
Introduction
Glioblastoma multiforme (GBM) is the most common primary brain tumor in adults, but despite multimodal treatment combining surgery, radiation, and chemotherapy, the median survival for patients remains under 15 months [1]. Pathologically, GBM is described as a heterogeneous tumor type with high levels of angiogenesis and invasion [2]. Recent studies provide evidence for a population of cancer stem cells (CSCs) within GBM contributing to this heterogeneity [3, 4]. These cells (hereafter called GSCs) carry neural stem cell markers such as CD133, self-renew over serial passages, and differentiate into multiple lineages. As with tumor-initiating cells described in other cancers [5-10], GSCs have been shown to be invasive and resistant to both radiation and chemotherapy [11-14]. As such, there is significant interest in understanding the biology of the GSC population to identify potential novel therapeutic targets to improve disease control.
NF-κB is a family of transcription factors consisting of five members: p65 (RelA), RelB, c-Rel, p105/p50, and p100/p52 that homo- and heterodimerize to regulate transcription of target genes. In the canonical pathway under basal conditions, p65-p50 dimers are bound to IκBα in the cytoplasm. Stimuli such as TNF-α or IL-1 lead to activation of the IKK complex, which consists of two catalytic subunits, IKKα and IKKβ, as well as a scaffolding subunit, IKKγ or NEMO. IKK phosphorylates IκBα, leading to its ubiquitination and proteasomal degradation, which allows NF-κB to accumulate in the nucleus. The non-canonical pathway is activated by stimuli crucial for lymphoid development such as BAFF or CD40. Here, the precursor p100 acts as an IκB molecule bound to RelB. Upon activation, IKKα phosphorylates p100, leading to its cleavage to produce p52. The active RelB-p52 dimer can then regulate transcription of target genes [15, 16]. Originally identified for its role in inflammatory signaling, the NF-κB pathway has since been demonstrated to be activated in various forms of cancer and is thought to contribute to the malignant phenotype through dysregulation of important biological processes such as proliferation, angiogenesis, apoptosis, and cell survival [16-19].
Within the nervous system, NF-κB is typically considered to be inactive, but activated in cases of injury or inflammation, consistent with its canonical function in other tissues [20, 21]. In normal neural stem cells (NSCs), TNFα has been shown to induce proliferation through NF-κB [22]. Kaus et al. [23] have shown that as NSCs acquire the ability to proliferate independent of exogenous growth factors, these cells demonstrate increased NF-κB activity. In GBM, NF-κB has been reported to regulate survival, invasion, and resistance to both radiation and chemotherapy [24-28]. PTEN deletion and EGFR amplification and/or mutation are two of the most common genetic alterations in GBM and both can lead to increased NF-κB activation [28, 29]. Additionally, TGF-β signaling has been demonstrated to contribute to GSC maintenance through the upregulation of LIF, Sox2, and Sox4 [30, 31]. While the TGF-β and NF-κB pathways are thought to antagonize each other in some settings, there is evidence of their cooperation within tumors, including GBM [32, 33]. Others have identified alterations in the NF-κB pathway itself, with a subset of GBMs harboring monoallelic NFKBIA (gene name for IκBα) deletions and others expressing high levels of miR-30e* which targets IκBα [34, 35]. Consistent with the involvement of NF-κB signaling in GBM, recent work demonstrated that treatment with a NEMO-binding domain (NBD) peptide that blocks interactions between NEMO and IKKα/β slowed tumor growth in both a human glioma xenograft and a genetic mouse model of glioma [36].
NF-κB activity has been associated with CSCs in several cancer [37-42], and in GBM the NF-κB targets IL-6 and A20 have been shown to contribute to the maintenance of GSCs [43, 44]. When cells are grown in CSC-permissive conditions instead of monolayers, there is an upregulation of NF-κB activity as seen through p65 phosphorylation and target gene expression [45]. Other data suggest that inducing differentiation of GSCs increases NF-κB activity. However, NF-κB inhibition accelerates differentiation, suggesting a role for this pathway in maintaining the cells in a more stem-like state [46].
In this study, we sought to investigate the role of the NF-κB pathway in GSCs directly. We find that phosphorylation of the p65 (RelA) subunit of NF-κB is elevated in CD133+ GBM cells as compared to CD133- cells. Targeting NF-κB signaling either genetically or pharmacologically impairs self-renewal in primary tumorsphere assays and in limiting dilution assays. Interestingly, both canonical and non-canonical NF-κB pathways contribute to the GSC phenotype. Our results indicate that one source of NF-κB activation in GSCs is a TGF-β signaling pathway acting through TAK1. Using an ex vivo brain slice co-culture model, we show that NF-κB contributes to the growth and survival of tumorspheres. Our findings indicate that NF-κB signaling is a key therapeutic target controlling GSCs.
Results
NF-κB is preferentially activated in CD133+ cells of GBM explants
Given that NF-κB is not highly active in normal brain tissue, we first compared the growth of normal neural stem cells and astrocytes to that of patient-derived GBM explant cultures. We treated cells with DMSO or with the selective IKKβ antagonist Compound A [47] daily for 5 days and measured cell viability by MTS assay (Figure 1A). Results demonstrate that growth was impaired in the GBM explant cultures by NF-κB inhibition but not in the normal neural stem cell or astrocyte cultures, suggesting that NF-κB inhibition preferentially targets tumor cells over normal brain cells. Since GBM was one of the first solid tumors in which CSCs were identified [3, 4] and based on our previous work implicating NF-κB signaling in breast CSCs [39], we next asked if these findings could be explained by a role for NF-κB signaling in the growth of GSCs. To study the stem cell-like vs. non-stem cell-like subpopulation of glioblastoma cells, we isolated CD133+ and CD133- fractions from the human glioblastoma explant cultures using magnetic beads. Interestingly, the CD133+ cells exhibited elevated levels of p65 phosphorylation, consistent with increased or altered activity of the canonical NF-κB pathway (Figure 1B).
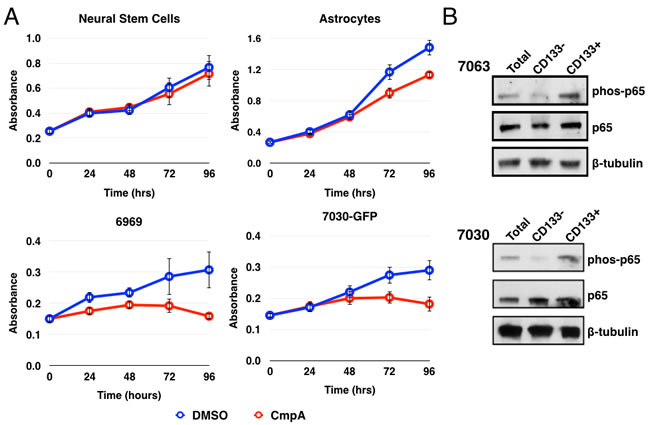
Figure 1: NF-κB is preferentially activated in CD133+ glioblastoma stem cells. A. MTS assay using normal neural stem cells, astrocytes, or two GBM explants: 6969 and 7030. Cells were treated daily with DMSO or 5 μM Compound A and analyzed every 24 hours for 96 hours. Data are represented as the mean ± s.d. and are representative of three independent experiments. B. Analysis of total cells, isolated CD133-, or CD133+ cells by immunoblot for phosphorylation of p65. Quantification of the immunoblots for the ratio of phospho-p65 to total p65 is: 7063 total: 1; CD133-: 0.23; CD133+: 1.77; 7030 total: 1; CD133-: 0.19; CD133+: 1.15.
Inhibition of NF-κB reduces tumorsphere formation
We next examined whether NF-κB was important in tumorsphere formation, which is an in vitro test for stem cell-like activity. After isolation, CD133+ cells were plated at a low density and allowed to form spheroids for one week prior to quantification. Daily treatment with Compound A abrogated tumorsphere formation completely. Importantly, a single treatment with Compound A at the beginning of the experiment was sufficient to reduce tumorsphere formation, suggesting that even transient loss of NF-κB activity could affect stem-like activity (Figure 2A, 2B). In order to further address the effects on self-renewal, primary tumorspheres from the first week of growth were dissociated, re-plated, and then subjected to the same treatments. Treatment with Compound A also reduced secondary tumorsphere formation, again consistent with a role for NF-κB activity in the stem-cell fraction (Figure 2C). Finally, we assayed tumorsphere formation by using a limiting dilution assay with a range of cell concentrations from 1-100 cell(s)/well. For both 7030 and GBM6 CD133+ cells, treatment with Compound A significantly reduced the ability of GBM stem cells to form tumorspheres (Figure 2D, 2E).
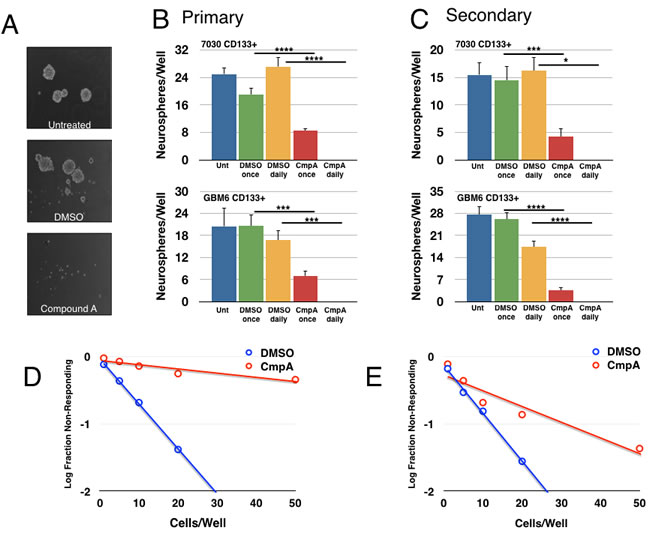
Figure 2: Pharmacological inhibition of the IKK/NF-κB pathway decreases tumorsphere formation. 7030 CD133+ cells plated for a tumorsphere assay and treated as indicated (DMSO or 5μM Compound A, once or daily). After one week of growth, tumorsphere formation was analyzed. A. Representative images of tumorsphere formation after daily treatment. B. Quantification of primary tumorspheres formed per well for two explants. Data are represented as mean ± SEM, ****p < 0.0001, ***p < 0.001, *p < 0.05 by t-test. C. Primary tumorspheres were dissociated, replated, and treated again as indicated. Secondary tumorsphere formation was quantified after another week of growth. Data are represented as mean ± SEM, ****p < 0.0001, ***p < 0.001, *p < 0.05 by t-test. D., E. Tumorsphere formation was measured through a limiting dilution assay with 7030 D. or GBM6 E. CD133+ cells plated at 100, 50, 20, 10, 5, or 1 cell(s)/well and treated with DMSO or 5μM Compound A (7030: n = 48 wells/condition; p = 2.02x10-47; GBM6: n≥116wells/condition; p = 2.23x10-11).
In order to validate the on-target activity of Compound A in inhibiting IKK, we repeated these assays using siRNA-mediated knockdown of IKKβ and/or p65. Knockdown of either protein resulted in a decrease in p65 phosphorylation, as well as a decrease in tumorsphere formation (Figure 3A-3C). The inhibitory effects of p65 knockdown were confirmed in the limiting dilution assay consistent with previous results (Figure 3D). Taken together, these results strongly implicate a role for the NF-κB pathway in GSC propagation and self-renewal.
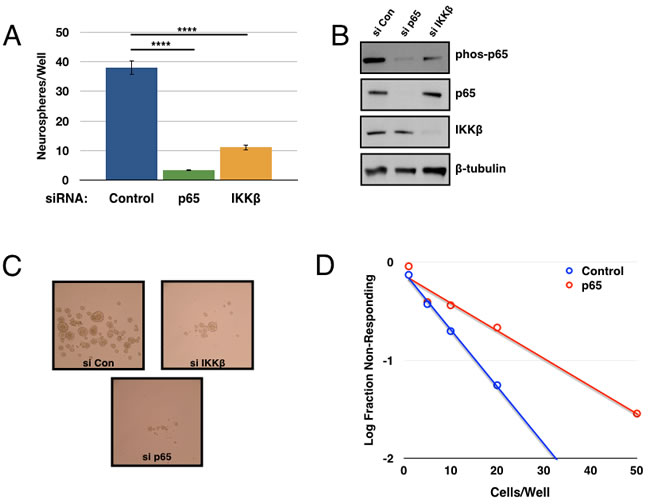
Figure 3: Genetic inhibition of the IKK/NF-κB pathway decreases tumorsphere formation. 7030 CD133+ cells were transfected with siRNA (control, p65, or IKKβ) and then plated for tumorsphere assays A. Quantification of tumorspheres formed per well after one week of growth. Data are represented as the mean ± SEM, ****p < 0.0001 by t-test. B. Transfected cells analyzed by immunoblot for p-p65, p65, IKKβ, and β-tubulin. C. Representative images of tumorsphere formation after daily treatment. D. Tumorsphere formation was measured through a limiting dilution assay with GBM6 CD133+ cells plated at 100, 50, 20, 10, 5, or 1 cell(s)/well following transfection with control or p65 siRNA (n≥70 wells; p = 1.76x10-6).
Multiple NF-κB subunits contribute to GSC maintenance
Up to this point, our studies have focused on the more highly studied canonical NF-κB pathway. We next sought to determine if the non-canonical NF-κB pathway, driven by a RelB-p52 dimer, could be contributing to GSC maintenance as well. Accordingly, GBM6 CD133+ cells were transfected with siRNAs targeting p65, p100/p52, or RelB. Each of these subunits produced a substantial decrease in tumorsphere formation in a limiting dilution assay (Figure 4A). Knockdown efficiency was confirmed through qPCR (Figure 4B). These results suggest that both the canonical and non-canonical NF-κB pathways contribute to GSC maintenance.
Consistent with the literature, knockdown of p65 resulted in decreased transcription of both RelB and p100 [48, 49]. We found that knockdown of p65 and IKKβ decreased expression of the non-canonical subunits at the protein level as well (Figure 4C). Interestingly, knockdown of IKKβ but not IKKα significantly decreased tumorsphere formation in a limiting dilution assay (Figure 4D, 4E). Under normal culture conditions, no NIK expression was detected even with the addition of the proteasome inhibitor MG132. Perhaps not surprisingly, we did not see consistent results when attempting to knockdown NIK and assay tumorsphere formation (data not shown). It is possible that the residual IKKα following siRNA knockdown is sufficient for p100 processing to p52 and that a more complete knockout would reveal a larger effect for IKKα. Alternatively, p100 could be functioning in a separate role rather than simply being the precursor to p52, which is what is being observed in these experiments. These possibilities are further explored in the discussion. Nonetheless, these results suggest that transcriptional regulation of the non-canonical subunits (RelB and p100) by the canonical subunit (p65) could be a significant aspect of non-canonical pathway regulation in GSCs.
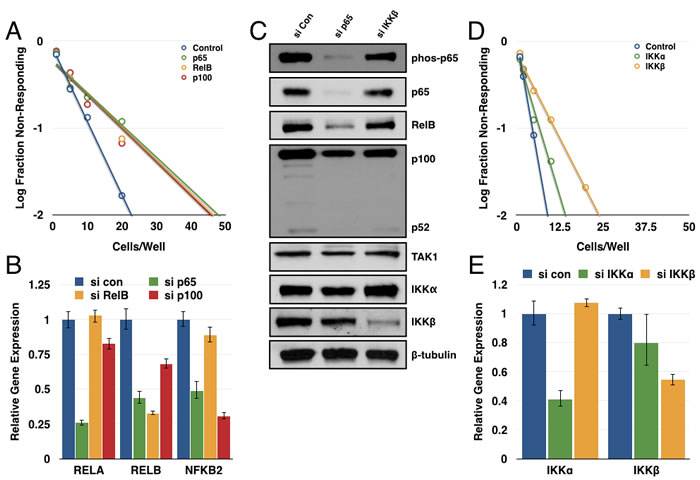
Figure 4: Multiple NF-κB subunits contribute to tumorsphere formation. A. GBM6 CD133+ cells were transfected with siRNA control or targeting p65, RelB, or p100. Subsequently, cells were plated out for limiting dilution assay at 100, 50, 20, 10, 5, or 1 cell(s)/well and scored for the presence or absence of tumorspheres following one week of growth (n≥95 wells/condition; p65 vs. control p = 2.26x10-5; RelB vs. control p = 6.44x10-5; p100 vs. control p = 3.82x10-5). B. Quantitative real-time PCR was performed to analyze expression of RELA, RELB, or NFKB2 in siRNA-transfected cells normalized to GUSB expression. C. Expression of canonical and non-canonical NF-κB components was analyzed by immunoblot in 7030 CD133+ cells transfected with siRNA (control, p65, or IKKβ). D. GBM6 CD133+ cells were transfected with siRNA (control, IKKα, or IKKβ). Subsequently, cells were plated out for limiting dilution assay at 50, 20, 10, 5, 2, or 1 cell(s)/well and scored for the presence or absence of tumorspheres following one week of growth (n≥48 wells/condition; IKKα vs. control p = 0.192; IKKβ vs. control p = 6.01x10-5). E. Quantitative real-time PCR was performed to analyze expression of CHUK and IKBKB in siRNA-transfected cells normalized to GUSB expression.
TAK1 activates the NF-κB pathway to promote GSC function
Next, we examined an upstream activator of the NF-κB pathway, transforming growth factor-β-activated kinase 1 (TAK1), which is known to activate the IKK complex following cytokine stimulation as well as in some oncogenic settings [50-54]. We first showed that use of two structurally distinct TAK1 inhibitors ((5Z)-7-oxozeanol and NG-25) decreased the expression of an NF-κB luciferase reporter in both GBM explant cultures (Figure 5A). In tumorsphere formation assays, treatment with either (5Z)-7-oxozeanol or NG-25 decreased tumorsphere formation in GBM 7030 CD133+ cells with a single treatment and to an even greater extent with daily treatment (Figure 5B). We then extended these studies in limiting dilution assays, finding that both siRNA against TAK1 and the TAK1 inhibitors resulted in significant decreases in tumorsphere formation (Figure 5C, 5D).
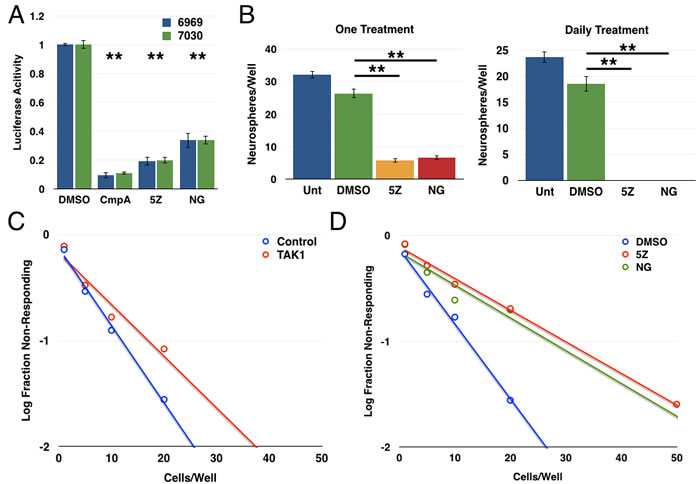
Figure 5: TAK1 activates the NF-κB pathway to promote glioblastoma stem cell function. A. 6969 or 7030 cells were transfected with 3x-κB luciferase reporter, treated with the indicated inhibitors for 24 hours, then harvest and analyzed for luciferase activity (n = 3; **p < 0.0001 by t-test, error bars represent SEM) B. Quantification of tumorsphere formation in GBM6 CD133+ cells following treatment with either (5Z)-7-oxozeaenol or NG25 either once or daily. Data are represented as the mean ± SEM, **p < 0.0001 by t-test. C. Limiting dilution assay with GBM6 CD133+ cells transfected with siRNA control or TAK1 (n = 72 wells/condition; p < 0.05). D. Limiting dilution assay following treatment of GBM6 CD133+ cells with structurally distinct TAK1 inhibitors: 2.5 μM (5Z)-7-oxozeaenol or 2 μM NG-25 (n≥90wells/condition; 5Z vs. DMSO p = 1.01x10-19, NG vs. DMSO p = 5.29x10-10).
TGF-β is one source of NF-κB activation
As the cognate activator of TAK1 is transforming growth factor-β (TGF-β) itself, and as the TGF-β pathway has previously been implicated in regulating GBM CSCs [30, 31], we next investigated a potential link between TGF-β and NF-κB in the setting of GBM. Studies showed that treatment with exogenous TGF-β led to an increase in Smad phosphorylation (as expected) and in p65 phosphorylation in GBM explant cultures (Figure 6A). Conversely, addition of a TGF-βR1 inhibitor (SB431542) led to a decrease in p65 phosphorylation (Figure 6B). Additionally, the TGF-βR1 inhibitor induced a small but consistent decrease in luciferase activity from an NF-κB reporter in GBM explant cultures (Figure 6C). Together, these results suggest an autocrine/paracrine role for TGF-β in maintaining NF-κB signaling in these GBM explant cultures. Use of the TGF-βR1 inhibitor in a tumorsphere assay leads to a significant decrease in tumorsphere formation in GBM6 CD133+ cells (Figure 6D). These results suggest that TGF-β can activate NF-κB signaling in these GBM explants. However, given the scale of these changes, it is likely only one of several sources of NF-κB activation, consistent with the pleiotropic nature of this pathway. Similarly, given the more drastic effects seen in the tumorsphere assay with the TGF-βR1 inhibitor, it is likely that the TGF-β pathway is also mediating additional factors involved in GSC biology.
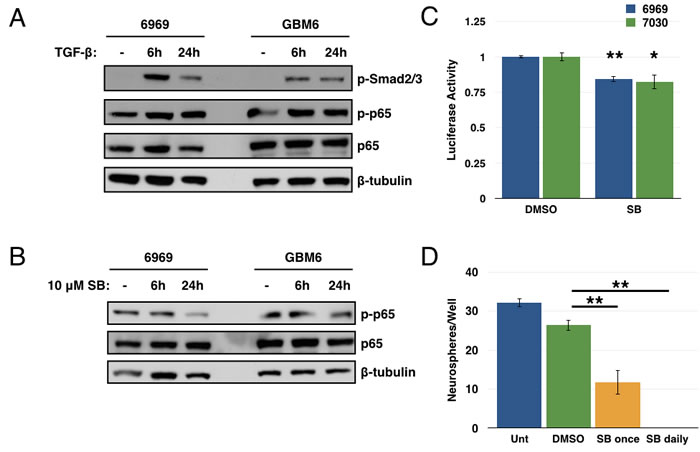
Figure 6: TGF-β is one source of NF-κB activation in GBM. A. 6969 and GBM6 explants were stimulated with 10ng/mL TGF-β for 6 or 24 hours, then analyzed by immunoblotting for phosphorylation of Smad and p65. B. 6969 and GBM6 explants were treated with 10 μM SB431542, a TGF-βR1 inhibitor, for 6 or 24 hours, then analyzed by immunoblotting for p65 phosphorylation. C. 6969 or 7030 cells were transfected with 3x-κB luciferase reporter and treated with DMSO or 10 μM SB431542 for 24 hours, then harvested and analyzed for luciferase activity (n = 3, **p < 0.0001, *p < 0.01 by t-test, error bars represent SEM). D. Quantification of tumorsphere formation in GBM6 CD133+ cells following treatment with 10 μM SB431542 either once or daily. Data are represented as the mean ± SEM, **p < 0.0001 by t-test.
Inhibition of the IKK/NF-κB pathway decreases glioblastoma growth ex vivo
Finally, we sought to validate the in vitro studies in a more biologically relevant setting, turning to an ex vivo organotypic brain slice preparation which has been used previously [55, 56]. This methodology provided the opportunity for higher throughput analysis, as well as a level of longitudinal imaging not typically available in vivo. For these experiments, neonatal rat brain tissues were sectioned into 250 μm coronal slices, then plated on top of an agar medium as previously described [57]. GBM6-GFP spheres were then engrafted onto the upper surfaces of these these brain slice explants shortly after slicing. A first round of imaging was then completed within a few hours of plating to establish a baseline for GBM tumor growth these brain slices. The brain slices were then imaged daily for up to eight days and the GFP-positive areas were quantified for each slice and on each day using ImageJ software. Representative images show that implanted GBM tumorspheres progressively grow and invade the surrounding brain tissues over the course of the experiment (Figure 7A).
To examine a role for NF-κB in GBM growth within these ex vivo orthotopic xenografts, GBM6-GFP cells were transfected with either control or p65 siRNA twenty-four hours prior to tumorsphere formation and brain slice implantation. Quantification of tumorsphere cross-sectional areas indicated that transfection with p65 siRNA significantly inhibited the growth rate of the implanted GBM tumorspheres over the course of the experiment (Figure 7B). Similarly, addition of the IKKβ inhibitor to the implanted brain slice cultures also led to significant inhibition of GBM tumorsphere growth (Figure 7C, 7D).
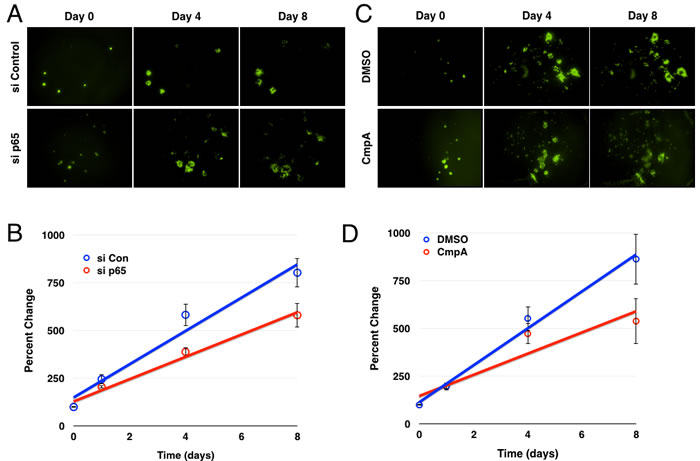
Figure 7: Inhibition of the IKK/NF-κB pathway decreases glioblastoma growth/survival ex vivo. A. Images of GFP+ GBM6 cells following siRNA transfection and brain slice culture. B. Average percent change in GFP+ area over the course of the experiment (n = 14 for control, 13 for p65; p < 0.005 by linear regression; error bars represent SEM) C. Images of GFP+ GBM6 cells over the course of eight days of culture on brain slices following treatment with DMSO or Compound A. D. Average percent change in GFP+ area over the course of the experiment (n = 12; p < 0.01 by linear regression; error bars represent SEM)
Discussion
As with many other tumor types, glioblastoma is characterized by a hierarchical organization of cells, including a subpopulation of so-called cancer stem cells (or tumor-initiating cells). These cells promote tumor initiation and recurrence, drive invasion and metastasis, and demonstrate increased resistance to radiation and chemotherapy [3, 4, 11-14]. It is crucial to investigate the signaling pathways responsible for these phenotypic differences from the bulk of the tumor, both to establish insight into mechanisms that promote these cells and to potentially identify therapeutic targets for disease control. In this study, we demonstrate the involvement of the IKK/NF-κB pathway in the function of GSCs. We identified a TGF-β/TAK1 axis as one source of NF-κB activation in these cells. TAK1 is a well-established activator of IKK and there is a precedent for cooperation between the TGF-β and NF-κB pathways in GBM [32, 33]. Nonetheless, it is likely that other sources for NF-κB activation exist, such as cytokines, which utilize TAK1 as a mediator of NF-κB activation. Indeed, common genetic alterations seen in GBM (PTEN deletion, EGFR amplification and mutation, and monoallelic NFKBIA deletion) have all been connected to enhanced NF-κB activation [28, 34, 58-63]. Our data from the ex vivo co-culture experiments show greater effects following IKK inhibition in the whole slice rather than just the cancer cells. Given the extensive number of cytokines and chemokines regulated by NF-κB, it is very likely that this pathway impacts the interactions between the tumor, microglia, and infiltrating immune cells, potentially related to the finding that GSCs promote tumor evasion via immunosuppression [64, 65]. Consistent with our studies overall, Verma and colleagues showed that treatment with an NBD peptide impaired tumor growth in both a human glioma xenograft and a genetic mouse model of glioma [36], focusing on inhibition of canonical NF-κB signaling through disruption of the IKK complex. Thus, the effects of IKK inhibition in GBM models may function, at least partly, at the level of GSCs.
As a family of transcription factors, the NF-κB pathway is likely mediating a variety of downstream effects in GBM cells. We found that both the canonical and non-canonical branches contribute to GSC biology. Our results show larger effects of targeting RelB and p100 compared to IKKα, as well as decreased expression of the non-canonical subunits following knockdown of p65. One possibility is that since siRNA knockdown is incomplete, the remaining IKKα is still capable of driving sufficient processing of p100 into p52 and that use of a CRISPR knockout would produce a more substantial phenotype. Alternatively, since p52 is generated from p100 protein, knockdown at the gene level affects both p100 and p52 levels. Thus, we could actually be observing an independent effect of loss of p100 that is not dependent on IKKα for processing into the active p52 subunit. For example, a recent report demonstrated that p100 can interact with ERK2 and inhibit its nuclear localization [66], providing a precedent for p100-specific activities.
At this point, it is still unknown whether these two pathways are acting on common or separate targets. The dimerization and DNA binding patterns of NF-κB subunits remain complex areas of study [67-72]. The non-canonical pathway has been shown to have distinct functions in oncogenesis, such as driving growth and invasion of mesenchymal glioma and regulation of the mutant C250T TERT promoter [73, 74]. Antagonism of p53 through NF-κB regulation of Mdm2 expression has been well-established and can contribute to chemotherapy resistance [75-77]. Additionally, NF-κB is known to regulate target genes related to several GSC functions including survival (BCL2, BCL2L1), invasion (IL6, IL8, CCL2, MMP2/3/9), and resistance to therapy (MGMT, TNFAIP3, TRADD) [78-85]. Gene expression analysis showed NF-κB regulation of some of these targets, however neither the expression nor the NF-κB-dependency was limited to the CD133+ population of cells. It is likely that the effects on GSCs observed following NF-κB inhibition result from a combination of these and other genes. Given its central position in GSC signaling, the IKK/NF-κB pathway is proposed as a target for therapeutic intervention in glioblastoma.
Materials and Methods
Cell culture, CD133+ isolation, and reagents
The human glioblastoma explants (6969, 7030, 7063, and GBM6) were obtained from UCSF and maintained in Neurobasal medium (Invitrogen), supplemented with B27 without Vitamin A, L-glutamine, 20ng/mL EGF, 40ng/mL FGF, and penicillin/streptomycin. To dissociate tumorspheres, cells were incubated with Accutase (Sigma) in a 37°C water bath for 10 minutes, then plated as desired. To isolate CD133+ cells, cells were dissociated with Accutase and passed over a pre-separation filter to achieve single cell suspension. Dead cells were removed using the Dead Cell Removal Kit according to manufacturer’s instructions (Miltenyi). Remaining cells were incubated with CD133 magnetic microbeads (Miltenyi) for 30 minutes, resuspended in MACS buffer and passed over two LS columns consecutively. Non-retained cells were saved for the CD133- fraction. After washing, the column was removed from the magnet and retained cells were expelled from the column, counted, and plated for experiments. Normal human astrocytes were a kind gift from Dr. Russell Pieper [86, 87] and were maintained in DMEM media with 10% FBS and 1% penicillin/streptomycin. Neural stem cells (Millipore) were maintained in RenCell NSC maintenance media and grown on laminin-coated plates.
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium cellular proliferation assay
Cells were seeded at 2000 or 3000 cells per well in 96-well plates, then treated with DMSO or Compound A daily as indicated. At each time point, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) compound (Promega) was added and absorbance was read at 490 nm on a Versamax Microplate Reader (Molecular Devices).
Luciferase assay
Cells were transfected with 3x-κB luciferase reporter plasmid [88] using FuGENE HD (Promega). Six hours post-transfection, cells were separated into 12-well plates and treated in duplicate with inhibitors as indicated for 24 hours. Cells were lysed in 150 μL of Passive Lysis Buffer, then 20 μL of lysate was used for analysis in triplicate using the Luciferase Assay System (Promega). Luciferase signal was read on a Synergy2 plate reader (Biotek), and then normalized to protein content of each well based on a Bradford assay.
Western blotting
Whole cell extracts were prepared by collecting cells, washing with cold PBS, then suspending in cold lysis buffer (1% NP-40, 20 mM Tris, 138 mM NaCl, 2mM EDTA, 10% glycerol) on ice for 10 minutes, followed by 10 minutes of centrifugation to remove insoluble components. Protein was quantitated by Bradford assay (Biorad). Equal amounts of lysate (25-50 μg) were separated by SDS-PAGE, transferred to nitrocellulose membranes, and blocked for 1 hour in 5% milk. Membranes were incubated with primary antibody overnight at 4°C, then incubated with secondary antibody for 1 hour at room temperature and developed using ECL reagent (GE). Antibodies used were phospho-Smad2/3 (S465, 467/S423, 425), phospho-p65 (S536), p65, IKKβ, RelB (Cell Signaling Technology), IKKα, p100/p52 (Millipore), TAK1 and β-tubulin (Santa Cruz Biotechnology).
siRNA transfection
Human siRNA targeting RELA (M-003533-02), RELB (M-004767-02), NFKB2 (M-003918-02), CHUK (M-003473-02), IKBKB (M-003503-03), MAP3K7 (M-003790-06), or control #3 (D001201-03) was purchased from Thermo/Dharmafect. DharmaFECT reagent 1 was used to transfect siRNA into cells according to the manufacturer’s instructions. Six hours post-transfection, the media was changed on the cells. Cells were harvested 48-72 hours post-transfection for analysis by quantitative real-time PCR or Western blot.
Quantitative real-time PCR
RNA extracts were obtained from cells using the RNeasy Plus Kit (Qiagen). Two micrograms of RNA were reverse transcribed using random primers and MMLV reverse transcriptase (Invitrogen). Real-time PCR was performed using Taqman Gene Expression Assay primer-probe sets for GUSB (Hs99999908_m1), RELA (Hs00153294_m1), RELB (Hs00232399_m1), NFKB2 (Hs01028901_g1), CHUK (Hs00989502_m1), and IKBKB (Hs00233287_m1) and relative quantification was determined using the ΔΔCt method.
Tumorsphere and limiting dilution assays
For tumorsphere assays, cells were dissociated into single cell suspension using Accutase. Cells were counted and plated at 100 cells per well in 24-well tissue culture plates. Cells were treated with inhibitors as indicated (once or daily for the duration of the experiment): 5 μM Compound A, 2.5 μM (5Z)-7-oxozeanol (Tocris), 2 μM NG25 (MedChem Express), and 10 μM SB431542 (Tocris). Following one week of growth, tumorspheres were viewed on the microscope and quantified. For limiting dilution assays, cells were serially diluted to be plated at 100, 50, 20, 10, 5, 2 or 1 cell(s)/well in 96-well plates. After one week of growth, wells were scored for the presence or absence of spheres. Extreme limiting dilution analysis was performed as previously described [89], using software available at http://bioinf.wehi.edu.au/software/elda/.
Brain slice explantation and tumorsphere implantation
Coronal brain slices (250 μm thick) from postnatal day 10 Sprague-Dawley rat pups of either sex (Charles River, Wilmington, MA) were prepared and explanted in organotypic culture as previously described [57]. Animals were sacrificed in accordance with NIH guidelines and under Duke IACUC approval and oversight. Briefly, brain tissues were sliced in ice-cold artificial cerebrospinal fluid and plated in interface configuration atop culture medium (Neurobasal A medium supplemented with 15% heat-inactivated horse serum, 10 mM KCl, 10 mM HEPES, 100 U/ml penicillin/streptomycin, 1 mM sodium pyruvate, and 1 mM L-glutamine) semi-solidified in 0.5% reagent-grade agarose in 12-well plates. Brain slice explants were incubated under 5% CO2 at 37 ºC for up to 8 days as indicated. Compound A or DMSO-only vehicle was added to culture medium at the time of brain slice explantation. Small groups of GBM tumorspheres were implanted shortly thereafter by direct application to the upper surfaces of each brain slice. An epifluorescence stereomicroscope was used to obtain images through the course of experiments. ImageJ was used to quantify the area of GFP+ cells.
Acknowledgments
We thank Dr. Russell Pieper for the generous gift of the normal human astrocytes. We also thank members of the Baldwin Lab for their feedback on this manuscript. Research support was provided by NIH grant R35 CA194687 to ASB and by a grant to ASB and DCL from Accelerate Brain Cancer Cure.
Conflicts of Interest
The authors state no conflict of interest.
References
1. Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2011 Nov 2;107:359-64.
2. Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010 May;10:319-31.
3. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003 Sep 15;63:5821-8.
4. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004 Nov 18;432:396-401.
5. Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008 May;133:704-15.
6. Long H, Xie R, Xiang T, Zhao Z, Lin S, Liang Z, Chen Z, Zhu B. Autocrine CCL5 signaling promotes invasion and migration of CD133+ ovarian cancer stem-like cells via NF-B-mediated MMP-9 upregulation. Stem Cells. 2012 Oct;30:2309-19.
7. Kumar M, Allison DF, Baranova NN, Wamsley JJ, Katz AJ, Bekiranov S, Jones DR, Mayo MW. NF-kappaB regulates mesenchymal transition for the induction of non-small cell lung cancer initiating cells. PLoS ONE. 2013 Jul 30;8:e68597.
8. Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G, Medema JP, Stassi G. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007 Oct 11;1:389-402.
9. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007 Sep;1:313-23.
10. Mueller MT, Hermann PC, Witthauer J, Rubio Viqueira B, Leicht SF, Huber S, Ellwart JW, Mustafa M, Bartenstein P, D’Haese JG, Schoenberg MH, Berger F, Jauch KW, Hidalgo M, Heeschen C. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology. 2009 Sep;137:1102-13.
11. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006 Oct 18;444:756-60.
12. Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67.
13. Cheng L, Wu Q, Guryanova OA, Huang Z, Huang Q, Rich JN, Bao S. Elevated invasive potential of glioblastoma stem cells. Biochem Biophys Res Commun. 2011 Mar;406:643-8.
14. Garcia JL, Perez-Caro M, Gomez-Moreta JA, Gonzalez F, Ortiz J, Blanco O, Sancho M, Hernandez-Rivas JM, Gonzalez-Sarmiento R, Sanchez-Martin M. Molecular analysis of ex-vivo CD133+ GBM cells revealed a common invasive and angiogenic profile but different proliferative signatures among high grade gliomas. BMC Cancer. 2010;10:454.
15. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008 Feb;132:344-62.
16. Bradford JW, Baldwin AS. IKK/nuclear factor-kappaB and oncogenesis: roles in tumor-initiating cells and in the tumor microenvironment. Adv Cancer Res. 2014;121:125-45.
17. DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012 Mar;246:379-400.
18. Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012 Jan 19;12:121-32.
19. Lim K-H, Yang Y, Staudt LM. Pathogenetic importance and therapeutic implications of NF-kappaB in lymphoid malignancies. Immunol Rev. 2012 Mar;246:359-78.
20. Kaltschmidt B, Widera D, Kaltschmidt C. Signaling via NF-kappaB in the nervous system. Biochim Biophys Acta. 2005 Sep 30;1745:287-99.
21. Widera D, Kaus A, Kaltschmidt C, Kaltschmidt B. Neural stem cells, inflammation and NF-B: basic principle of maintenance and repair or origin of brain tumours? Journal of Cellular and Molecular Medicine. 2008 Apr;12:459-70.
22. Widera D, Mikenberg I, Elvers M, Kaltschmidt C, Kaltschmidt B. Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci. 2006;7:64.
23. Kaus A, Widera D, Kassmer S, Peter J, Zaenker K, Kaltschmidt C, Kaltschmidt B. Neural stem cells adopt tumorigenic properties by constitutively activated NF-kappaB and subsequent VEGF up-regulation. Stem Cells Dev. 2010 Jul;19:999-1015.
24. Raychaudhuri B, Han Y, Lu T, Vogelbaum MA. Aberrant constitutive activation of nuclear factor B in glioblastoma multiforme drives invasive phenotype. J Neurooncol. 2007 May 4;85:39-47.
25. Kesanakurti D, Chetty C, Rajasekhar Maddirela D, Gujrati M, Rao JS. Essential role of cooperative NF-kappaB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma. Oncogene. 2012 Nov 26;32:5144-55.
26. Thaker NG, Zhang F, McDonald PR, Shun TY, Lewen MD, Pollack IF, Lazo JS. Identification of survival genes in human glioblastoma cells by small interfering RNA screening. Mol Pharmacol. 2009 Nov 20;76:1246-55.
27. Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L, James JD, Goodman LD, Conroy S, Long L, Lelic N, Wang S, Gumin J, Raj D, Kodama Y, Raghunathan A, Olar A, Joshi K, Pelloski CE, Heimberger A, Kim SH, Cahill DP, Rao G, Dunnen Den WFA, Boddeke HWGM, Phillips HS, Nakano I, Lang FF, Colman H, Sulman EP, Aldape K. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell. 2013 Aug;24:331-46.
28. Tanaka K, Babic I, Nathanson D, Akhavan D, Guo D, Gini B, Dang J, Zhu S, Yang H, De Jesus J, Amzajerdi AN, Zhang Y, Dibble CC, Dan H, Rinkenbaugh A, Yong WH, Vinters HV, Gera JF, Cavenee WK, Cloughesy TF, Manning BD, Baldwin AS, Mischel PS. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer Discovery. 2011 Nov;1:524-38.
29. Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, Kelly MO, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN, Network TCGAR. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010 Jan 19;17:98-110.
30. Peñuelas S, Anido J, Prieto-Sanchez RM, Folch G, Barba I, Cuartas I, Garcia-Dorado D, Poca MA, Sahuquillo J, Baselga J, Seoane J. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009 Apr 7;15:315-27.
31. Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-Box factors. Stem Cell. 2009 Nov 6;5:504-14.
32. Wang H, Pan J-Q, Luo L, Ning X-J, Ye Z-P, Yu Z, Li W-S. NF-kappaB induces miR-148a to sustain TGF-beta/Smad signaling activation in glioblastoma. Mol Cancer. 2015;14:2.
33. Song L, Liu L, Wu Z, Li Y, Ying Z, Lin C, Wu J, Hu B, Cheng S-Y, Li M, Li J. TGF-beta induces miR-182 to sustain NF-kappaB activation in glioma subsets. J Clin Invest. 2012 Sep 24;122:3563-2578.
34. Bredel M, Scholtens DM, Yadav AK, Alvarez AA, Renfrow JJ, Chandler JP, Yu ILY, Carro MS, Dai F, Tagge MJ, Ferrarese R, Bredel C, Phillips HS, Lukac PJ, Robe PA, Weyerbrock A, Vogel H, Dubner S, Mobley B, He X, Scheck AC, Sikic BI, Aldape KD, Chakravarti A, Harsh GR IV. NFKBIA deletion in glioblastomas. N Engl J Med. 2011 Feb 17;364:627-37.
35. Jiang L, Lin C, Song L, Wu J, Chen B, Ying Z, Fang L, Yan X, He M, Li J, Li M. MicroRNA-30e* promotes human glioma cell invasiveness in an orthotopic xenotransplantation model by disrupting the NF-kappaB/IkappaBalpha negative feedback loop. J Clin Invest. 2012 Jan 3;122:33-47.
36. Friedmann-Morvinski D, Narasimamurthy R, Xia Y, Myskiw C, Soda Y, Verma IM. Targeting NF-kappaB in glioblastoma: A therapeutic approach. Sci Adv. 2016 Jan;2:e1501292.
37. Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, Luger SM, Jordan CT. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001 Oct 15;98:2301-7.
38. Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat Commun. 2011 Jan 18;2:162.
39. Kendellen MF, Bradford JW, Lawrence CL, Clark KS, Baldwin AS. Canonical and non-canonical NF-kappaB signaling promotes breast cancer tumor-initiating cells. Oncogene. 2013 Mar 11;33:1297-305.
40. Murohashi M, Hinohara K, Kuroda M, Isagawa T, Tsuji S, Kobayashi S, Umezawa K, Tojo A, Aburatani H, Gotoh N. Gene set enrichment analysis provides insight into novel signalling pathways in breast cancer stem cells. Br J Cancer. 2010 Jan 5;102:206-12.
41. Leizer AL, Alvero AB, Fu HH, Holmberg JC, Cheng Y-C, Silasi D-A, Rutherford T, Mor G. Regulation of inflammation by the NF-kappaB Pathway in ovarian cancer stem cells. Am J Reprod Immunol. 2010 Sep 6;65:438-47.
42. Pratt MAC, Tibbo E, Robertson SJ, Jansson D, Hurst K, Perez-Iratxeta C, Lau R, Niu MY. The canonical NF-kappaB pathway is required for formation of luminal mammary neoplasias and is activated in the mammary progenitor population. Oncogene. Nature Publishing Group. Nature Publishing Group; 2009 Jun 1;28:2710-22.
43. Wang H, Lathia JD, Wu Q, Wang J, Li Z, Heddleston JM, Eyler CE, Elderbroom J, Gallagher J, Schuschu J, MacSwords J, Cao Y, McLendon RE, Wang X-F, Hjelmeland AB, Rich JN. Targeting interleukin-6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 2009 Oct;27:2393-404.
44. Hjelmeland AB, Wu Q, Wickman S, Eyler C, Heddleston J, Shi Q, Lathia JD, MacSwords J, Lee J, McLendon RE, Rich JN. Targeting A20 decreases glioma stem cell survival and tumor growth. PLoS Biol. 2010 Feb 23;8:e1000319.
45. Garner JM, Fan M, Yang CH, Du Z, Sims M, Davidoff AM, Pfeffer LM. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor-kappaB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J Biol Chem. 2013 Sep 6;288:26167-76.
46. Nogueira L, Ruiz-Ontañon P, Vazquez-Barquero A, Lafarga M, Berciano MT, Aldaz B, Grande L, Casafont I, Segura V, Robles EF, Suarez D, Garcia LF, Martinez-Climent JA, Fernandez-Luna JL. Blockade of the NFB pathway drives differentiating glioblastoma-initiating cells into senescence both in vitro and in vivo. Oncogene. 2011 Aug 11;30:3537-48.
47. Ziegelbauer K, Gantner F, Lukacs NW, Berlin A, Fuchikami K, Niki T, Sakai K, Inbe H, Takeshita K, Ishimori M, Komura H, Murata T, Lowinger T, Bacon KB. A selective novel low-molecular-weight inhibitor of IkappaB kinase-beta (IKK-beta) prevents pulmonary inflammation and show broad anti-inflammatory activity. Br J Pharmacol. 2005 May;145:178-92.
48. Lombardi L, Ciana P, Cappellini C, Trecca D, Guerrini L, Migliazza A, Maiolo AT, Neri A. Structural and functional characterization of the promoter regions of the NFKB2 gene. Nucleic Acids Res. 1995 Jun 25;23:2328-36.
49. Bren GD, Solan NJ, Miyoshi H, Pennington KN, Pobst LJ, Paya CV. Transcription of the RelB gene is regulated by NF-B. Oncogene. 2001;20:7722-33.
50. Takaesu G, Surabhi RM, Park K-J, Ninomiya-Tsuji J, Matsumoto K, Gaynor RB. TAK1 is critical for IkappaB kinase-mediated activation of the NF-kappaB pathway. J Mol Biol. 2003 Feb;326:105-15.
51. Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999 Mar 18;398:252-6.
52. Mizukami J, Takaesu G, Akatsuka H, Sakurai H, Ninomiya-Tsuji J, Matsumoto K, Sakurai N. Receptor activator of NF-kappaB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol Cell Biol. 2002;22:992-1000.
53. Melisi D, Xia Q, Paradiso G, Ling J, Moccia T, Carbone C, Budillon A, Abbruzzese JL, Chiao PJ. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J Natl Cancer Inst. 2011 Aug 3;103:1190-204.
54. Bosman MCJ, Schepers H, Jaques J, Brouwers-Vos AZ, Quax WJ, Schuringa JJ, Vellenga E. The TAK1-NF-kappaB axis as therapeutic target for AML. Blood. 2014 Nov 13;124:3130-40.
55. Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XHF, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, Massagué J. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014 Feb;156:1002-16.
56. Miao H, Gale NW, Guo H, Qian J, Petty A, Kaspar J, Murphy AJ, Valenzuela DM, Yancopoulos G, Hambardzumyan D, Lathia JD, Rich JN, Lee J, Wang B. EphA2 promotes infiltrative invasion of glioma stem cells in vivo through cross-talk with Akt and regulates stem cell properties. Oncogene. 2014 Feb 3;34:558-67.
57. Dunn DE, He DN, Yang P, Johansen M, Newman RA, Lo DC. In vitro and in vivo neuroprotective activity of the cardiac glycoside oleandrin from Nerium oleander in brain slice-based stroke models. J Neurochem. 2011 Sep 26;119:805-14.
58. Gustin JA, Maehama T, Dixon JE, Donner DB. The PTEN tumor suppressor protein inhibits tumor necrosis factor-induced nuclear factor-kappaB Activity. J Biol Chem. 2001 Jul 20;276:27740-4.
59. Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SAG. The PI3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene. 2004 Sep 27;23:8571-80.
60. Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H, Fletcher-Sananikone E, Zhang H, Liu Y, Wang W, Ren X, Zheng H, Kimmelman AC, Paik JH, Lim C, Perry SR, Jiang S, Malinn B, Protopopov A, Colla S, Xiao Y, Hezel AF, Bardeesy N, Turley SJ, Wang YA, Chin L, Thayer SP, DePinho RA. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discovery. 2011 Jul 17;1:158-69.
61. Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang L, You MJ, Koh MY, Cote G, Aldape K, Li Y, Verma IM, Chiao PJ, Lu Z. EGFR-induced and PKC monoubiquitylation-dependent NF-kappaB activation upregulates PKM2 expression and promotes tumorigenesis. Mol Cell. 2012 Dec;48:771-84.
62. Bonavia R, Inda MM, Vandenberg S, Cheng S-Y, Nagane M, Hadwiger P, Tan P, Sah DWY, Cavenee WK, Furnari FB. EGFRvIII promotes glioma angiogenesis and growth through the NF-B, interleukin-8 pathway. Oncogene. 2011 Dec 5;31:4054-66.
63. Rinkenbaugh A, Baldwin AS. Monoallelic deletion of NFKBIA in glioblastoma: when less is more. Cancer Cell. 2011 Feb 15;19:163-5.
64. Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, Gumin J, Henry V, Colman H, Priebe W, Sawaya R, Lang FF, Heimberger AB. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010 Jan 12;9:67-78.
65. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology. 2010 Oct 21;12:1113-25.
66. Wang Y, Xu J, Gao G, Li J, Huang H, Jin H, Zhu J, Che X, Huang C. Tumor-suppressor NFB2 p100 interacts with ERK2 and stabilizes PTEN mRNA via inhibition of miR-494. Oncogene. 2015 Dec 21;35:4080-90.
67. Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, Ragoussis J, Udalova IA, Smale ST, Bulyk ML. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappaB family DNA binding. Nature Immunol. 2011 Nov 20;13:95-102.
68. Wang VY-F, Huang W, Asagiri M, Spann N, Hoffmann A, Glass C, Ghosh G. The transcriptional specificity of NF-kappaB dimers is coded within the kappaB DNA response elements. 2012 Oct;2:824-39.
69. Wang J, Wang X, Hussain S, Zheng Y, Sanjabi S, Ouaaz F, Beg AA. Distinct roles of different NF-kappaB subunits in regulating inflammatory and T cell stimulatory gene expression in dendritic cells. J Immunol. 2007 May 18;178:6777-88.
70. Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004 Aug 20;118:453-64.
71. Fusco AJ, Huang D-B, Miller D, Wang VY-F, Vu D, Ghosh G. NF-kappaB p52:RelB heterodimer recognizes two classes of kappaB sites with two distinct modes. EMBO Rep. 2008 Dec 19;10:152-9.
72. Fujita T, Nolan GP, Ghosh S, Baltimore D. Independent modes of transcriptional activation by the p50 and p65 subunits of NF-kappa B. Genes Dev. 1992 May;6:775-87.
73. Lee DW, Ramakrishnan D, Valenta J, Parney IF, Bayless KJ, Sitcheran R. The NF-kappaB RelB protein is an oncogenic driver of mesenchymal glioma. PLoS ONE. 2013 Feb 25;8:e57489.
74. Li Y, Zhou Q-L, Sun W, Chandrasekharan P, Cheng HS, Ying Z, Lakshmanan M, Raju A, Tenen DG, Cheng S-Y, Chuang K-H, Li J, Prabhakar S, Li M, Tergaonkar V. Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol. 2015 Sep 21;17:1327-38.
75. Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NF-kappaB activation. Cancer Cell. 2002;1:493-503.
76. Kashatus D. Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev. 2006 Jan 15;20:225-35.
77. Park S, Hatanpaa KJ, Xie Y, Mickey BE, Madden CJ, Raisanen JM, Ramnarain DB, Xiao G, Saha D, Boothman DA, Zhao D, Bachoo RM, Pieper RO, Habib AA. The receptor interacting protein 1 inhibits p53 induction through NF-kappaB activation and confers a worse prognosis in glioblastoma. Cancer Res. 2009 Mar 24;69:2809-16.
78. Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor B and its significance in prostate cancer. Oncogene. 2001;20:7342-51.
79. Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappaB transcription factor. Mol Cell Biol. 1990 May;10:2327-34.
80. Kunsch C, Rosen CA. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol. 1993 Oct;13:6137-46.
81. Farina AR, Tacconelli A, Vacca A, Maroder M, Gulino A, Mackay AR. Transcriptional up-regulation of matrix metalloproteinase-9 expression during spontaneous epithelial to neuroblast phenotype conversion by SK-N-SH neuroblastoma cells, involved in enhanced invasivity, depends upon GT-box and nuclear factor kappaB elements. Cell Growth Differ. 1999 May;10:353-67.
82. Han YP, Tuan TL, Wu H, Hughes M, Garner WL. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-(kappa)B mediated induction of MT1-MMP. J Cell Sci. 2001 Jan;114:131-9.
83. Lavon I, Fuchs D, Zrihan D, Efroni G, Zelikovitch B, Fellig Y, Siegal T. Novel mechanism whereby nuclear factor-kappaB mediates DNA damage repair through regulation of O6-methylguanine-DNA-methyltransferase. Cancer Res. 2007 Sep 15;67:8952-9.
84. Bredel M, Bredel C, Juric D, Duran GE, Yu RX, Harsh GR, Vogel H, Recht LD, Scheck AC, Sikic BI. Tumor necrosis factor-alpha-induced protein 3 as a putative regulator of nuclear factor-kappaB-mediated resistance to O6-alkylating agents in human glioblastomas. J Clin Oncol. 2006 Jan 10;24:274-87.
85. Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappaB elements. J Biol Chem. 1992 Sep 5;267:17971-6.
86. Sonoda Y, Ozawa T, Aldape KD, Deen DF, Berger MS, Pieper RO. Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res. 2001 Sep 15;61:6674-8.
87. Sonoda Y, Ozawa T, Hirose Y, Aldape KD, McMahon M, Berger MS, Pieper RO. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 2001 Jul 1;61:4956-60.
88. Baldwin AS, Azizkhan JC, Jensen DE, Beg AA, Coodly LR. Induction of NF-kappa B DNA-binding activity during the G0-to-G1 transition in mouse fibroblasts. Mol Cell Biol. 1991 Oct;11:4943-51.
89. Hu Y, Smyth GK. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009 Aug;347:70-8.