiotherapy in locally advanced soft tissue sarcoma [106]. While none of the ten patients showed a volume reduction after radiotherapy, a high pathologic complete response rate (>95%) was observed in four patients. The overall findings of this trial showed that pazopanib and radiotherapy is tolerable in the neoadjuvant setting. Currently, a phase II/III trial (PAZNTIS) is assessing pre-operative chemoradiation or radiation with or without pazopanib for non-rhabdomyosarcoma soft tissue sarcomas (NCT02180867). Moreover, a study protocol recently published by the German Interdisciplinary Sarcoma Group described an ongoing phase II clinical trial (GISG-04/NOPASS) that is assessing the role of pazopanib in high-risk, resectable soft tissue sarcoma patients treated with radiotherapy [107]. Interestingly, in this trial, the primary endpoint is defined as metabolic response rate, measured as a reduction in the uptake value in post- versus pre-treatment using positron emission tomography (PET-CT) (NCT01543802).
RANKL inhibitor (denosumab)
Denosumab is a monoclonal antibody that targets the receptor activator of nuclear factor-kappa b ligand (RANKL), which is normally expressed on osteoblasts and functions to activate osteoclasts to control bone regeneration and remodeling. This agent has exhibited a particularly significant clinical benefit in giant cell tumors, which highly express RANKL [108-111]. While the current treatment options available for this benign but locally aggressive bone tumor are mainly surgical, depending on site, this can be associated with severe morbidity [108,112]. However, a recent phase II trial conducted on 222 patients with technically-resectable giant cell tumors treated with neoadjuvant denosumab for a median duration of 15.3 months not only exhibited down-staging of the tumor, but also showed activity in restoring the bone with increased cortical thickness [113]. Moreover, 48% (106/222) of the patients in this trial either delayed their need for surgery or underwent less morbid interventions than had appeared necessary prior to denosumab treatment [113]. These promising results support the role of denosumab in achieving disease control and favorable clinical outcomes without exposing patients to potentially high-morbidity surgical interventions.
Epigenetic Therapy
Epigenomics
Epigenetic and epigenomic modifications are emerging as key mechanisms in the pathogenesis of many sarcomas. Some subtypes could be described as exhibiting an “epigenomic mutator phenotype,” wherein a single missense mutation at an epigenetic modification target site, generates an “oncohistone,” resulting in aberrant repression and de-repression of genes at the global transcriptome level. An example of one such mutation, investigated by Lu et al [114], is seen in ~95% of chondroblastomas, in which a lysine-to-methionine mutation in histone 3.3 (H3.3K36M) has a dominant negative effect over the thirty other H3 histone alleles, inhibiting the normal methylation of wild-type H3K36. This results in aberrant expression of genes associated with mesenchymal differentiation, leading to the development not only of chondroblastoma, but also of some cases of undifferentiated sarcoma. These oncohistones not only reduced (activating) H3K36 methylation, but also increased (repressive) H3K27 methylation. Although the H3.3K36M mutation is specific to chondroblastomas [115], this study identified an H3.1K36M mutation in a pediatric undifferentiated soft tissue sarcoma, suggesting that K36M mutations in other H3 histones might play a role in poorly differentiated sarcomas. In follow-up papers investigating the mechanism by which K-to-M oncohistones inhibit global histone lysine methylation [116,117], it was found that the mutant histones occupy the active sites of histone methyltransferases, inhibiting their function and sequestering them from wild-type H3 histones.
In 2014, MPNST saw major breakthroughs in understanding the epigenetic mechanisms by which somatic mutations bring about malignant transformation, which have since led to practical diagnostic advances. Lee et al [118] identified loss-of-function mutations in EED and SUZ12, key components of the PRC2 polycomb repressive complex, in 12 of 15 (80%) cases of MPNST. Similarly, Zhang et al [119] conducted whole-genome sequencing on MPNSTs and found mutations critical to PRC2 functioning (SUZ12, EED, EZH2) in nearly half (24/50) of samples (alterations not seen in 11 sequenced neurofibroma samples). Given that the primary function of PRC2 is methylation of H3K27, these studies also identified complete loss of trimethylated H3K27 (H3K27me3) in samples with PRC2-related mutations. Lee et al [118] further showed that H3K27 trimethylation could be recovered in vitro by re-introducing the lost PRC2 component, which concomitantly decreased cell growth. This loss of H3K27 methylation was subsequently confirmed in three independent studies. Schaefer et al [120] evaluated immunohistochemistry for H3K27me3 in 100 MPNSTs and found that 51 (51%) were negative, correlating with higher grade. Among 200 MPNST mimics also evaluated, only 4 (2%) were negative for H3K27me3. Cleven et al [121] also performed H3K27me3 immunohistochemistry, and observed loss of H3K27me3 in 34% (55/162) of MPNSTs, while expression was retained in neurofibromas (n = 32). Within other tumors, H3K27me3 loss was seen in 24 of 341 (7%) cases, notably 9 of 15 (60%) synovial sarcomas and 3 of 8 (38%) fibrosarcomatous-DFSP. In this sample set, H3K27me3 loss correlated with inferior survival. Rorich et al [122] used DNA methylation arrays to characterize 171 peripheral nerve sheath tumors, and found that 21 of 41 (51%) MPNSTs had loss of H3K27 trimethylation. This study also confirmed PRC2 loss-of-function mutations among H3K27me3-deficient MPNSTs, identifying 15 (71%) and 4 (19%) cases with SUZ12 and EED mutations, respectively. Taken together, these studies show that loss of H3K27me3 is highly specific for MPNST and may be a useful diagnostic marker, particularly for its histologically-difficult distinction from neurofibroma, but not for distinction from malignant mimics synovial sarcoma and FS-DFSP. Further, loss of H3K27me3 was associated with higher grade and poorer survival in MPNST, so loss of PRC2 function may be contributing to increased tumor aggressiveness.
In rhabdomyosarcoma, the DNA methylome was characterized for 37 tumors and 10 cell lines [123], and PAX3-FOXO1 fusion-positive tumors showed distinctly lower global methylation than in fusion-negative tumors. Fusion-negative rhabdomyosarcoma bore more resemblance to normal skeletal muscle, suggesting that fusion-positive rhabdomyosarcoma has aberrant DNA methylation. Sites of abnormal methylation were associated with changes in mRNA expression patterns, and these sites included an augmented number of PAX3-FOXO1 target gene binding sites. This finding suggests that the rhabdomyosarcoma fusion protein modifies DNA methylation to regulate target gene expression.
Translational science
Bromodomain
Bromodomain and extra terminal domain (BET) proteins are “readers” of histone acetylation marks, facilitating the transcription of genes in marked areas. In cancer, BET proteins are key translators of aberrant acetylomes, and as such, BET inhibitors are emerging as promising treatments for some cancers, including bone sarcomas.
In osteosarcoma, Lee et al [124] and Baker et al [125] investigated the use of BET protein BRD4 inhibitor JQ1. In vitro, JQ1 treatment inhibited cell line proliferation and survival; however, in vivo, JQ1 alone was ineffective against mouse cell line xenografts [124]. Combination with the mTOR inhibitor rapamycin was able to overcome resistance to JQ1 treatment in both models. Because BET inhibitors like JQ1 function by decreasing transcription of genes with aberrantly acetylated histones, the authors also explored changes in transcription induced by treatment. They identified RUNX2 [124] - an osteoblast differentiation transcription factor - and FOSL1 [125] - part of the AP-1 differentiation/proliferation/apoptosis transcription factor complex - as abnormally expressed genes that are directly modified by JQ1 treatment.
In Ewing sarcoma, BET inhibition was shown to directly block transcription of the fusion oncoprotein in two independent studies [126,127], which observed a strong down-regulation of EWS-FLI1 in cell lines following treatment with JQ1. Chromatin immunoprecipitation demonstrated that BRD4 becomes depleted in the fusion oncogene’s promoter [127], and RNA microarray analysis revealed down-regulation of Ewing sarcoma-associated expression programs [126]. Cell line proliferation and migration were inhibited with JQ1 treatment, which induced both cell cycle arrest and apoptosis. In mouse xenografts, treatment suppressed tumor development. Taken together, these studies suggest that BET inhibition interferes with EWS-FLI1 activity and may prove a promising targeted strategy for treatment of Ewing sarcoma.
EZH2
EZH2 is the catalytic subunit of the PRC2 transcriptionally repressive chromatin remodeling complex. Lv et al [128] found that EZH2 is overexpressed in osteosarcoma and correlates with poor prognosis. RNA silencing of EZH2 inhibited tumor growth in cell lines, induced apoptosis, and enhanced sensitivity to cisplatin. In vivo, EZH2 knockdown impaired xenograft growth and metastasis. These results suggest that EZH2 is important for tumor growth and metastasis in osteosarcoma, implying that EZH2 inhibition may prove a worthwhile treatment strategy as such drugs become available [129-133].
Synovial sarcoma
Laporte et al [134] employed proximity ligation assay to validate the previously proposed [135] association of synovial sarcoma fusion oncoprotein SS18-SSX with TLE1 cofactor. TLE1 co-localized in the nucleus with SS18-SSX (but not with wild-type SS18) in cell lines and in human tumor tissue, and these interactions were disruptable by treatment with histone deacetlyase inhibitors. This technique could be useful for identifying agents that disrupt the oncoprotein complex in high-throughput drug screens for this disease. EZH2 inhibitors may also have activity in this disease [136,137].
Clinical trials
HDAC Inhibitors
In pre-clinical studies, histone deacetylase (HDAC) inhibitors have been shown to be particularly active in translocation-associated sarcomas such as synovial sarcoma and Ewing sarcoma, through reversing aberrant transcriptional repression induced by the underlying fusion proteins in these sarcomas [135,138,139]. Clinical studies in other types of cancer have demonstrated an enhanced efficacy of HDAC inhibitors when combined with topoisomerase II inhibitors, such as anthracyclines, leading to more transcriptionally-active chromatin that is primarily observed with the administration of HDAC inhibitors prior to anthracyclines [140,141]. This observation was the basis for the design of a phase I trial assessing the HDAC inhibitor panobinostat followed by the topoisomerase II inhibitor of epirubicin in patients with advanced soft tissue sarcoma [142]. Among 20 patients included in this trial, 60% (n = 12) achieved a clinical benefit, suggesting that the combination of panobinostat and epirubicin might have value in overcoming anthracycline resistance. Currently, different epigenetic treatment strategies, including HDAC inhibitors, are being evaluated in a number of active sarcoma clinical trials (NCT01136499), (NCT01879085), (NCT00937495).
Immune Therapy
Immune microenvironment of sarcomas
The immune microenvironment of sarcomas is poorly characterized to date, leaving open the question of which sarcoma subtypes are immunogenic. D’Angelo et al [143] conducted an immunohistochemistry survey of 50 soft tissue sarcomas to evaluate the presence of tumor-infiltrating lymphocytes (TILs), tumor-associated macrophages, and immune checkpoint receptor and ligand, PD1 and PD-L1. Immunohistochemical staining examined CD3 (TILs), CD4 (helper T-cells), CD8 (cytotoxic T-cells), FOXP3 (regulatory T-cells), PD1, and PD-L1 expression, and multiplex IHC was performed for CD3/PD1, CD3/CD8, and CD3/CD4/FOXP3. The presence of macrophages was evaluated histologically. Lymphocyte and macrophage infiltration were observed in 98% and 90% of cases, respectively. Defining “low” or “high” density TILs as below or above 5%, they noted that 27 (54%) had low-density TILs, mainly leiomyosarcoma (3/4), synovial sarcoma (4/5), and chondrosarcoma (1/1), and 22 (44%) had high-density TILs, mainly GIST (9/14). Tumor, lymphocyte, and macrophage PD-L1 expression were 12%, 30%, and 58%, respectively, with the highest frequency of PD-L1 positivity seen in GIST (4/14). They observed no clear correlation between marker expression and clinical outcomes in this small study. Movva et al [18] also assessed PD-L1 expression by immunohistochemistry across 221 sarcomas, and found that 57% expressed PD-L1 and 54.8% had PD-1+ TILs. Significantly high expressors of PD-L1 included 19 of 60 (32%) leiomyosarcomas, 12 of 16 (75%) chondrosarcomas, 23 of 30 (77%) liposarcomas, and 7 of 10 (70%) undifferentiated pleomorphic sarcomas.
Smaller studies focusing on specific subtypes revealed broadly similar results. A study of 35 well- and de- differentiated liposarcomas [144] found TILs in all samples by flow cytometry, with a greater prevalence of CD4+ (80%) than CD8+ (20%) T-cells. Among CD8 T cells, 65% expressed PD-1. They also found mature dendritic cells in close proximity with CD4+ T-cells, suggesting intra-tumoral antigen presentation. Feng et al [145] examined 78 chordomas by immunohistochemistry and found that 75% have TILs present. While PD-L1 expression was seen in 95% of samples, 43% were classified as “PD-L1-high” due to moderate or strong staining intensities. While presence of these TILs correlated with PD-L1 expression, there was no clear correlation with survival. In osteosarcoma, Fritzsching et al [146] surveyed 135 samples for CD8+ and FOXP3+ T-cell presence by immunohistochemistry. 95% of cases had both CD8+ and FOXP3+ T-cells, and a high CD8:FOXP3 ratio correlated with improved survival. It is clear that the immune microenvironment of sarcomas is highly variable; however, given the strong immune presence in some subtypes, there is promise for immune therapy in many of these malignancies. Table 3 summarizes the clinical trials in immune therapy described below.
Table 3: A survey of recently published immune therapy studies in sarcoma
Immune Therapy |
Study Analyzed |
Design |
Stage |
Subtype |
N |
Median PFS (months) |
Median OS (months) |
Ipilimumab |
Merchant et al. Clin Cancer Res. 2016 |
Phase I |
Advanced |
CCS OSa RMS SS |
17 |
NA |
NA |
Pembrolizumab |
Tawbi et al. ASCO Meeting Abstracts. 2016 |
Phase II |
Advanced |
CSa DDLPS Ewing LMS OSa SS |
40 |
Not reached |
Not reached |
Dendritic cell training, tumor lysate, reinfusion |
Merchant et al. Clin Cancer Res. 2016 |
Phase II |
Advanced |
STS |
29 |
24 |
42 |
Tumor cell transduction with GM-CSF, radiation, reinfusion |
Goldberg et al. Clin Cancer Res. 2015 |
Phase I |
Advanced |
ASPS CCS |
11 |
NA |
NA |
HER-2 expressing CAR-T-cells |
Ahmed et al. J Clin Oncol. 2015 |
Phase I/II |
Advanced |
DSRCT Ewing OSa |
16 |
1.5 |
10.3 |
MAGE-A1, MAGE-A3, and NY-ESO-1 dendritic cell vaccine (with decitabine) |
Krishnadas et al. Cancer Immunol Immunother. 2015 |
Phase I |
Advanced |
Ewing RMS |
2 |
0 |
NA |
Abbreviations: PFS = progression-free survival, OS = overall survival, ASPS = alveolar soft part sarcoma, CCS = clear cell sarcoma, CSa = chondrosarcoma, DDLPS = dedifferentiated liposarcoma, DSRCT = desmoplastic small round cell tumor, LMS = leiomyosarcoma, OSa = osteosarcoma, RMS = rhabdomyosarcoma, SS = synovial sarcoma, STS = soft tissue sarcoma, UPS = undifferentiated pleomorphic sarcoma, NA = not available
“Hot” tumors: checkpoint inhibitor strategies
Hot tumors are those that are immunogenic, associated with high numbers of TILs and tumor associated macrophages, but that are actively modulating the immune response to survive, for example by expressing immune checkpoint ligands that suppress anti-tumor immune responses. Hot tumors are the most likely to benefit from immunomodulatory therapies such as checkpoint inhibitors.
Translational science
Lussier et al [147] showed that metastatic osteosarcomas (but not primary tumors) express PD-L1 and are infiltrated by PD1+ T-cells. Upon treatment with an anti-PD-L1 monoclonal antibody in a mouse cell-line xenograft model of metastatic osteosarcoma, they observed improved cytotoxic T-cell functioning, decreased tumor burden, and increased survival. However, in a follow-up study [148], they noted that xenografts quickly become resistant to PD-L1 blockade through the up-regulation of additional checkpoints, including CTLA-4. Treatment with a combination of anti-PD-L1 and anti-CTLA-4 monoclonal antibodies was able to control tumors completely and also conferred immunity to further tumor inoculation. Together, these pre-clinical results demonstrate that checkpoint blockade might be a worthwhile strategy for metastatic osteosarcoma patients.
Clinical trials
Ipilimumab, an anti-CTLA-4 therapeutic monoclonal antibody, was tested in a phase I trial on 33 pediatric patients with refractory solid tumors, including 17 sarcomas (8 osteosarcomas, 2 rhabdomyosarcomas, 2 clear cell sarcomas, 2 synovial sarcomas) [149]. At 3 weeks, researchers observed increased numbers of circulating activated T-cells, predominately CD4+ helper T-cells. Toxicities were similar to those reported in adult patients. Although no objective tumor responses were observed, 6 subjects had stable disease at 6 weeks, including cases of osteosarcoma, clear cell sarcoma, and synovial sarcoma. Furthermore, subjects with immune-related toxicities exhibited improved overall survival over those who showed no evidence of immune stimulation.
In an abstract recently published for ASCO, Tawbi et al [150] presented the first results from SARC028, a much-anticipated phase II study of the PD1 inhibitor pembrolizumab in 40 soft tissue (leiomyosarcoma, dedifferentiated liposarcoma, undifferentiated pleomorphic sarcoma, synovial sarcoma) and 40 bone (osteosarcoma, Ewing sarcoma, chondrosarcoma) sarcomas. At 8 weeks, partial responses by RECIST 1.1 were observed for undifferentiated pleomorphic sarcoma (4/9), dedifferentiated liposarcoma (2/9), synovial sarcoma (1/9), chondrosarcoma (1/6), and osteosarcoma (1/19). Stable disease was observed in some cases for all subtypes enrolled. The PFS rate at 8 weeks was 50% in leiomyosarcoma, 63% in liposarcoma, 30% in synovial sarcoma, 67% in UPS, 24% in osteosarcoma, 9% in Ewing sarcoma, and 67% in chondrosarcoma. A new phase II study (NCT02500797) for ipilimumab (anti-CTLA-4) with or without nivolumab (anti-PD1) is currently accruing patients with unresectable or metastatic bone or soft tissue sarcoma. Together, these studies suggest select and limited use for checkpoint blockade in these diseases; however, because immune therapy trials are only just beginning for sarcomas, it is difficult to draw concrete conclusions from the evidence available.
“Cold” tumors: immune augmentation strategies
Cold tumors are those that appear not to have been recognized by the immune system much - if at all - and theoretically would benefit best from stimulatory immune therapy such as cytokine treatment, immune cell engineering, and/or cancer vaccines.
Translational science
Yang et al [151] investigated STAT3 inhibitors as an adjuvant to conventional chemotherapy. STAT3 is an oncogenic transcription factor that functions to enhance cell growth, inhibit apoptosis, and mediate immunosuppression. Knockout or pharmacologic inhibition of STAT3 in syngeneic fibrosarcoma tumors in mice enhanced growth inhibition by anthracycline-based chemotherapy. This response was only found in immunocompetent, not immunodeficient mice, likely due to immune activation from STAT3 inhibition. There was increased tumor infiltration by dendritic and cytotoxic T cells and an increase in the expression of interferon-responsive genes. Reintroduction of wild-type STAT3 inhibited this expression pattern and eliminated the improved response to chemotherapy. These pre-clinical results suggest that STAT3 inhibitors may improve the outcome of chemotherapy via activation of the immune system.
Dendritic cell strategies involve removal of a patient’s own immune cells, dendritic cell training with or without tumor antigen exposure/engineering, and re-introduction of the trained dendritic cells. In osteosarcoma, dendritic cells from an osteosarcoma rat model were electrically fused with an osteosarcoma cell line to generate a fusion tumor vaccine [152]. This fusion vaccine was reintroduced, resulting in T lymphocyte proliferation. CD8+/CD4+ T-cell ratio increased, and CD4+ T-cell percentage dropped. Following vaccination, tumors shrunk or disappeared entirely, leading to improved survival of the rats. Also in osteosarcoma, Kawano et al [153] found that a combination of a dendritic cell strategy with an anti-GITR antibody inhibited tumor growth in a mouse xenograft model. Therapy constituted treatment with tumor lysate-pulsed dendritic cells, combined with an agonist for GITR, a co-stimulatory receptor for CD4/CD8 T-cell proliferation and effector functions. With combination treatment, they observed tumor growth inhibition and improved survival. Furthermore, this regimen increased the numbers of cytotoxic and reduced the numbers of regulatory T-cells. The same group found that combining doxorubicin with the same dendritic cell therapy in that murine osteosarcoma model induces immunogenic cell death and tumor inhibition [154]. This combinatorial approach is particularly appealing, as it suggests that dendritic cell therapy may serve to significantly enhance responses to conventional chemotherapy. Taken together, these studies suggest a promising future for dendritic cell therapies in osteosarcoma.
The innate immune system’s natural killer (NK) cells are newer targets for immune therapy interventions. NKG2D is an activating immune receptor on NK and cytotoxic T cells whose ligands are frequently present on tumor cell surfaces but rarely detectable on normal cells. Fernández et al [155] found moderate to high levels of NKG2D ligand expression by flow cytometry on all of 22 human osteosarcoma cell lines. Using a mouse osteosarcoma xenograft model injected with human natural killer cells, they showed that treatment of mouse osteosarcoma xenografts with IL-2 and the diuretic spironolactone resulted in enhanced receptor-ligand interactions of NKG2D, leading to NK cell activation, expansion, and targeting of osteosarcoma tumor-initiating cells. Zhu et al [156] found that osteosarcoma expression of NKG2D receptor and ligands were enhanced by treatment with the HDAC inhibitor entinostat, an effect not seen in normal human fibroblasts. In a mouse cell line xenograft model, mice treated with both entinostat and NK cells had a significantly reduced tumor burden, supporting the hypothesis that HDAC treatment sensitizes osteosarcoma cells to NK-mediated cell death. Finally, Jemitzky et al [157] show that anti-IGF1 receptor therapeutic antibodies not only diminish Ewing cell line viability, but also promote in vitro expansion of human NK cells. When co-incubated with Ewing sarcoma cell lines, NK cells exhibit potent degranulation responses. These studies together suggest that combining adoptive transfer of activated NK cells with IL-2, HDAC, anti-IGF1R, or other agents may have therapeutic benefit in sarcomas.
Clinical trials
A phase II cancer vaccine trial for 29 sarcomas by Merchant et al [158] employed a particularly complex protocol involving autologous monocyte extraction, dendritic cell training, tumor lysate-pulsing, and reinfusion. They observed a 62% 5-year overall survival, which is a significant improvement over the subjects’ expected 25% survival (based on previous studies, not a randomized control group). Benefits were seen chiefly in Ewing sarcoma and rhabdomyosarcoma (63%), whereas no benefit was seen in synovial sarcoma or desmoplastic small round cell tumor (0%). T-cell responses were identified in 62% subjects, and these responses were associated with improved survival.
In a phase I trial for alveolar soft part sarcoma and clear cell sarcoma by Goldberg et al [159], tumor cells from subject metastases (n = 11) were transduced with GM-CSF, irradiated, and re-infused. Vaccination enhanced infiltration of local dendritic cells and stimulated T cell reactions to tumor cells. Tumor biopsies showed PD1-positive T cells and PD-L1-expressing sarcoma cells, in correlation with each other. No tumor regressions were observed, but authors suggest concomitant treatment with checkpoint blockade to improve antitumor immunity.
Immune therapy against specific tumor antigens may prove a particularly useful strategy in sarcomas, due to their comparatively low genomic complexity (and presumed low burden of tumor neoantigens). Chimeric antigen receptor T-cells (CAR-T-cells) are T-cells, usually autologous cells extracted from the patient receiving therapy, that have been engineered to target a specific tumor antigen. Ahmed et al [160] conducted a phase I/II trial using CAR-T-cells targeting HER-2-expressing sarcomas. At 6 weeks, stable disease was attained in 3 of 14 osteosarcomas, 1 of 1 desmoplastic small round cell tumor, and 0 of 1 Ewing sarcoma. No partial or complete responses were observed. They analysed peripheral-blood mononuclear cells to confirm presence of HER2-CAR-T-cells, which were detected in 14 of 16 treated patients. In a related phase I veterinary trial, 18 dogs presenting with osteosarcoma were treated with HER2 Listeria vaccine [161]. Treatment induced a HER2-specific interferon-γ response 15 of 18 dogs within 6 months. Additionally, they saw reduced rates of metastasis and improved 1-, 2- and 3- year survival rates, compared to a historical control group treated with amputation and chemotherapy alone.
Dendritic cell vaccines can also serve to target specific tumor antigens, as seen in a phase I trial by Krishnadas et al [162] in children with relapsed or refractory solid tumors, including Ewing sarcoma (n = 2), osteosarcoma (n = 2), and rhabdomyosarcoma (n = 1). The protocol involved treatment with decitabine (a cytotoxic therapy), followed by a vaccine of autologous dendritic cells pulsed with peptides derived from tumor antigens MAGE-A1, MAGE-A3, and NY-ESO-1. Only one Ewing sarcoma patient and one rhabdomyosarcoma patient received the dendritic cell vaccine, and while neither had any objective response to therapy, both developed an antigen-specific response in their CD4+ T-cells. Given the limited number of sarcoma subjects in this study and the presence of a tumor-specific immune response, this strategy warrants further exploration for these and other sarcoma subtypes, such as myxoid liposarcoma, in which a strong correlation between poor prognosis and high expression (protein and mRNA) of PRAME and/or NY-ESO-1 has been reported [163].
Summary
Recent publications have shown that significant, albeit incremental progress can still be made using cytotoxic chemotherapy agents, including eribulin, trabectedin, and aldoxorubicin. Efforts to conduct larger international studies have been somewhat successful, and there has been a partial move toward more histology-specific studies rather than the lumping together of completely disparate entities. Targeted therapies have shown fewer advances at the level of clinical practice, despite numerous trials involving various receptor tyrosine kinase inhibitors; however, advances in basic science, driven by spectacular technological advances in genomics, highlight targetable pathways, including mTOR, Notch, Wnt, Hedgehog, and MDM2, and other targetable proteins, such as Hsp90, RNA helicase, and FOXM1. Epigenetic approaches have had some preclinical success with BET and EZH2 inhibitors, and immuno-oncology approaches are advancing from pre-clinical to phase I- and II- level studies in both immunostimulatory and immune checkpoint therapies.
Acknowledgments
We thank Neal Poulin for assistance with the literature review.
Conflicts of Interest
None applicable.
Grant Support
This work was supported by grants from the Canadian Cancer Society Research Institute (Grant # 701582), the Terry Fox Research Institute (TFF 105265 New Frontiers in Cancer), the Liddy Shriver Sarcoma Initiative (ImmunoSarc), and the Sarcoma Cancer Foundation of Canada (Beth England’s Sarcoma Research Fund).
References
1. Fletcher CDM. WHO Classification of Tumours of Soft Tissue and Bone. World Health Organization; 2013. 1 p.
2. Chibon F, Aurias A, Coindre JM. Sarcomas Genetics: From Point Mutation to Complex Karyotype, from Diagnosis to Therapies. Cancer Genomics. Dordrecht: Springer Netherlands; 2013. pp. 429–52. doi: 10.1007/978-94-007-5842-1_13.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66: 7–30. doi: 10.3322/caac.21332.
4. Canadian Cancer Statistics. 2015; : 1–151.
5. Young RJ, Natukunda A, Litière S, Woll PJ, Wardelmann E, van der Graaf WTA. First-line anthracycline-based chemotherapy for angiosarcoma and other soft tissue sarcoma subtypes: Pooled analysis of eleven European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group trials. European Journal of Cancer. 2014; 50: 3178–86. doi: 10.1016/j.ejca.2014.10.004.
6. Kanojia D, Nagata Y, Garg M, Lee D, Sato A, Yoshida K, Sato Y, Sanada M, Mayakonda A, Bartenhagen C, Klein H, Doan N, Said J, et al. Genomic landscape of liposarcoma. Oncotarget. 2015; 6: 42429–44. doi: 10.18632/oncotarget.6464.
7. Jamshidi F, Bashashati A, Shumansky K, Dickson B, Gokgoz N, Wunder JS, Andrulis IL, Lazar AJ, Shah SP, Huntsman DG, Nielsen TO. The genomic landscape of epithelioid sarcoma cell lines and tumours. The Journal of Pathology. 2015; 238: 63–73. doi: 10.1002/path.4636.
8. Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, Tolman C, Hurd L, Liao H, et al. Comprehensive Genomic Analysis of Rhabdomyosarcoma Reveals a Landscape of Alterations Affecting a Common Genetic Axis in Fusion-Positive and Fusion-Negative Tumors. Cancer Discovery. American Association for Cancer Research; 2014; 4: 216–31. doi: 10.1158/2159-8290.CD-13-0639.
9. Williamson D, Missiaglia E, de Reyniès A, Pierron G, Thuille B, Palenzuela G, Thway K, Orbach D, Laé M, Fréneaux P, Pritchard-Jones K, Oberlin O, Shipley J, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. American Society of Clinical Oncology; 2010; 28: 2151–8. doi: 10.1200/JCO.2009.26.3814.
10. Skapek SX, Anderson J, Barr FG, Bridge JA, Gastier Foster JM, Parham DM, Rudzinski ER, Triche T, Hawkins DS. PAX‐FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatric Blood & Cancer. 2013; 60: 1411–7. doi: 10.1002/pbc.24532.
11. Missiaglia E, Williamson D, Chisholm J, Wirapati P, Pierron G, Petel F, Concordet J-P, Thway K, Oberlin O, Pritchard-Jones K, Delattre O, Delorenzi M, Shipley J. PAX3/FOXO1 Fusion Gene Status Is the Key Prognostic Molecular Marker in Rhabdomyosarcoma and Significantly Improves Current Risk Stratification. JCO. American Society of Clinical Oncology; 2012; 30: JCO.2011.38.5591–1677. doi: 10.1200/JCO.2011.38.5591.
12. Italiano A, Di Mauro I, Rapp J, Pierron G, Auger N, Alberti L, Chibon F, Escande F, Voegeli A-C, Ghnassia J-P, Keslair F, Laé M, Ranchere-Vince D, et al. Clinical effect of molecular methods in sarcoma diagnosis (GENSARC): a prospective, multicentre, observational study. Lancet Oncol. 2016; 17: 532–8. doi: 10.1016/S1470-2045(15)00583-5.
13. Mody RJ, Wu Y-M, Lonigro RJ, Cao X, Roychowdhury S, Vats P, Frank KM, Prensner JR, Asangani I, Palanisamy N, Dillman JR, Rabah RM, Kunju LP, et al. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA. American Medical Association; 2015; 314: 913–25. doi: 10.1001/jama.2015.10080.
14. Harris MH, DuBois SG, Bender JLG, Kim A, Crompton BD, Parker E, Dumont IP, Hong AL, Guo D, Church A, Stegmaier K, Roberts CWM, Shusterman S, et al. Multicenter Feasibility Study of Tumor Molecular Profiling to Inform Therapeutic Decisions in Advanced Pediatric Solid Tumors: The Individualized Cancer Therapy (iCat) Study. JAMA Oncol. American Medical Association; 2016; 2: 608–15. doi: 10.1001/jamaoncol.2015.5689.
15. Chang W, Brohl AS, Patidar R, Sindiri S, Shern JF, Wei JS, Song YK, Yohe ME, Gryder B, Zhang S, Calzone KA, Shivaprasad N, Wen X, et al. MultiDimensional ClinOmics for Precision Therapy of Children and Adolescent Young Adults with Relapsed and Refractory Cancer: A Report from the Center for Cancer Research. Clinical Cancer Research. Clinical Cancer Research; 2016. doi: 10.1158/1078-0432.CCR-15-2717.
16. Andersson C, Fagman H, Hansson M, Enlund F. Profiling of potential driver mutations in sarcomas by targeted next generation sequencing. Cancer Genetics. 2016; 209: 154–60. doi: 10.1016/j.cancergen.2016.02.004.
17. Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, Kerstein RA, Gutierrez S, Petersen AK, Bavle A, Lin FY, López-Terrada DH, Monzon FA, et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol. American Medical Association; 2016; 2: 616–24. doi: 10.1001/jamaoncol.2015.5699.
18. Movva S, Wen W, Chen W, Millis SZ, Gatalica Z, Reddy S, Mehren von M, Van Tine BA. Multi-platform profiling of over 2000 sarcomas: Identification of biomarkers and novel therapeutic targets. Oncotarget. 2015; 6: 12234–47. doi: 10.18632/oncotarget.3498.
19. Hofvander J, Tayebwa J, Nilsson J, Magnusson L, Brosjö O, Larsson O, Steyern von FV, Domanski HA, Mandahl N, Mertens F. RNA sequencing of sarcomas with simple karyotypes: identification and enrichment of fusion transcripts. Laboratory Investigation. Nature Publishing Group; 2015; 95: 603–9. doi: 10.1038/labinvest.2015.50.
20. van IJzendoorn DGP, de Jong D, Romagosa C, Picci P, Benassi MS, Gambarotti M, Daugaard S, van de Sande M, Szuhai K, Bovée JVMG. Fusion events lead to truncation of FOSin epithelioid hemangioma of bone. Genes, Chromosomes and Cancer. 2015; 54: 565–74. doi: 10.1002/gcc.22269.
21. Le Loarer F, Watson S, Pierron G, de Montpreville VT, Ballet S, Firmin N, Auguste A, Pissaloux D, Boyault S, Paindavoine S, Dechelotte PJ, Besse B, Vignaud JM, et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nature Genetics. Nature Publishing Group; 2015; 47: 1200–5. doi: 10.1038/ng.3399.
22. Judson I, Verweij J, Gelderblom H, Hartmann JT, Schöffski P, Blay JY, Kerst JM, Sufliarsky J, Whelan J, Hohenberger P, Krarup-Hansen A, Alcindor T, Marreaud S, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014; 15: 415–23. doi: 10.1016/S1470-2045(14)70063-4.
23. Schöffski P, Cornillie J, Wozniak A, Li H, Hompes D. Soft tissue sarcoma: an update on systemic treatment options for patients with advanced disease. Oncol Res Treat. Karger Publishers; 2014; 37: 355–62. doi: 10.1159/000362631.
24. Le Cesne A, Judson I, Crowther D, Rodenhuis S, Keizer HJ, Van Hoesel Q, Blay JY, Frisch J, Van Glabbeke M, Hermans C, Van Oosterom A, Tursz T, Verweij J. Randomized phase III study comparing conventional-dose doxorubicin plus ifosfamide versus high-dose doxorubicin plus ifosfamide plus recombinant human granulocyte-macrophage colony-stimulating factor in advanced soft tissue sarcomas: A trial of the European Organization for Research and Treatment of Cancer/Soft Tissue and Bone Sarcoma Group. JCO. 2000; 18: 2676–84.
25. Ravi V, Patel S, Benjamin RS. Chemotherapy for soft-tissue sarcomas. Oncology (Williston Park, NY). 2015; 29: 43–50.
26. Dickson MA, D’Adamo DR, Keohan ML, D’Angelo SP, Carvajal RD, Gounder MM, Maki RG, Qin L-X, Lefkowitz RA, McKennon OR, Hirst CM, Schwartz GK, Tap WD. Phase II Trial of Gemcitabine and Docetaxel with Bevacizumab in Soft Tissue Sarcoma. Sarcoma. Hindawi Publishing Corporation; 2015; 2015: 532478–7. doi: 10.1155/2015/532478.
27. Hensley ML, Miller A, O’Malley DM, Mannel RS, Behbakht K, Bakkum-Gamez JN, Michael H. Randomized phase III trial of gemcitabine plus docetaxel plus bevacizumab or placebo as first-line treatment for metastatic uterine leiomyosarcoma: an NRG Oncology/Gynecologic Oncology Group study. J Clin Oncol. American Society of Clinical Oncology; 2015; 33: 1180–5. doi: 10.1200/JCO.2014.58.3781.
28. Zucali PA, Bertuzzi A, Parra HJS, Campagnoli E, Quagliuolo V, Santoro A. The “old drug” dacarbazine as a second/third line chemotherapy in advanced soft tissue sarcomas. Invest New Drugs. Springer US; 2008; 26: 175–81. doi: 10.1007/s10637-007-9086-z.
29. García-Del-Muro X, López-Pousa A, Maurel J, Martín J, Martínez-Trufero J, Casado A, Gómez-España A, Fra J, Cruz J, Poveda A, Meana A, Pericay C, Cubedo R, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol. American Society of Clinical Oncology; 2011; 29: 2528–33. doi: 10.1200/JCO.2010.33.6107.
30. Edmonson JH, Ryan LM, Blum RH, Brooks JS, Shiraki M, Frytak S, Parkinson DR. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. JCO. 1993; 11: 1269–75.
31. Antman K, Crowley J, Balcerzak SP, Rivkin SE, Weiss GR, Elias A, Natale RB, Cooper RM, Barlogie B, Trump DL. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. JCO. 1993; 11: 1276–85.
32. Dybdal-Hargreaves NF, Risinger AL, Mooberry SL. Eribulin mesylate: mechanism of action of a unique microtubule-targeting agent. Clinical Cancer Research. Clinical Cancer Research; 2015; 21: 2445–52. doi: 10.1158/1078-0432.CCR-14-3252.
33. Smith JA, Wilson L, Azarenko O, Zhu X, Lewis BM, Littlefield BA, Jordan MA. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry. American Chemical Society; 2010; 49: 1331–7. doi: 10.1021/bi901810u.
34. Funahashi Y, Okamoto K, Adachi Y, Semba T, Uesugi M, Ozawa Y, Tohyama O, Uehara T, Kimura T, Watanabe H, Asano M, Kawano S, Tizon X, et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014; 105: 1334–42. doi: 10.1111/cas.12488.
35. Yoshida T, Ozawa Y, Kimura T, Sato Y, Kuznetsov G, Xu S, Uesugi M, Agoulnik S, Taylor N, Funahashi Y, Matsui J. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br J Cancer. Nature Publishing Group; 2014; 110: 1497–505. doi: 10.1038/bjc.2014.80.
36. Schöffski P, Chawla S, Maki RG, Italiano A, Gelderblom H, Choy E, Grignani G, Camargo V, Bauer S, Rha SY, Blay JY, Hohenberger P, D’Adamo D, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016; 387: 1629–37. doi: 10.1016/S0140-6736(15)01283-0.
37. D’Incalci M, Badri N, Galmarini CM, Allavena P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br J Cancer. Nature Publishing Group; 2014; 111: 646–50. doi: 10.1038/bjc.2014.149.
38. Minuzzo M, Ceribelli M, Pitarque-Martì M, Borrelli S, Erba E, DiSilvio A, D’Incalci M, Mantovani R. Selective effects of the anticancer drug Yondelis (ET-743) on cell-cycle promoters. Mol Pharmacol. American Society for Pharmacology and Experimental Therapeutics; 2005; 68: 1496–503. doi: 10.1124/mol.105.013615.
39. Demetri GD, Mehren von M, Jones RL, Hensley ML, Schuetze SM, Staddon A, Milhem M, Elias A, Ganjoo K, Tawbi H, Van Tine BA, Spira A, Dean A, et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J Clin Oncol. American Society of Clinical Oncology; 2016; 34: 786–93. doi: 10.1200/JCO.2015.62.4734.
40. Kawai A, Araki N, Sugiura H, Ueda T, Yonemoto T, Takahashi M, Morioka H, Hiraga H, Hiruma T, Kunisada T, Matsumine A, Tanase T, Hasegawa T, et al. Trabectedin monotherapy after standard chemotherapy versus best supportive care in patients with advanced, translocation-related sarcoma: a randomised, open-label, phase 2 study. Lancet Oncol. 2015; 16: 406–16. doi: 10.1016/S1470-2045(15)70098-7.
41. Araki N, Takahashi S, Sugiura H, Ueda T, Yonemoto T, Takahashi M, Morioka H, Hiraga H, Hiruma T, Kunisada T, Matsumine A, Kawai A. Retrospective inter- and intra-patient evaluation of trabectedin after best supportive care for patients with advanced translocation-related sarcoma after failure of standard chemotherapy. Eur J Cancer. 2016; 56: 122–30. doi: 10.1016/j.ejca.2015.12.014.
42. Pautier P, Floquet A, Chevreau C, Penel N, Guillemet C, Delcambre C, Cupissol D, Selle F, Isambert N, Piperno-Neumann S, Thyss A, Bertucci F, Bompas E, et al. Trabectedin in combination with doxorubicin for first-line treatment of advanced uterine or soft-tissue leiomyosarcoma (LMS-02): a non-randomised, multicentre, phase 2 trial. Lancet Oncol. 2015; 16: 457–64. doi: 10.1016/S1470-2045(15)70070-7.
43. Bui-Nguyen B, Butrynski JE, Penel N, Blay JY, Isambert N, Milhem M, Kerst JM, Reyners AKL, Litière S, Marréaud S, Collin F, van der Graaf WTA, European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group (EORTC/STBSG) and the Sarcoma Alliance for Research through Collaboration (SARC). A phase IIb multicentre study comparing the efficacy of trabectedin to doxorubicin in patients with advanced or metastatic untreated soft tissue sarcoma: the TRUSTS trial. Eur J Cancer. 2015; 51: 1312–20. doi: 10.1016/j.ejca.2015.03.023.
44. Kratz F, Warnecke A, Scheuermann K, Stockmar C, Schwab J, Lazar P, Drückes P, Esser N, Drevs J, Rognan D, Bissantz C, Hinderling C, Folkers G, et al. Probing the cysteine-34 position of endogenous serum albumin with thiol-binding doxorubicin derivatives. Improved efficacy of an acid-sensitive doxorubicin derivative with specific albumin-binding properties compared to that of the parent compound. J Med Chem. 2002; 45: 5523–33. doi: 10.1021/jm020276c.
45. Mita MM, Natale RB, Wolin EM, Laabs B, Dinh H, Wieland S, Levitt DJ, Mita AC. Pharmacokinetic study of aldoxorubicin in patients with solid tumors. Invest New Drugs. Springer US; 2015; 33: 341–8. doi: 10.1007/s10637-014-0183-5.
46. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000; 65: 271–84.
47. Chawla SP, Papai Z, Mukhametshina G, Sankhala K, Vasylyev L, Fedenko A, Khamly K, Ganjoo K, Nagarkar R, Wieland S, Levitt DJ. First-Line Aldoxorubicin vs Doxorubicin in Metastatic or Locally Advanced Unresectable Soft-Tissue Sarcoma: A Phase 2b Randomized Clinical Trial. JAMA Oncol. American Medical Association; 2015; 1: 1272–80. doi: 10.1001/jamaoncol.2015.3101.
48. Seddon B, Scurr M, Jones RL, Wood Z, Propert-Lewis C, Fisher C, Flanagan A, Sunkersing J, A’Hern R, Whelan J, Judson I. A phase II trial to assess the activity of gemcitabine and docetaxel as first line chemotherapy treatment in patients with unresectable leiomyosarcoma. Clin Sarcoma Res. BioMed Central; 2015; 5: 13. doi: 10.1186/s13569-015-0029-8.
49. Davis EJ, Chugh R, Zhao L, Lucas DR, Biermann JS, Zalupski MM, Feng M, Wong SL, Jacobson J, Zyczynski L, Reinke D, Metko G, Baker LH, et al. A randomised, open-label, phase II study of neo/adjuvant doxorubicin and ifosfamide versus gemcitabine and docetaxel in patients with localised, high-risk, soft tissue sarcoma. Eur J Cancer. 2015; 51: 1794–802. doi: 10.1016/j.ejca.2015.05.010.
50. D’Adamo DR, Anderson SE, Albritton K, Yamada J, Riedel E, Scheu K, Schwartz GK, Chen H, Maki RG. Phase II study of doxorubicin and bevacizumab for patients with metastatic soft-tissue sarcomas. JCO. American Society of Clinical Oncology; 2005; 23: 7135–42. doi: 10.1200/JCO.2005.16.139.
51. Agulnik M, Yarber JL, Okuno SH, Mehren von M, Jovanovic BD, Brockstein BE, Evens AM, Benjamin RS. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. Oxford University Press; 2013; 24: 257–63. doi: 10.1093/annonc/mds237.
52. Maki RG, Wathen JK, Patel SR, Priebat DA, Okuno SH, Samuels B, Fanucchi M, Harmon DC, Schuetze SM, Reinke D, Thall PF, Benjamin RS, Baker LH, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. American Society of Clinical Oncology; 2007; 25: 2755–63. doi: 10.1200/JCO.2006.10.4117.
53. Kasper B, Gruenwald V, Reichardt P, Bauer S, Hohenberger P, Haller F. Correlation of CTNNB1 Mutation Status with Progression Arrest Rate in RECIST Progressive Desmoid-Type Fibromatosis Treated with Imatinib: Translational Research Results from a Phase 2 Study of the German Interdisciplinary Sarcoma Group (GISG-01). Ann Surg Oncol. Springer International Publishing; 2016; 23: 1924–7. doi: 10.1245/s10434-016-5132-4.
54. Lazar A, Tuvin D, Hajibashi S, Habeeb S. Specific mutations in the β-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. The American Journal of Pathology. 2008; 173: 1518–27. doi: 10.2353/ajpath.2008.080475.
55. Colombo C, Miceli R, Lazar AJ, Perrone F, Pollock RE, Cesne A, Hartgrink HH, Cleton Jansen AM, Domont J, Bovée JVMG, Bonvalot S, Lev D, Gronchi A. CTNNB1 45F mutation is a molecular prognosticator of increased postoperative primary desmoid tumor recurrence. Cancer. 2013; 119: 3696–702. doi: 10.1002/cncr.28271.
56. van Broekhoven DLM, Verhoef C, Grünhagen DJ, van Gorp JMHH, Bakker den MA, Hinrichs JWJ, de Voijs CMA, van Dalen T. Prognostic Value of CTNNB1 Gene Mutation in Primary Sporadic Aggressive Fibromatosis. Ann Surg Oncol. Springer US; 2015; 22: 1464–70. doi: 10.1245/s10434-014-4156-x.
57. Eastley N, McCulloch T, Esler C, Hennig I, Fairbairn J, Gronchi A, Ashford R. Extra-abdominal desmoid fibromatosis: A review of management, current guidance and unanswered questions. Eur J Surg Oncol. 2016. doi: 10.1016/j.ejso.2016.02.012.
58. Liu J, Wu J, Zhou L, Pan C, Zhou Y, Du W, Chen J-M, Zhu X, Shen J, Chen S, Liu R-Y, Huang W. ZD6474, a new treatment strategy for human osteosarcoma, and its potential synergistic effect with celecoxib. Oncotarget. 2015; 6: 21341–52. doi: 10.18632/oncotarget.4179.
59. Holtkamp N, Okuducu AF, Mucha J, Afanasieva A, Hartmann C, Atallah I, Estevez-Schwarz L, Mawrin C, Friedrich RE, Mautner V-F, Deimling von A. Mutation and expression of PDGFRA and KIT in malignant peripheral nerve sheath tumors, and its implications for imatinib sensitivity. Carcinogenesis. Oxford University Press; 2006; 27: 664–71.
60. Chugh R, Wathen JK, Maki RG, Benjamin RS, Patel SR, Meyers PA, Myers PA, Priebat DA, Reinke DK, Thomas DG, Keohan ML, Samuels BL, Baker LH. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J Clin Oncol. American Society of Clinical Oncology; 2009; 27: 3148–53. doi: 10.1200/JCO.2008.20.5054.
61. Patwardhan PP, Surriga O, Beckman MJ, de Stanchina E, Dematteo RP, Tap WD, Schwartz GK. Sustained Inhibition of Receptor Tyrosine Kinases and Macrophage Depletion by PLX3397 and Rapamycin as a Potential New Approach for the Treatment of MPNSTs. Clinical Cancer Research. Clinical Cancer Research; 2014; 20: 3146–58. doi: 10.1158/1078-0432.CCR-13-2576.
62. Shang H, Braggio D, Lee YJ, Sannaa Al GA, Creighton CJ, Bolshakov S, Lazar AJF, Lev D, Pollock RE. Targeting the Notch pathway: A potential therapeutic approach for desmoid tumors. Cancer. 2015; 121: 4088–96. doi: 10.1002/cncr.29564.
63. Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds JM, English PA, McLachlan KR, Kern KA, et al. A Phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin Cancer Res. Clinical Cancer Research; 2015; 21: 60–7. doi: 10.1158/1078-0432.CCR-14-0607.
64. Hughes DPM, Kummar S, Lazar AJ. New, Tolerable γ-Secretase Inhibitor Takes Desmoid Down a Notch. Clinical Cancer Research. Clinical Cancer Research; 2015; 21: 7–9. doi: 10.1158/1078-0432.CCR-14-1660.
65. Kephart JJG, Tiller RGJ, Crose LES, Slemmons KK, Chen P-H, Hinson AR, Bentley RC, Chi J-TA, Linardic CM. Secreted Frizzled-Related Protein 3 (SFRP3) Is Required for Tumorigenesis of PAX3-FOXO1-Positive Alveolar Rhabdomyosarcoma. Clinical Cancer Research. American Association for Cancer Research; 2015; 21: 4868–80. doi: 10.1158/1078-0432.CCR-14-1797.
66. Hirotsu M, Setoguchi T, Sasaki H, Matsunoshita Y, Gao H, Nagao H, Kunigou O, Komiya S. Smoothened as a new therapeutic target for human osteosarcoma. Molecular Cancer 2010 9:1. BioMed Central; 2010; 9: 1. doi: 10.1186/1476-4598-9-5.
67. Yang W, Liu X, Choy E, Mankin H, Hornicek FJ, Duan Z. Targeting hedgehog‐GLI‐2 pathway in osteosarcoma. Journal of Orthopaedic Research. Wiley Subscription Services, Inc., A Wiley Company; 2013; 31: 502–9. doi: 10.1002/jor.22230.
68. Nagao-Kitamoto H, Nagata M, Nagano S, Kitamoto S, Ishidou Y, Yamamoto T, Nakamura S, Tsuru A, Abematsu M, Fujimoto Y, Yokouchi M, Kitajima S, Yoshioka T, et al. GLI2 is a novel therapeutic target for metastasis of osteosarcoma. International Journal of Cancer. 2015; 136: 1276–84. doi: 10.1002/ijc.29107.
69. Saitoh Y, Setoguchi T, Nagata M, Tsuru A, Nakamura S, Nagano S, Ishidou Y, Nagao-Kitamoto H, Yokouchi M, Maeda S, Tanimoto A, Furukawa T, Komiya S. Combination of Hedgehog inhibitors and standard anticancer agents synergistically prevent osteosarcoma growth. International Journal of Oncology. Spandidos Publications; 2016; 48: 235–42. doi: 10.3892/ijo.2015.3236.
70. Bekki H, Kohashi K, Maekawa A, Yamada Y, Yamamoto H, Harimaya K, Hakozaki M, Nabeshima K, Iwamoto Y, Oda Y. Elevated expression of HSP90 and the antitumor effect of an HSP90 inhibitor via inactivation of the Akt/mTOR pathway in undifferentiated pleomorphic sarcoma. BMC Cancer 2015 15:1. 7 ed. BioMed Central; 2015; 15: 1. doi: 10.1186/s12885-015-1830-8.
71. Ernst A, Anders H, Kapfhammer H, Orth M, Hennel R, Seidl K, Winssinger N, Belka C, Unkel S, Lauber K. HSP90 inhibition as a means of radiosensitizing resistant, aggressive soft tissue sarcomas. Cancer Letters. 2015; 365: 211–22. doi: 10.1016/j.canlet.2015.05.024.
72. Safavi S, Järnum S, Vannas C, Udhane S, Jonasson E, Tomic TT, Grundevik P, Fagman H, Hansson M, Kalender Z, Jauhiainen A, Dolatabadi S, Stratford EW, et al. HSP90 inhibition blocks ERBB3 and RET phosphorylation in myxoid/round cell liposarcoma and causes massive cell death in vitro and in vivo. Oncotarget. 2016; 7: 433–45. doi: 10.18632/oncotarget.6336.
73. Bill KLJ, Garnett J, Meaux I, Ma X, Creighton CJ, Bolshakov S, Barriere C, Debussche L, Lazar AJ, Prudner BC, Casadei L, Braggio D, Lopez G, et al. SAR405838: A Novel and Potent Inhibitor of the MDM2:p53 Axis for the Treatment of Dedifferentiated Liposarcoma. Clin Cancer Res. Clinical Cancer Research; 2016; 22: 1150–60. doi: 10.1158/1078-0432.CCR-15-1522.
74. Ou W-B, Zhu J, Eilers G, Li X, Kuang Y, Liu L, Mariño-Enríquez A, Yan Z, Li H, Meng F, Zhou H, Sheng Q, Fletcher JA. HDACi inhibits liposarcoma via targeting of the MDM2-p53 signaling axis and PTEN, irrespective of p53 mutational status. Oncotarget. 2015; 6: 10510–20. doi: 10.18632/oncotarget.3230.
75. Lamhamedi-Cherradi S-E, Menegaz BA, Ramamoorthy V, Aiyer RA, Maywald RL, Buford AS, Doolittle DK, Culotta KS, O’Dorisio JE, Ludwig JA. An Oral Formulation of YK-4-279: Preclinical Efficacy and Acquired Resistance Patterns in Ewing Sarcoma. Mol Cancer Ther. Molecular Cancer Therapeutics; 2015; 14: 1591–604. doi: 10.1158/1535-7163.MCT-14-0334.
76. Wilky BA, Kim C, McCarty G, Montgomery EA, Kammers K, DeVine LR, Cole RN, Raman V, Loeb DM. RNA helicase DDX3: a novel therapeutic target in Ewing sarcoma. Oncogene. Nature Publishing Group; 2016; 35: 2574–83. doi: 10.1038/onc.2015.336.
77. Eisinger-Mathason TSK, Mucaj V, Biju KM, Nakazawa MS, Gohil M, Cash TP, Yoon SS, Skuli N, Park KM, Gerecht S, Simon MC. Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc Natl Acad Sci USA. National Acad Sciences; 2015; 112: E3402–11. doi: 10.1073/pnas.1420005112.
78. Maekawa A, Kohashi K, Kuda M, Iura K, Ishii T, Endo M, Nakatsura T, Iwamoto Y, Oda Y. Prognostic significance of FOXM1 expression and antitumor effect of FOXM1 inhibition in synovial sarcomas. BMC Cancer 2015 15:1. 5 ed. BioMed Central; 2016; 16: 511. doi: 10.1186/s12885-016-2542-4.
79. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998; 279: 577–80.
80. Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, Silberman S, Capdeville R, Dimitrijevic S, Druker B, Demetri GD. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. Massachusetts Medical Society; 2001; 344: 1052–6. doi: 10.1056/NEJM200104053441404.
81. Heinrich MC, Corless CL, Demetri GD, Blanke CD, Mehren von M, Joensuu H, McGreevey LS, Chen C-J, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. JCO. American Society of Clinical Oncology; 2003; 21: 4342–9. doi: 10.1200/JCO.2003.04.190.
82. Rubin BP, Schuetze SM, Eary JF, Norwood TH, Mirza S, Conrad EU, Bruckner JD. Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofibrosarcoma protuberans. JCO. 2002; 20: 3586–91.
83. Eilers G, Czaplinski JT, Mayeda M, Bahri N, Tao D, Zhu M, Hornick JL, Lindeman NI, Sicinska E, Wagner AJ, Fletcher JA, Mariño-Enríquez A. CDKN2A/p16 Loss Implicates CDK4 as a Therapeutic Target in Imatinib-Resistant Dermatofibrosarcoma Protuberans. Mol Cancer Ther. Molecular Cancer Therapeutics; 2015; 14: 1346–53. doi: 10.1158/1535-7163.MCT-14-0793.
84. Bowne WB, Antonescu CR, Leung DH, Katz SC, Hawkins WG, Woodruff JM, Brennan MF, Lewis JJ. Dermatofibrosarcoma protuberans: A clinicopathologic analysis of patients treated and followed at a single institution. Cancer. 2000; 88: 2711–20.
85. Llombart B, Monteagudo C, Sanmartín O, López-Guerrero JA, Serra-Guillén C, Poveda A, Jorda E, Fernandez-Serra A, Pellín A, Guillén C, Llombart-Bosch A. Dermatofibrosarcoma protuberans: a clinicopathological, immunohistochemical, genetic (COL1A1-PDGFB), and therapeutic study of low-grade versus high-grade (fibrosarcomatous) tumors. J Am Acad Dermatol. 2011; 65: 564–75. doi: 10.1016/j.jaad.2010.06.020.
86. Abbott JJ, Oliveira AM, Nascimento AG. The prognostic significance of fibrosarcomatous transformation in dermatofibrosarcoma protuberans. Am J Surg Pathol. 2006; 30: 436–43.
87. Stacchiotti S, Pantaleo MA, Negri T, Astolfi A, Tazzari M, Dagrada GP, Urbini M, Indio V, Maestro R, Gronchi A, Fiore M, Dei Tos AP, Conca E, et al. Efficacy and Biological Activity of Imatinib in Metastatic Dermatofibrosarcoma Protuberans (DFSP). Clin Cancer Res. Clinical Cancer Research; 2016; 22: 837–46. doi: 10.1158/1078-0432.CCR-15-1243.
88. Lemm D, Mügge L-O, Mentzel T, Höffken K. Current treatment options in dermatofibrosarcoma protuberans. J Cancer Res Clin Oncol. Springer-Verlag; 2009; 135: 653–65. doi: 10.1007/s00432-009-0550-3.
89. Hindi N, Casali PG, Morosi C, Messina A, Palassini E, Pilotti S, Tamborini E, Radaelli S, Gronchi A, Stacchiotti S. Imatinib in advanced chordoma: A retrospective case series analysis. Eur J Cancer. 2015; 51: 2609–14. doi: 10.1016/j.ejca.2015.07.038.
90. Stacchiotti S, Longhi A, Ferraresi V, Grignani G, Comandone A, Stupp R, Bertuzzi A, Tamborini E, Pilotti S, Messina A, Spreafico C, Gronchi A, Amore P, et al. Phase II study of imatinib in advanced chordoma. J Clin Oncol. American Society of Clinical Oncology; 2012; 30: 914–20. doi: 10.1200/JCO.2011.35.3656.
91. Brämswig K, Ploner F, Martel A, Bauernhofer T, Hilbe W, Kühr T, Leitgeb C, Mlineritsch B, Petzer A, Seebacher V, Stöger H, Girschikofsky M, Hochreiner G, et al. Sorafenib in advanced, heavily pretreated patients with soft tissue sarcomas. Anticancer Drugs. 2014; 25: 848–53. doi: 10.1097/CAD.0000000000000108.
92. Santoro A, Comandone A, Basso U, Soto Parra H, De Sanctis R, Stroppa E, Marcon I, Giordano L, Lutman FR, Boglione A, Bertuzzi A. Phase II prospective study with sorafenib in advanced soft tissue sarcomas after anthracycline-based therapy. Ann Oncol. Oxford University Press; 2013; 24: 1093–8. doi: 10.1093/annonc/mds607.
93. Maki RG, D’Adamo DR, Keohan ML, Saulle M, Schuetze SM, Undevia SD, Livingston MB, Cooney MM, Hensley ML, Mita MM, Takimoto CH, Kraft AS, Elias AD, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. American Society of Clinical Oncology; 2009; 27: 3133–40. doi: 10.1200/JCO.2008.20.4495.
94. Bompas E, Le Cesne A, Tresch-Bruneel E, Lebellec L, Laurence V, Collard O, Saada-Bouzid E, Isambert N, Blay JY, Amela EY, Salas S, Chevreau C, Bertucci F, et al. Sorafenib in patients with locally advanced and metastatic chordomas: a phase II trial of the French Sarcoma Group (GSF/GETO). Ann Oncol. Oxford University Press; 2015; 26: 2168–73. doi: 10.1093/annonc/mdv300.
95. Villacis RAR, Silveira SM, Barros-Filho MC, Marchi FA, Domingues MAC, Scapulatempo-Neto C, Aguiar S, Lopes A, Cunha IW, Rogatto SR. Gene expression profiling in leiomyosarcomas and undifferentiated pleomorphic sarcomas: SRC as a new diagnostic marker. Angelucci A, editor. PLoS ONE. Public Library of Science; 2014; 9: e102281. doi: 10.1371/journal.pone.0102281.
96. Schrage YM, Briaire-de Bruijn IH, de Miranda NFCC, van Oosterwijk J, Taminiau AHM, van Wezel T, Hogendoorn PCW, Bovée JVMG. Kinome profiling of chondrosarcoma reveals SRC-pathway activity and dasatinib as option for treatment. Clinical Cancer Research. American Association for Cancer Research; 2009; 69: 6216–22. doi: 10.1158/0008-5472.CAN-08-4801.
97. Shor AC, Keschman EA, Lee FY, Muro-Cacho C, Letson GD, Trent JC, Pledger WJ, Jove R. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Clinical Cancer Research. American Association for Cancer Research; 2007; 67: 2800–8. doi: 10.1158/0008-5472.CAN-06-3469.
98. Timeus F, Crescenzio N, Fandi A, Doria A, Foglia L, Cordero di Montezemolo L. In vitro antiproliferative and antimigratory activity of dasatinib in neuroblastoma and Ewing sarcoma cell lines. Oncol Rep. 2008; 19: 353–9.
99. Michels S, Trautmann M, Sievers E, Kindler D, Huss S, Renner M, Friedrichs N, Kirfel J, Steiner S, Endl E, Wurst P, Heukamp L, Penzel R, et al. SRC signaling is crucial in the growth of synovial sarcoma cells. Clinical Cancer Research. American Association for Cancer Research; 2013; 73: 2518–28. doi: 10.1158/0008-5472.CAN-12-3023.
100. Schuetze SM, Wathen JK, Lucas DR, Choy E, Samuels BL, Staddon AP, Ganjoo KN, Mehren von M, Chow WA, Loeb DM, Tawbi HA, Rushing DA, Patel SR, et al. SARC009: Phase 2 study of dasatinib in patients with previously treated, high-grade, advanced sarcoma. Cancer. 2016; 122: 868–74. doi: 10.1002/cncr.29858.
101. van der Graaf WTA, Blay JY, Chawla SP, Kim D-W, Bui-Nguyen B, Casali PG, Schöffski P, Aglietta M, Staddon AP, Beppu Y, Le Cesne A, Gelderblom H, Judson IR, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012; 379: 1879–86. doi: 10.1016/S0140-6736(12)60651-5.
102. Kawai A, Araki N, Hiraga H, Sugiura H, Matsumine A, Ozaki T, Ueda T, Ishii T, Esaki T, Machida M, Fukasawa N. A randomized, double-blind, placebo-controlled, Phase III study of pazopanib in patients with soft tissue sarcoma: results from the Japanese subgroup. Jpn J Clin Oncol. Oxford University Press; 2016; 46: 248–53. doi: 10.1093/jjco/hyv184.
103. Benson C, Ray-Coquard I, Sleijfer S, Litière S, Blay JY, Le Cesne A, Papai Z, Judson I, Schöffski P, Chawla S, Gil T, Piperno-Neumann S, Marréaud S, et al. Outcome of uterine sarcoma patients treated with pazopanib: A retrospective analysis based on two European Organisation for Research and Treatment of Cancer (EORTC) Soft Tissue and Bone Sarcoma Group (STBSG) clinical trials 62043 and 62072. Gynecol Oncol. 2016; 142: 89–94. doi: 10.1016/j.ygyno.2016.03.024.
104. Martin-Liberal J, Benson C, McCarty H, Thway K, Messiou C, Judson I. Pazopanib is an active treatment in desmoid tumour/aggressive fibromatosis. Clin Sarcoma Res. BioMed Central; 2013; 3: 13. doi: 10.1186/2045-3329-3-13.
105. Senan S, Smit EF. Design of clinical trials of radiation combined with antiangiogenic therapy. Oncologist. AlphaMed Press; 2007; 12: 465–77. doi: 10.1634/theoncologist.12-4-465.
106. Haas RLM, Gelderblom H, Sleijfer S, van Boven HH, Scholten A, Dewit L, Borst G, van der Hage J, Kerst JM, Nout RA, Hartgrink HH, de Pree I, Verhoef C, et al. A phase I study on the combination of neoadjuvant radiotherapy plus pazopanib in patients with locally advanced soft tissue sarcoma of the extremities. Acta Oncol. Taylor & Francis; 2015; 54: 1195–201. doi: 10.3109/0284186X.2015.1037404.
107. Ronellenfitsch U, Dimitrakopoulou-Strauss A, Jakob J, Kasper B, Nowak K, Pilz LR, Attenberger U, Gaiser T, Egerer G, Fröhling S, Derigs H-G, Schwarzbach M, Hohenberger P. Preoperative therapy with pazopanib in high-risk soft tissue sarcoma: a phase II window-of-opportunity study by the German Interdisciplinary Sarcoma Group (GISG-04/NOPASS). BMJ Open. British Medical Journal Publishing Group; 2016; 6: e009558. doi: 10.1136/bmjopen-2015-009558.
108. van der Heijden L, Dijkstra PDS, van de Sande MAJ, Kroep JR, Nout RA, van Rijswijk CSP, Bovée JVMG, Hogendoorn PCW, Gelderblom H. The clinical approach toward giant cell tumor of bone. Oncologist. AlphaMed Press; 2014; 19: 550–61. doi: 10.1634/theoncologist.2013-0432.
109. Thomas D, Henshaw R, Skubitz K, Chawla S, Staddon A, Blay JY, Roudier M, Smith J, Ye Z, Sohn W, Dansey R, Jun S. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010; 11: 275–80. doi: 10.1016/S1470-2045(10)70010-3.
110. Branstetter DG, Nelson SD, Manivel JC, Blay JY, Chawla S, Thomas DM, Jun S, Jacobs I. Denosumab induces tumor reduction and bone formation in patients with giant-cell tumor of bone. Clin Cancer Res. Clinical Cancer Research; 2012; 18: 4415–24. doi: 10.1158/1078-0432.CCR-12-0578.
111. Chawla S, Henshaw R, Seeger L, Choy E, Blay JY, Ferrari S, Kroep J, Grimer R, Reichardt P, Rutkowski P, Schuetze S, Skubitz K, Staddon A, et al. Safety and efficacy of denosumab for adults and skeletally mature adolescents with giant cell tumour of bone: interim analysis of an open-label, parallel-group, phase 2 study. Lancet Oncol. 2013; 14: 901–8. doi: 10.1016/S1470-2045(13)70277-8.
112. Balke M, Schremper L, Gebert C, Ahrens H, Streitbuerger A, Koehler G, Hardes J, Gosheger G. Giant cell tumor of bone: treatment and outcome of 214 cases. J Cancer Res Clin Oncol. 2008; 134: 969–78. doi: 10.1007/s00432-008-0370-x.
113. Rutkowski P, Ferrari S, Grimer RJ, Stalley PD, Dijkstra SPD, Pienkowski A, Vaz G, Wunder JS, Seeger LL, Feng A, Roberts ZJ, Bach BA. Surgical downstaging in an open-label phase II trial of denosumab in patients with giant cell tumor of bone. Ann Surg Oncol. Springer US; 2015; 22: 2860–8. doi: 10.1245/s10434-015-4634-9.
114. Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, Wunder JS, Dickson BC, Lundgren SM, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. American Association for the Advancement of Science; 2016; 352: 844–9. doi: 10.1126/science.aac7272.
115. Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, Nik-Zainal S, Martin S, McLaren S, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nature Genetics. Nature Publishing Group; 2013; 45: 1479–82. doi: 10.1038/ng.2814.
116. Jayaram H, Hoelper D, Jain SU, Cantone N, Lundgren SM, Poy F, Allis CD, Cummings R, Bellon S, Lewis PW. S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proceedings of the National Academy of Sciences. National Acad Sciences; 2016; : 201605523. doi: 10.1073/pnas.1605523113.
117. Fang D, Gan H, Lee J-H, Han J, Wang Z, Riester SM, Jin L, Chen J, Zhou H, Wang J, Zhang H, Yang N, Bradley EW, et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science. American Association for the Advancement of Science; 2016; 352: 1344–8. doi: 10.1126/science.aae0065.
118. Lee W, Teckie S, Wiesner T, Ran L, Granada CNP, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, Tap WD, Fletcher JA, Huberman KH, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nature Genetics. Nature Publishing Group; 2014; 46: 1227–32. doi: 10.1038/ng.3095.
119. Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, Wang Q, Belzberg AJ, Chaichana K, Gallia GL, Gokaslan ZL, Riggins GJ, Wolinksy J-P, et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nature Genetics. Nature Publishing Group; 2014; 46: 1170–2. doi: 10.1038/ng.3116.
120. Schaefer I-M, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Modern Pathology. Nature Publishing Group; 2015; 29: 4–13. doi: 10.1038/modpathol.2015.134.
121. Cleven AH, Sannaa GAA, Briaire-de Bruijn I, Ingram DR, van de Rijn M, Rubin BP, de Vries MW, Watson KL, Torres KE, Wang W-L, van Duinen SG, Hogendoorn PCW, Lazar AJ, et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Modern Pathology. Nature Publishing Group; 2016. doi: 10.1038/modpathol.2016.45.
122. Röhrich M, Koelsche C, Schrimpf D, Capper D, Sahm F, Kratz A, Reuss J, Hovestadt V, Jones DTW, Bewerunge-Hudler M, Becker A, Weis J, Mawrin C, et al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathologica. Springer Berlin Heidelberg; 2016; 131: 877–87. doi: 10.1007/s00401-016-1540-6.
123. Sun W, Chatterjee B, Wang Y, Stevenson HS, Edelman DC, Meltzer PS, Barr FG. Distinct methylation profiles characterize fusion-positive and fusion-negative rhabdomyosarcoma. Modern Pathology. Nature Publishing Group; 2015; 28: 1214–24. doi: 10.1038/modpathol.2015.82.
124. Lee DH, Qi J, Bradner JE, Said JW, Doan NB, Forscher C, Yang H, Koeffler HP. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. International Journal of Cancer. 2015; 136: 2055–64. doi: 10.1002/ijc.29269.
125. Baker EK, Taylor S, Gupte A, Sharp PP, Walia M, Walsh NC, Zannettino ACW, Chalk AM, Burns CJ, Walkley CR. BET inhibitors induce apoptosis through a MYC independent mechanism and synergise with CDK inhibitors to kill osteosarcoma cells. Sci Rep. 2015; 5: 10120. doi: 10.1038/srep10120.
126. Hensel T, Giorgi C, Schmidt O, Calzada-Wack J, Neff F, Buch T, Niggli FK, Schäfer BW, Burdach S, Richter GHS. Targeting the EWS-ETS transcriptional program by BET bromodomain inhibition in Ewing sarcoma. Oncotarget. 2016; 7: 1451–63. doi: 10.18632/oncotarget.6385.
127. Jacques C, Lamoureux F, Baud’huin M, Calleja LR, Quillard T, Amiaud J, Tirode F, Rédini F, Bradner JE, Heymann D, Ory B. Targeting the epigenetic readers in Ewing Sarcoma inhibits the oncogenic transcription factor EWS/Fli1. Oncotarget. 2016; 7: 24125-40. doi: 10.18632/oncotarget.8214.
128. Lv Y-F, Yan G-N, Meng G, Zhang X, Guo Q-N. Enhancer of zeste homolog 2 silencing inhibits tumor growth and lung metastasis in osteosarcoma. Sci Rep. 2015; 5: 12999. doi: 10.1038/srep12999.
129. Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med. 2016; 22: 128–34. doi: 10.1038/nm.4036.
130. Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang K-C, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. Molecular Cancer Therapeutics; 2014; 13: 842–54. doi: 10.1158/1535-7163.MCT-13-0773.
131. Tiffen JC, Gunatilake D, Gallagher SJ, Gowrishankar K, Heinemann A, Cullinane C, Dutton-Regester K, Pupo GM, Strbenac D, Yang JY, Madore J, Mann GJ, Hayward NK, et al. Targeting activating mutations of EZH2 leads to potent cell growth inhibition in human melanoma by derepression of tumor suppressor genes. Oncotarget. 2015; 6: 27023–36. doi: 10.18632/oncotarget.4809.
132. Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, Wang W, Haswell JR, Walensky LD, Hahn WC, Orkin SH, Roberts CWM. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015; 21: 1491–6. doi: 10.1038/nm.3968.
133. Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, Allison DF, Cai L, Rockowitz S, Liu S, Liu Y, Li F, Vedadi M, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. American Society of Hematology; 2015; 125: 346–57. doi: 10.1182/blood-2014-06-581082.
134. Laporte AN, Ji JX, Ma L, Nielsen TO, Brodin BA. Identification of cytotoxic agents disrupting synovial sarcoma oncoprotein interactions by proximity ligation assay. Oncotarget. 2016; 7: 34384-94. doi: 10.18632/oncotarget.8882.
135. Su L, Sampaio AV, Jones KB, Pacheco M, Goytain A, Lin S, Poulin N, Yi L, Rossi FM, Kast J, Capecchi MR, Underhill TM, Nielsen TO. Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell. 2012; 21: 333–47. doi: 10.1016/j.ccr.2012.01.010.
136. Shen JK, Cote GM, Gao Y, Choy E, Mankin HJ, Hornicek FJ, Duan Z. Targeting EZH2-mediated methylation of H3K27 inhibits proliferation and migration of Synovial Sarcoma in vitro. Sci Rep. Nature Publishing Group; 2016; 6: 25239. doi: 10.1038/srep25239.
137. Kawano S, Grassian AR, Tsuda M, Knutson SK, Warholic NM, Kuznetsov G, Xu S, Xiao Y, Pollock RM, Smith JS, Kuntz KK, Ribich S, Minoshima Y, et al. Preclinical Evidence of Anti-Tumor Activity Induced by EZH2 Inhibition in Human Models of Synovial Sarcoma. Rota R, editor. PLoS ONE. Public Library of Science; 2016; 11: e0158888. doi: 10.1371/journal.pone.0158888.
138. Su L, Cheng H, Sampaio AV, Nielsen TO, Underhill TM. EGR1 reactivation by histone deacetylase inhibitors promotes synovial sarcoma cell death through the PTEN tumor suppressor. Oncogene. Nature Publishing Group; 2010; 29: 4352–61. doi: 10.1038/onc.2010.204.
139. Lubieniecka JM, de Bruijn DRH, Su L, van Dijk AHA, Subramanian S, van de Rijn M, Poulin N, van Kessel AG, Nielsen TO. Histone deacetylase inhibitors reverse SS18-SSX-mediated polycomb silencing of the tumor suppressor early growth response 1 in synovial sarcoma. Clinical Cancer Research. American Association for Cancer Research; 2008; 68: 4303–10. doi: 10.1158/0008-5472.CAN-08-0092.
140. Munster P, Marchion D, Bicaku E, Lacevic M, Kim J, Centeno B, Daud A, Neuger A, Minton S, Sullivan D. Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: phase I/II trial of valproic acid and epirubicin/FEC. Clin Cancer Res. Clinical Cancer Research; 2009; 15: 2488–96. doi: 10.1158/1078-0432.CCR-08-1930.
141. Munster P, Marchion D, Bicaku E, Schmitt M, Lee JH, DeConti R, Simon G, Fishman M, Minton S, Garrett C, Chiappori A, Lush R, Sullivan D, et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol. American Society of Clinical Oncology; 2007; 25: 1979–85. doi: 10.1200/JCO.2006.08.6165.
142. Thomas S, Aggarwal R, Jahan T, Ryan C, Troung T, Cripps AM, Raha P, Thurn KT, Chen S, Grabowsky JA, Park J, Hwang J, Daud A, et al. A phase I trial of panobinostat and epirubicin in solid tumors with a dose expansion in patients with sarcoma. Ann Oncol. 2016; 27: 947–52. doi: 10.1093/annonc/mdw044.
143. D’Angelo SP, Shoushtari AN, Agaram NP, Kuk D, Qin L-X, Carvajal RD, Dickson MA, Gounder M, Keohan ML, Schwartz GK, Tap WD. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum Pathol. Elsevier; 2015; 46: 357–65. doi: 10.1016/j.humpath.2014.11.001.
144. Tseng WW, Malu S, Zhang M, Chen J, Sim GC, Wei W, Ingram D, Somaiah N, Lev DC, Pollock RE, Lizée G, Radvanyi L, Hwu P. Analysis of the intratumoral adaptive immune response in well differentiated and dedifferentiated retroperitoneal liposarcoma. Sarcoma. 2015; 2015: 547460–9. doi: 10.1155/2015/547460.
145. Feng Y, Shen J, Gao Y, Liao Y, Cote G, Choy E, Chebib I, Mankin H, Hornicek F, Duan Z. Expression of programmed cell death ligand 1 (PD-L1) and prevalence of tumor-infiltrating lymphocytes (TILs) in chordoma. Oncotarget. 2015; 6: 11139–49. doi: 10.18632/oncotarget.3576.
146. Fritzsching B, Fellenberg J, Moskovszky L, Sápi Z, Krenacs T, Machado I, Poeschl J, Lehner B, Szendrõi M, Bosch AL, Bernd L, Csóka M, Mechtersheimer G, et al. CD8(+)/FOXP3(+)-ratio in osteosarcoma microenvironment separates survivors from non-survivors: a multicenter validated retrospective study. Oncoimmunology. 2015; 4: e990800. doi: 10.4161/2162402X.2014.990800.
147. Lussier DM, O’Neill L, Nieves LM, McAfee MS, Holechek SA, Collins AW, Dickman P, Jacobsen J, Hingorani P, Blattman JN. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J Immunother. 2015; 38: 96–106. doi: 10.1097/CJI.0000000000000065.
148. Lussier DM, Johnson JL, Hingorani P, Blattman JN. Combination immunotherapy with α-CTLA-4 and α-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J Immunother Cancer. BioMed Central; 2015; 3: 21. doi: 10.1186/s40425-015-0067-z.
149. Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, Delbrook C, Lodish M, Bishop R, Wolchok JD, Streicher H, Mackall CL. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin Cancer Res. Clinical Cancer Research; 2016; 22: 1364–70. doi: 10.1158/1078-0432.CCR-15-0491.
150. Tawbi HA-H, Burgess MA, Crowley J, Van Tine BA, Hu J, Schuetze S, D’Angelo SP, Attia S, Priebat DA, Okuno SH, Riedel RF, Davis LE, Movva S, et al. Safety and efficacy of PD-1 blockade using pembrolizumab in patients with advanced soft tissue (STS) and bone sarcomas (BS): Results of SARC028--A multicenter phase II study. ASCO Meeting Abstracts. 2016; 34: 11006.
151. Yang H, Yamazaki T, Pietrocola F, Zhou H, Zitvogel L, Ma Y, Kroemer G. STAT3 Inhibition Enhances the Therapeutic Efficacy of Immunogenic Chemotherapy by Stimulating Type 1 Interferon Production by Cancer Cells. Clinical Cancer Research. 2015; 75: 3812–22. doi: 10.1158/0008-5472.CAN-15-1122.
152. Fang X, Jiang C, Xia Q. Effectiveness evaluation of dendritic cell immunotherapy for osteosarcoma on survival rate and in vitro immune response. Genet Mol Res. 2015; 14: 11763–70. doi: 10.4238/2015.October.2.10.
153. Kawano M, Tanaka K, Itonaga I, Iwasaki T, Miyazaki M, Ikeda S, Tsumura H. Dendritic cells combined with anti-GITR antibody produce antitumor effects in osteosarcoma. Oncol Rep. Spandidos Publications; 2015; 34: 1995–2001. doi: 10.3892/or.2015.4161.
154. Kawano M, Tanaka K, Itonaga I, Iwasaki T, Miyazaki M, Ikeda S, Tsumura H. Dendritic cells combined with doxorubicin induces immunogenic cell death and exhibits antitumor effects for osteosarcoma. Oncol Lett. Spandidos Publications; 2016; 11: 2169–75. doi: 10.3892/ol.2016.4175.
155. Fernández L, Valentín J, Zalacain M, Leung W, Patiño-García A, Pérez-Martínez A. Activated and expanded natural killer cells target osteosarcoma tumor initiating cells in an NKG2D-NKG2DL dependent manner. Cancer Letters. Elsevier; 2015; 368: 54–63. doi: 10.1016/j.canlet.2015.07.042.
156. Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, Hughes DPM, Kleinerman ES, Lee DA. The narrow-spectrum HDAC inhibitor entinostat enhances NKG2D expression without NK cell toxicity, leading to enhanced recognition of cancer cells. Pharm Res. Springer US; 2015; 32: 779–92. doi: 10.1007/s11095-013-1231-0.
157. Jamitzky S, Krueger A-C, Janneschuetz S, Piepke S, Kailayangiri S, Spurny C, Rossig C, Altvater B. Insulin-like growth factor-1 receptor (IGF-1R) inhibition promotes expansion of human NK cells which maintain their potent antitumor activity against Ewing sarcoma cells. Pediatric Blood & Cancer. 2015; 62: 1979–85. doi: 10.1002/pbc.25619.
158. Merchant MS, Bernstein D, Amoako M, Baird K, Fleisher TA, Morre M, Steinberg SM, Sabatino M, Stroncek DF, Venkatasan AM, Wood BJ, Wright M, Zhang H, et al. Adjuvant Immunotherapy to Improve Outcome in High-Risk Pediatric Sarcomas. Clin Cancer Res. Clinical Cancer Research; 2016. doi: 10.1158/1078-0432.CCR-15-2550.
159. Goldberg JM, Fisher DE, Demetri GD, Neuberg D, Allsop SA, Fonseca C, Nakazaki Y, Nemer D, Raut CP, George S, Morgan JA, Wagner AJ, Freeman GJ, et al. Biologic Activity of Autologous, Granulocyte-Macrophage Colony-Stimulating Factor Secreting Alveolar Soft-Part Sarcoma and Clear Cell Sarcoma Vaccines. Clin Cancer Res. Clinical Cancer Research; 2015; 21: 3178–86. doi: 10.1158/1078-0432.CCR-14-2932.
160. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, Gray T, Wu M-F, Liu H, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol. 2015; 33: 1688–96. doi: 10.1200/JCO.2014.58.0225.
161. Mason N, Gnanandarajah J, Engiles J, Gray F, Laughlin D, Gaurnier-Hausser A, Wallecha A, Huebner M, Paterson Y. Immunotherapy with a HER2 targeted Listeria induces HER2-specific immunity and demonstrates potential therapeutic effects in a phase I trial in canine osteosarcoma. Clin Cancer Res. 2016. doi: 10.1158/1078-0432.CCR-16-0088.
162. Krishnadas DK, Shusterman S, Bai F, Diller L, Sullivan JE, Cheerva AC, George RE, Lucas KG. A phase I trial combining decitabine/dendritic cell vaccine targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol Immunother. Springer Berlin Heidelberg; 2015; 64: 1251–60. doi: 10.1007/s00262-015-1731-3.
163. Iura K, Kohashi K, Hotokebuchi Y, Ishii T, Maekawa A, Yamada Y, Yamamoto H, Iwamoto Y, Oda Y. Cancer-testis antigens PRAME and NY-ESO-1 correlate with tumour grade and poor prognosis in myxoid liposarcoma. J Pathol Clin Res. 2015; 1: 144–59. doi: 10.1002/cjp2.16.