
INTRODUCTION
The majority of colorectal cancers up-regulate the Wnt signalling pathway [1]. This is commonly due to mutation of the APC gene which is uncommonly mutated in BRAF mutant serrated pathway cancers [2]. Serrated neoplasia accounts for 25-30% of all colorectal cancers and these arise from sessile serrated adenomas (SSAs) or traditional serrated adenomas (TSAs) [3, 4]. Approximately half of all BRAF mutant serrated pathway cancers will methylate the mismatch repair gene MLH1 and develop microsatellite instability (MSI) [5, 6], whilst the remainder are microsatellite stable (MSS). A recent study reported germline mutation of the upstream Wnt inhibitor RNF43 in a minority of subjects with serrated polyposis [7, 8], providing a potential alternative to APC mutation as a mechanism for altering the Wnt signal in serrated neoplasia.
RNF43 is a transmembrane E3 ubiquitin ligase that is up-regulated in response to accumulation of β-catenin following increased Wnt signalling [8, 9]. Together with its functional homologue, ZNRF3, it localizes to the plasma membrane and targets Frizzled (FZD), a key receptor of the Wnt ligand, for ubiquitination and lysosomal degradation [8]. RNF43 and ZNRF3 are highly expressed in murine intestinal stem cells, and deletion of these renders cells hypersensitive to Wnt ligand secretion, resulting in intestinal adenomas with strong expression of β-catenin [8].
Previous studies have found RNF43 to be functionally mutated in pancreatic cysts [10, 11] and mucinous ovarian cancer [12]. Frequent frameshift mutations in RNF43 have been associated with MSI gastric, colorectal and endometrial cancers [13–15]. Loss of heterozygosity at the RNF43 locus, 17q22, has been observed in the pancreatic cyst, intraductal papillary mucinous neoplasms (IPMNs) [10, 16] and copy number variations of ZNRF3, predominantly consisting of homozygous deletions at its 22q12.1 locus, have been found in adrenocortical carcinoma and osteoblastoma [17–19].
Release of the Wnt ligand from a signalling cell is dependent on the molecule Porcupine (PORCN). PORCN facilitates post-translational palmitoylation of the Wnt ligand which is necessary for its transport out of the cell and subsequent recognition by a FZD receptor on a receiving cell. Treatment with a Porcupine inhibitor reduces growth of RNF43 mutant pancreatic cancer cell lines, where proliferation is dependent on endogenous Wnt signalling [20]. The porcupine inhibitor LGK974 has been shown to dramatically reduce growth of pancreatic cancer cells that carry an RNF43 mutation [20].
We analysed RNF43 and ZNRF3 for mutation, copy number variation and expression in a large number of colorectal cancers stratified for molecular subtype. We hypothesized that in serrated pathway cancers where APC mutation is uncommon, inactivation of RNF43 and/or ZNRF3 would present an alternate mechanism for activating the Wnt signal.
RESULTS
Clinicopathological data of cancer cohorts
Cancers displayed typical clinicopathological features when stratified for molecular subtype as previously described [21–23]. Patients with BRAF mutant/MSI cancers presented at significantly older ages compared to both MSS subtypes. BRAF mutant cancers were predominantly present in the proximal colons of females and displayed the methylator phenotype more commonly than BRAF wild type cancers (Table 1).
Table 1: Clinical and Molecular data of Cancers
BRAF mutant/MSI |
BRAF mutant/MSS |
BRAF wild type /MSS |
p value |
P ValueA |
P ValueB |
P ValueC |
|
|---|---|---|---|---|---|---|---|
RBWH cancers: |
|||||||
n |
54 |
33 |
79 |
- |
- |
- |
- |
Average age (yrs) |
74.6 |
68.4 |
68.2 |
0.005 |
0.026 |
0.003 |
0.900 |
Female gender |
42/54 (77.8%) |
17/33 (51.5%) |
33/79 (18.4%) |
0.0002 |
0.017 |
<0.0001 |
0.407 |
Proximal site |
41/43 (95.3%) |
17/24 (70.8%) |
15/63 (23.8%) |
<0.0001 |
0.008 |
<0.0001 |
0.0001 |
CIMP high |
54/54 (100%) |
32/33 (96.7%) |
12/79 (15.1%) |
<0.0001 |
0.379 |
<0.0001 |
<0.0001 |
Envoi cancers: |
|||||||
n |
63 |
37 |
44 |
- |
- |
- |
- |
Average age (yrs) |
77.9 |
72.6 |
63.6 |
<0.0001 |
0.01 |
<0.0001 |
0.004 |
Female gender |
37/57 (64.9%) |
26/37 (70.2%) |
13/28 (46.4%) |
0.12 |
0.66 |
0.16 |
0.07 |
Proximal site |
53/57 (93.0%) |
30/37 (81.1%) |
9/28 (32.1%) |
<0.0001 |
0.10 |
<0.0001 |
<0.0001 |
CIMP high |
59/63 (93.7%) |
23/35 (65.7%) |
0/35 (0%) |
<0.0001 |
0.001 |
<0.0001 |
<0.0001 |
P ValueA: P value between BRAF mutant/MSI and BRAF mutant/MSS cohorts; P ValueB: P value between BRAF mutant/MSI and BRAF wild type cohorts; P ValueC: P value between BRAF mutant/MSS and BRAF wild type cohorts
Mutation frequencies of RNF43 and ZNRF3 vary by molecular subtype
The presence of RNF43 mutations was examined in 54 BRAF mutant/MSI, 33 BRAF mutant/MSS and 79 BRAF wild type cancers. RNF43 was frequently mutated in BRAF mutant/MSI cancers (47/54; 87.0%). BRAF mutant/MSS cancers had a moderate proportion of RNF43 mutations (8/33; 24.2%), whilst BRAF wild type cancers had the least frequent mutations compared to the BRAF mutant subgroups (3/79, 3.8%) (p<0.0001) (Table 2).
Table 2: Frequency and type of RNF43 mutations
Cohort |
n |
Number of cancers mutated |
Total number of mutations |
X659 frameshift mutations |
X117 frameshift mutations |
Other frameshift mutations |
Mis/nonsense mutations |
|---|---|---|---|---|---|---|---|
BRAF mutant /MSI |
54 |
47 (87.0%) |
65 |
43 (79.6%) |
6 (11.1%) |
9 (16.7%) |
7 (13.0%) |
BRAF mutant /MSS |
33 |
8 (24.2%) |
8 |
1 (3.0%) |
0 |
5 (15.2%) |
2 (6.1%) |
BRAF wild type /MSS |
79 |
3 (3.8%) |
3 |
0 |
0 |
0 |
3 (3.8%) |
P value for number of mutations: Overall p<0.0001; BRAF mutant/MSI vs BRAF mutant/MSS p<0.0001; BRAF mutant/MSI vs BRAF wild type p<0.0001; BRAF mutant/MSS vs BRAF wild type p=0.002
The most common type of RNF43 mutation was a frameshift at nucleotide 659 (G7 repeat tract) in exon 9 that occurred in 43/54 (79.6%) BRAF mutant/MSI, in 1/33 (3.0%) BRAF mutant/MSS and 0/79 BRAF wild type cancers (Table 2). The high frequency of this particular mutation was further validated in the second series of formalin fixed cancers where it was found in 25/35 (71.4%) BRAF mutant/MSI, 1/12 (8.3%) BRAF mutant/MSS and 0/22 BRAF wild type cancers (p<0.0001).
The prevalence of other mutations is shown in Table 2 and Figure 1A. MSI cancers tended to show mutations in repeat tracts. In addition to the G659 (G7 deletion), recurrent frameshift mutations were present at R117 (C6 repeat tract) in exon 3 and affected 6/54 (11.1%) of BRAF mutant/MSI cancers. Other recurrent mutations in BRAF mutant/MSI cancers were X441fs (n=3), X370fs (n=2) and G363D (n=2). Eight BRAF mutant/MSS cancers (8/33, 24.2%) had RNF43 mutations including 2 point mutations and 6 frameshift mutations that were located throughout the gene. BRAF wild type cancers had a minimal mutation rate of 3/79 (3.8%) and all were missense mutations in exon 9 (Table 2, Figure 1A).
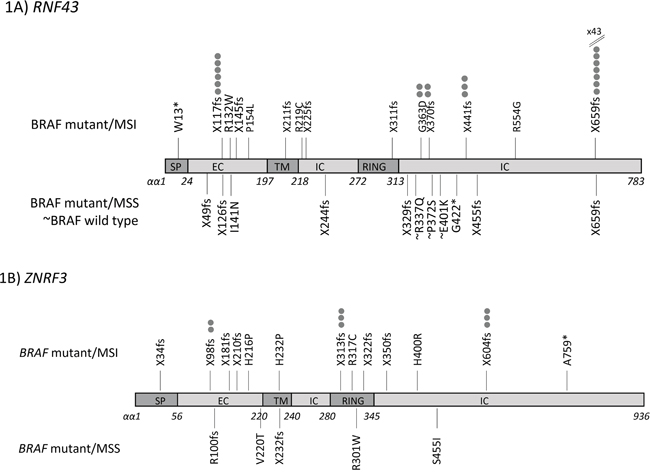
Figure 1: Mutation location map across coding sequences for A. RNF43 and B. ZNRF3 in colorectal cancer subtypes.
ZNRF3 mutations also associated with the BRAF V600E mutation (MSI: 16/54, 29.6%; MSS: 5/33, 15.2%, p=0.2) (Table 3, Figure 1B). There were no ZNRF3 mutations in the BRAF wild type cohort (vs BRAF mutant/MSI p=0.0008; vs BRAF mutant/MSS p=0.06) (Table 3). Common ZNRF3 frameshift mutations were seen at P313 (C5 deletion) and G604 (G6 deletion) with 3 of each seen in BRAF mutant/MSI cancers (Table 3). Point mutations were found in 5 BRAF mutant/MSI and 4 BRAF mutant/MSS cancers, and these were located along the length of the gene (Figure 1B). RNF43 or ZNRF3 mutations were not associated with other clinical or molecular parameters (Supplementary Table S1).
Table 3: Frequency and type of ZNRF3 mutations
Cohort |
n |
Number of cancers mutated |
Total number of mutations |
Frameshift mutations |
Mis/nonsense (%A) (%B) |
|---|---|---|---|---|---|
BRAF mutant /MSI |
54 |
16 (29.6%) |
18 |
13 (24.1%) |
5 (9.3%) |
BRAF mutant /MSS |
33 |
5 (15.2%) |
5 |
1 (3.0%) |
4 (9.1%) |
BRAF wild type /MSS |
27 |
0 |
0 |
0 |
0 |
P value for number of mutations: Overall p=0.004; BRAF mutant/MSI vs BRAF mutant/MSS p=0.2; BRAF mutant/MSI vs BRAF wild type p=0.008; BRAF mutant/MSS vs BRAF wild type p=0.06
Ten colorectal cancer cell lines were analysed for presence of an RNF43 and ZNRF3 mutation. RNF43 mutations were found in five cell lines (RKO, SW48, DLD1, HCT116, LS174T) which are all MSI. RKO, SW48 and DLD1 harboured the X659fs mutation, with RKO showing a X659fs homozygous mutation. Additional mutations were seen in SW48 (X299fs) and DLD1 (L214M). HCT116 had a homozygous X117fs and a heterozygous X606fs mutation. LS174T had two missense mutations (K108E, R389H). RKO and LISP1 were the only cell lines harbouring ZNRF3 mutations. RKO had 1 frameshift and 1 missense mutation (X249fs, G481W), whilst LISP1 had a missense mutation (H461R).
RNF43 and ZNRF3 transcript expression is upregulated in BRAF wild type compared to BRAF mutant colorectal cancers
From those cancers that were assessed for presence of mutation, a subset of cancers were examined for RNF43 and ZNRF3 transcript expression levels. Analysis revealed significantly higher expression for both RNF43 and ZNRF3 in BRAF wild type compared to BRAF mutant cancers and normal colorectal mucosa samples (p<0.0001 for both genes) (Figure 2A and 2B). RNF43 transcript expression was lower in RNF43 mutant compared to RNF43 wild type cancers (mean of 6.91 and 8.49 respectively; p=0.0002) (Supplementary Table S2A). Cancers with an X659fs mutation had lower expression compared to cancers harbouring other mutations (average of 6.5 and 8.15 respectively; p=0.01) and compared to RNF43 wild type cancers (p<0.0001) (Supplementary Table S2A). Similarly, transcript expression of ZNRF3 mutant cancers was significantly lower than ZNRF3 wild type cancers (average of 6.16 and 7.02 respectively; p=0.009) (Supplementary Table S2B).
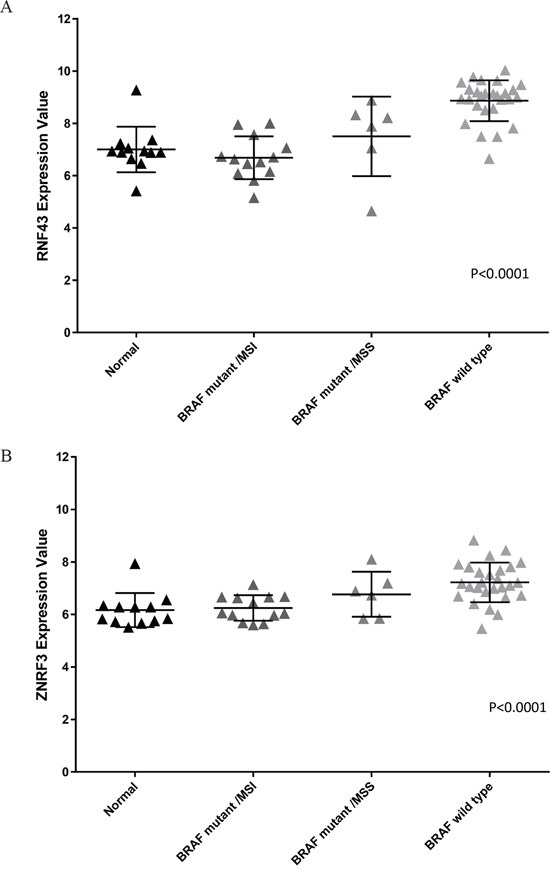
Figure 2: Transcript expression for A. RNF43 and B. ZNRF3 between cancer cohorts. BRAF wild type cancers have increased expression of both RNF43 and ZNRF3 compared to BRAF mutant/MSI and BRAF mutant/MSS subgroups and normals (p<0.0001 for RNF43 and ZNRF3).
Structural variations in RNF43 and ZNRF3 between cohorts
At the 17q22 RNF43 locus, BRAF mutant/MSS cancers had significantly more frequent deletion events (at 18/33, 54.5%) than BRAF wild type cancers (4/18, 22.2%; p=0.04). BRAF mutant/MSI cancers had a low rate of deletion (4/30, 13.3%), which was expected since MSI cancers are known to be diploid [22].
At the ZNRF3 locus, 22q12.1, there was a high frequency of deletion events in MSS cancers (BRAF mutant/MSS: 16/33, 48.5%; BRAF wild type: 10/18, 55.6%). In addition to frequent loss of heterozygosity of ZNRF3 occurring in the BRAF mutant/MSS cohort, one of these cancers harboured a homozygous deletion spanning approximately 170kb and including the C terminal half of the ZNRF3 locus. Again BRAF mutant/MSI cancers rarely showed deletion events at this locus (1/30, 3.3%).
RNF43 cytoplasmic expression is reduced in BRAF mutant/MSI cancers
Immunohistochemical staining was performed to detect presence of RNF43 protein expression for 63 BRAF mutant/MSI, 37 BRAF mutant/MSS and 44 BRAF wild type cancers from the formalin fixed series. Cytoplasmic staining was least frequent in the BRAF mutant/MSI compared to BRAF mutant/MSS and BRAF wild type cancers (p=0.009) (Table 4A, Figure 3A and 3B). Cytoplasmic RNF43 staining was significantly less common in cancers harbouring the X659fs mutation compared to that in wild type cancers (RNF43 cytoplasmic positive cancers: 11/27, 41% vs 35/49, 71% respectively, p=0.01). No difference in nuclear RNF43 expression was found between the X659fs mutant and wild type cancers (Table 4B).
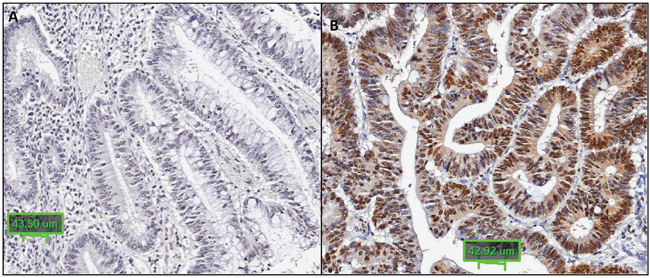
Figure 3: Representative images of RNF43 immunohistochemistry. RNF43 immunohistochemistry showing: A. negative cytoplasmic and nuclear staining in a BRAF mutant/MSI cancer; B. positive cytoplasmic and nuclear staining in a BRAF wild type cancer (x20).
Table 4: Immunohistochemical analysis of RNF43 expression between cohorts
A. Immunohistochemical analysis of RNF43 cytoplasmic and nuclear expression between cohorts
BRAF mutant/MSI |
BRAF mutant/MSS |
BRAF wild type |
p valueA |
p valueB |
p valueC |
p valueD |
|
|---|---|---|---|---|---|---|---|
Cytoplasm |
31/63 (49.2%) |
27/37 (73.0%) |
33/44 (75.0%) |
0.009 |
0.02 |
0.009 |
1.00 |
Nucleus |
36/63 (57.1%) |
30/36 (83.3%) |
26/43 (60.5%) |
0.02 |
0.008 |
0.84 |
<0.05 |
A p value overall; B p value between BRAF mutant/MSI and BRAF mutant/MSS; C p value between BRAF mutant/MSI and BRAF wild type; D p value between BRAF mutant/MSS and BRAF wild type
B. Immunohistochemical analysis of RNF43 in cancers harbouring an X659fs mutation
RNF43 IHC |
X659fs mutant n=27 |
Wild type n=49 |
P Value |
|---|---|---|---|
cytoplasmic positive (2-3) |
11/27 (40.7%) |
35/49 (71.4%) |
p=0.01 |
nuclear positive (2-3) |
16/27 (59.3%) |
33/27 (67.3%) |
p=0.6 |
β-catenin immunohistochemical staining was assessed, with nuclear accumulation indicative of elevated Wnt signal. Overall, β-catenin was more likely to be nuclear in BRAF wildtype compared to BRAF mutant cancers (27/28, 96.4% versus 36/92, 39.1%, P<0.0001). In BRAF wild type cancers with nuclear β-catenin staining, 19/27 (70.4%) showed a concomitant increase in RNF43 cytoplasmic expression, which may represent a futile attempt to dampen the Wnt signal by RNF43-mediated degradation of the Frizzled receptor. In BRAF mutant/MSI cancers with normal staining for β-catenin, 21/33 (63.6%) also maintained normal RNF43 staining compared to only 4/23 (17.4%) BRAF mutant/MSS cancers (P<0.001). This likely reflects the higher RNF43 mutation rate in MSI cancers and therefore the inability to upregulate RNF43 in response to elevated Wnt signal, as is common in BRAF wild type cancers.
Response to Porcupine inhibitor, LGK974, for RNF43 and ZNRF3 mutant cell lines
Cell lines were seeded at varying densities according to growth rate to promote long term colony formation. Treatment with the porcupine inhibitor, LGK974, at various concentrations (at 5uM, 10uM and 20uM) was replenished every 48 hours for approximately 2 weeks and colonies were subsequently fixed, stained and counted by two independent examiners. In all RNF43 and ZNRF3 mutant cell lines, colony formation was inhibited in a dose-dependent manner of up to 53% with LGK974 treatment compared to growth seen in the DMSO treated control cells (Figure 4A; Supplementary Figure S1A). HCT116 had the least growth inhibition with LGK974 treatment, whilst RKO and LS174T demonstrated the most. There was no overall decrease of growth with LGK974 treatment observed in any of the RNF43 / ZNRF3 wild type cell lines (Figure 4A).
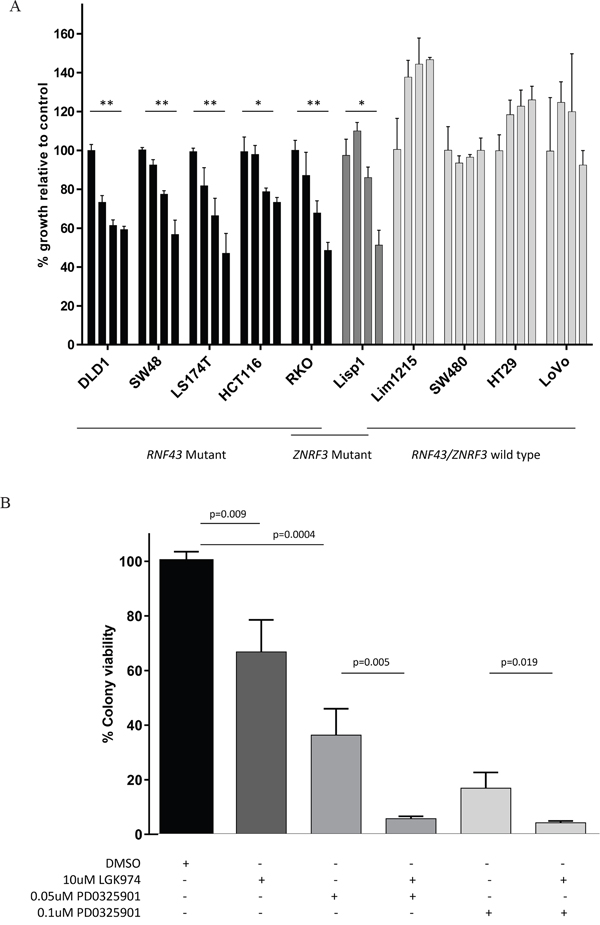
Figure 4: Cell colony growth of colorectal cancer cell lines following treatment with Porcupine inhibitor, LGK974 (A); and in combination with MEK inhibitor, PD0325901 (B). A. Cell lines harbouring an RNF43 and/or ZNRF3 mutation showed significantly reduced cell colony formation following treatment with the Porcupine inhibitor, LGK974 in a dose dependent manner. There was no decrease of cell colony formation in wild type cell lines with LGK974 treatment. The graph shows the percentage of cell growth relative to the control. The bars per cell line correspond to treatment with 1) control (DMSO), 2)5 uM LGK974, 3)10 uM LGK974 and 4)20 uM LGK974. Significance of decreased cell line growth following LGK974 treatment is shown (** p<0.001; *p<0.01). Standard deviation error bars are shown. B. When LGK974 treatment was combined with a MEK inhibitor, PD0325901 in the RNF43/ZNRF3 mutant cell line, RKO, there was further abrogation of cell colony growth in a dose dependent manner compared to either single treatment. Standard deviation error bars are shown.
Combination of Porcupine and MEK inhibitors accentuate growth inhibition in RKO cells
The RKO cell line most closely resembles primary cancers of the serrated pathway as it is BRAF mutant, MSI and CIMP high [24, 25]. RKO harbours a homozygous RNF43 X659 frameshift mutation and two heterozygous ZNRF3 mutations (X249fs, G481W). Treatment with 10μM LGK974 decreased colony growth by approximately 30% (Figure 4A and 4B), and treatment with the MEK inhibitor, PD0325901, reduced colony growth by 64% at 0.05uM and 84% at 0.1uM (Figure 4B). The combination of LGK974 and PD0325901 synergised to further reduce RKO’s cell colony growth (Figure 4B), and this was in line with markedly reduced phosphorylation of ERK1/2 and downstream c-MYC (Supplementary Figure S2). These results demonstrate the potential effectiveness of this novel combination therapy in treating RNF43 / BRAF mutant serrated pathway cancers. (Images of cells treated with 0.002uM – 0.5uM with and without LGK974 application are shown in Supplementary Figure S1B).
DISCUSSION
The E3 ubiquitin ligases RNF43 and ZNRF3 normally cooperate to downregulate the Wnt signal by targeting the Frizzled receptor for degradation. This study provides a comprehensive analysis of RNF43 and ZNRF3 in cohorts of colorectal cancers stratified by BRAF mutation and MSI status. The high rate of mutation and structural variation in BRAF mutant cancers suggests alteration of these genes is important for progression of serrated pathway cancers, where Wnt activation by APC mutation is uncommon [2].
RNF43 was commonly mutated in BRAF mutant cancers compared to BRAF wild type cancers. The functional significance of mutations occurring in the proximal region of the gene have previously been established, where Schwank et al [26] demonstrated R-spondin independent growth in murine colonic organoids. Here we report 71-80% of MSI cancers from two independent series had an x659 frameshift mutation in the distal region of the gene. This is predicted to result in a premature stop codon and was also reported by Giannakis et al to occur in 64% of MSI cancers [18]. The high mutation rate suggests this mutations confers a positive selective advantage [13]. This was confirmed by Yan et al where biallelic RNF43 X659fs mutation, along with targeting of ZNRF3, in a murine colon organoid model conferred R-spondin independence [27].
Serrated neoplasia may originate in sessile serrated adenomas (SSA) or traditional serrated adenomas (SSA), the former being the precursor of MSI cancers and the latter preceding the development of BRAF mutant / MSS cancers [28, 29]. Sekine et al [30] reported RNF43 mutations in 6% SSA and 24% TSA, compared to our findings of 87% in BRAF mutant MSI and 24% BRAF mutant MSS cancers. This suggests that RNF43 may assist progression of SSA to MSI cancers, whereas RNF43 mutation may be involved at an earlier stage of TSA development. ZNRF3 mutations were not observed in SSAs and in only 1% of TSAs [30] compared to 30% BRAF mutant/MSI cancers and 15% BRAF mutant /MSS cancers, suggesting that this less common mutation is involved in progression to cancer rather than precursor development. BRAF mutant/MSS cancers also commonly showed deletion of the RNF43 locus at 17q22, which may contribute to gene silencing and compensate for the lower RNF43 / ZNRF3 mutation rate in this cancer subgroup.
Cytoplasmic RNF43 expression was least frequently observed in the BRAF mutant/MSI cancers. Because RNF43 abrogates the Wnt signal at the level of the Frizzled receptor, it may be expected to be localised predominantly in the cytoplasm or membrane rather than the nucleus. Whether its expression is transient and is degraded soon after functioning, or whether it is maintained within the cell for a period of time is unknown. RNF43 has been reportedly expressed in both the cytoplasm and nucleus of gastric cancer cells [14], the cytoplasm of gastric cancer and glioma cells [31, 32] as well as in the nuclear membrane and endoplasmic reticulum of cervical cancer cells [33]. A recent study described nuclear localization of RNF43 where it functioned to inhibit the Wnt pathway through its sequestering of TCF4, a transcription factor activated by beta-catenin target gene, to the nuclear membrane which prevented TCF4’s mediated transcription of genes involved in downstream Wnt signalling [34]. RNF43 may have various time and cell type specific roles each requiring a unique subcellular localization. In the current study, nuclear localisation of β-catenin was used as a surrogate for elevated Wnt signalling. This was observed in the majority of BRAF wildtype cancers where elevated cytoplasmic RNF43 staining was also observed, likely as a futile attempt to dampen the Wnt signal by RNF43-mediated degradation of the Frizzled receptor. BRAF wildtype cancers also showed a significant increase in level of transcript expression. MSI cancers were more likely to maintain a normal staining pattern for β-catenin and this was correlated with a lower incidence of abnormal cytoplasmic staining for RNF43, most likely due to the high RNF43 mutation rate in this subgroup.
The RNF43 and ZNRF3 mutant colorectal cancer cell lines were all MSI, and all harbour a mutation within the EGFR-MAPK signalling pathway. Treatment of these cells lines with the Porcupine inhibitor, LGK974, resulted in consistently and significantly reduced cellular proliferation compared to wild type cell lines. The RNF43 wild type cancer cell lines all harbour either APC or β-catenin mutations which would be predicted to render them insensitive to upstream inhibition of the Wnt signal, as was observed. The approximate 50% growth reduction for the RNF43 and ZNRF3 mutant cell lines was not as dramatic as was observed for an RNF43 mutant organoid model, where the colonoid was sensitive to the similar porcupine inhibitor, IWP2 [35]. This difference may reflect a different magnitude of reliance on endogenous Wnt signalling compared to the colorectal cell lines. Organoid models are more representative of in vivo cancer growth due to their three dimensional structure and shorter time in culture. The epigenetic instability of BRAF mutant cancers in particular can induce DNA methylation and silencing of other Wnt pathway molecules that may alter the sensitivity of the cells to an upstream porcupine inhibitor. The novel combination of Porcupine and MEK inhibition synergistically inhibited RKO cell growth by more than 90%, which suggests this may be a promising therapeutic strategy to treat BRAF / RNF43 / ZNRF3 mutant serrated pathway cancers.
Overall this study has identified that the E3 ubiquitin ligases, RNF43 and ZNRF3, are frequently mutated in BRAF mutant cancers of the serrated pathway, particularly those that are MSI. Heterozygous loss of the RNF43 and ZNRF3 loci were identified in BRAF mutant/MSS cancers. BRAF mutant cancers did not upregulate RNF43 or ZNRF3 expression at the transcript or protein level, unlike the BRAF wild type cancers that attempt to mitigate the Wnt signal. Functionally, it was shown that RNF43 as well as ZNRF3 mutant colorectal cancers may be Wnt ligand dependent and that inhibition of the Wnt signal by targeting Porcupine may facilitate decreased growth of a proportion of MSI serrated pathway cancers. Furthermore, the novel combination of Porcupine and MEK inhibition resulted in considerable growth reduction, which indicates this may be a promising treatment approach for patients with serrated pathway cancers harbouring RNF43 and/or ZNRF3 mutations.
MATERIALS AND METHODS
Patient demography and sample selection
Cancer and matched normal samples were obtained either as fresh frozen tissue from patients undergoing surgery at the Royal Brisbane and Women’s Hospital, or as formalin-fixed paraffin embedded (FFPE) tissue from Envoi Specialist Pathologists, Brisbane, Australia. This study was approved by the Royal Brisbane and Women’s Hospital and Bancroft Human Research Ethics Committees.
Clinicopathological data including gender, age at diagnosis and anatomical site of cancer (with proximal defined as proximal to the splenic flexure) were collected. DNA from fresh cancer and matched normal tissue was extracted using AllPrep DNA mini kit (Qiagen, Dusseldorf, Germany). DNA from the FFPE cancers was extracted by the Chelex-100 method (Bio-Rad Laboratories, CA, USA). The presence of MSI had been previously analysed for the RBWH cancer samples using the National Cancer Institute’s 5 marker panel [21, 36, 37]. Cancers from Envoi Specialist Pathologists were evaluated for immunohistochemical loss of MLH1 mismatch repair protein expression as a surrogate for MSI. Presence of the BRAF V600E (a1796t) mutation, p53 mutation (over exons 4-8) and KRAS mutation (over codons 2 and 3) had been previously investigated for the fresh RBWH samples [21] whilst BRAF V600E (a1796t) and KRAS (codons 2 and 3) mutations were analysed for the formalin fixed Envoi samples as previously described [21, 38–40].
Cell LinesA panel of colorectal cancer cell lines: DLD1, HCT116, HT29, Lim1215, LISP1, LoVo, LS174T, RKO, SW48 and SW480 were maintained in RPMI media supplemented with 10% fetal bovine serum (Bovogen Biologicals, Victoria, Australia) and 1% penicillin-streptomycin (Life Technologies, Carlsbad, USA). Cell lines were authenticated by short tandem repeat profiling to confirm authenticity of ATCC cell lines.
RNF43 and ZNRF3 mutation analysis
Sanger sequencing was analysed across the coding region of RNF43 (performed by Macrogen Incorporated, South Korea) in 87 BRAF mutant (54 MSI and 33 MSS) and 79 BRAF wild type cancers from the RBWH series. ZNRF3 was sequenced in all 87 BRAF mutant and an unselected subset of 27 BRAF wild type cancers. The second series of FFPE cancers from Envoi Specialist Pathologists consisting of 48 BRAF mutant (36 MSI and 12 MSS) and 29 BRAF wild type cancers were sequenced to further validate the frequency of the X659fs mutation (sequencing primers: F 5’GTCCAGGCCTCCTATTCCTC; R 5’ CTGGTAGCAGCCTCTTGTCC). Only mutations predicted to elicit a functional response (frameshift, missense, nonsense and splice mutations) were included in analyses, and mutations found intronically, in untranslated regions or silent mutations were excluded from further investigation.
Transcript expression
19 BRAF mutant (13 MSI, 6 MSS), 27 BRAF wild type and 12 matched normal mucosa samples were randomly chosen from the freshly collected RBWH series and analysed for transcript expression on the HumanHT-12 v4 Expression BeadChip arrays (Illumina, San Diego, CA). Total mRNA (500 ng) was reverse-transcribed, amplified and biotinylated using the Illumina TotalPrep-96 RNA Amp Kit (Ambion, Austin, TX). The labelled cRNA (750 ng) was hybridized to the BeadChip arrays followed by washing, blocking, and staining with streptavidin-Cy3 according to the manufacturer’s specifications. Fluorescence intensity for each probe on the array chips were measured on the iScan system and the data was extracted using GenomeStudio software (Illumina). All arrays passed quality control criteria in the GenomeStudio package as described by the manufacturer. A sample probe profile file generated in GenomeStudio without background subtraction or normalisation was used for subsequent analysis in R. The data was background corrected and quantile normalised using the limma package function. Probes with detection p-values P>0.05 in >95% of all samples were excluded from further analysis.
Single nucleotide polymorphism arrays
33 BRAF mutant/MSS, 30 BRAF mutant/MSI, 18 BRAF wild type/MSS cancers and matched normal samples all randomly chosen from the RBWH series were analysed for genome-wide copy number aberrations (CNAs) with HumanCytoSNP-12v2.1 Single Nucleotide Polymorphism (SNP) arrays (Illumina, San Diego, Ca.) according to the manufacturer’s instructions. The beadchips were scanned using Illumina’s iScan system and the image data was analysed with Illumina’s GenomeStudio version 2011.1.0.24550 and as previously described [22].
Immunohistochemical analysis
Tissue sections obtained from FFPE blocks from the Envoi series underwent antigen retrieval at low pH (pH6, Reveal decloaker; Biocare Medical, CA, USA) for 15mins at 105°C. H2O2 and Sniper were used to facilitate endogenous peroxidase and protein blocks respectively. Primary antibodies were manually applied and incubated: RNF43 antibody (anti-RNF43 HPA-008079; Sigma, St Louis, MO, USA) at 1/500 dilution for 1 hour and beta-catenin (anti-beta-catenin 224M16 [14] Cell Marque, California, USA) at 1/600 for 1 hour. MACH3 rabbit or mouse secondary antibody probe and polymer were applied for 15 and 30 minutes respectively (Biocare Medical, CA, USA), and DAB chromagen (Biocare Medical, CA, USA) was applied for 5-8 minutes.
Observation of samples was performed by an expert gastrointestinal pathologist (MB) and scored on a scale of 0-3 with 0 representing absent and 3 representing maximal staining. A score of 0-1 was considered negative and a score of 2-3 as positive. RNF43 staining within the cancer region of a sample section was compared to that observed within normal lymphocytes, which served as an internal control.
Cell colony assays
All cell lines were authenticated using short tandem repeat (STR) profiling in accordance with ATCC standards. Cell lines were last tested in June 2015.
Cell lines were seeded at varying densities in 6 well plates (HCT116, SW480 at 500 cells/well; DLD1, RKO, Lim1215 at 750 cells/well; SW48, Lisp1, LS174T, LoVo, HT29 at 1000 cells/well) and maintained in 2mls RPMI media as described above. 24 hours later, cells were treated in triplicate with either DMSO in control wells or the Porcuine inhibitor LGK974 (at 5uM, 10uM or 20uM in DMSO) (Cayman Chemicals, Michigan, USA) every 48 hours. At approximately 2 weeks, cells were fixed with ice-cold 100% methanol and stained with 0.5% crystal violet in 80% methanol. For LGK974 treatments, cell colonies were counted by 2 examiners and the counts were averaged. For combination treatments, RKO cells were treated at 24 hours post plating and then every 48 hours with either DMSO, 10uM LGK974, and/or the MEK inhibitor PD0325901 (Selleck Chemicals, Texus, USA) (at 0.002uM, 0.05uM, 0.1uM, 0.3uM or 0.5uM). Drug combination cell colony assays were quantitated using Aperio ImageScope v12.1.0.5019 with application of the Positive Pixel Count v9.1 algorithm.
Western blotting1X106 RKO cells were either treated singly or in-combination with LGK974 and PD0325901. DMSO used as a vehicle control. 48 hr post-treatment, cells were collected and lysed with 7M urea buffer (7M Urea, 1% SDS, 20-30 mM Tris pH 8, 100-150 mM NaCl). Western blotting was then performed according to standard protocol as described previously [41] using indicated antibodies. C-MYC (AB32072, Abcam), pERK1/2 (#4370), ERK1/2 (#4695), β-catenin (#9582, cell signalling) and Cox-IV (PN926-42214, Li-COR). The Super Signal chemiluminescent ECL-plus (Amersham) was used.
Statistical analysis
Categorical data was analysed for significance using Fisher’s exact test or Pearson’s chi-squared test where appropriate. Continuous data was analysed with either a student’s t-test or ANOVA as appropriate. P values <0.05 were considered significant.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare for this study or in the production of this manuscript.
ACKNOWLEDGMENTS AND GRANT SUPPORT
This study was funded by a National Health and Medical Research Council grant (NHMRC: 1050455) and by Pathology Queensland. Vicki Whitehall is supported by a Gastroenterological Society of Australia Senior Research Fellowship. Murugan Kalimutho is supported by a Cancer Council Queensland (CCQ) Project Grant (1087363).
REFERENCES
1. TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330-7.
2. Samowitz WS, Slattery ML, Sweeney C, Herrick J, Wolff RK, Albertsen H. APC mutations and other genetic and epigenetic changes in colon cancer. Mol Cancer Res. 2007; 5:165-70.
3. Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010; 138:2088-100.
4. Bettington M, Walker N, Clouston A, Brown I, Leggett B, Whitehall V. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013; 62:367-86.
5. Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer research. 1997; 57:808-11.
6. Koinuma K, Shitoh K, Miyakura Y, Furukawa T, Yamashita Y, Ota J, Ohki R, Choi YL, Wada T, Konishi F, Nagai H, Mano H. Mutations of BRAF are associated with extensive hMLH1 promoter methylation in sporadic colorectal carcinomas. Int J Cancer. 2004; 108:237-42.
7. Gala MK, Mizukami Y, Le LP, Moriichi K, Austin T, Yamamoto M, Lauwers GY, Bardeesy N, Chung DC. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology. 2014; 146:520-9.
8. Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJ, Maurice MM, Clevers H. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012; 488:665-9.
9. Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, Lei H, Mickanin C, Liu D, Ruffner H, Mao X, Ma Q, Zamponi R, et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature. 2012; 485:195-200.
10. Wu J, Jiao Y, Dal Molin M, Maitra A, de Wilde RF, Wood LD, Eshleman JR, Goggins MG, Wolfgang CL, Canto MI, Schulick RD, Edil BH, Choti MA, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A. 2011; 108:21188-93.
11. Sakamoto H, Kuboki Y, Hatori T, Yamamoto M, Sugiyama M, Shibata N, Shimizu K, Shiratori K, Furukawa T. Clinicopathological significance of somatic RNF43 mutation and aberrant expression of ring finger protein 43 in intraductal papillary mucinous neoplasms of the pancreas. Modern pathology. 2015; 28:261-7.
12. Ryland GL, Hunter SM, Doyle MA, Rowley SM, Christie M, Allan PE, Bowtell DD, Australian Ovarian Cancer Study G, Gorringe KL, Campbell IG. RNF43 is a tumour suppressor gene mutated in mucinous tumours of the ovary. The Journal of pathology. 2013; 229:469-76.
13. Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, Saksena G, Lawrence MS, Qian ZR, Nishihara R, Van Allen EM, Hahn WC, Gabriel SB, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014; 46:1264-6.
14. Jo YS, Kim MS, Lee JH, Lee SH, An CH, Yoo NJ. Frequent frameshift mutations in 2 mononucleotide repeats of RNF43 gene and its regional heterogeneity in gastric and colorectal cancers. Hum Pathol. 2015.
15. Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, Chan KH, Chan AS, Tsui WY, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014; 46:573-82.
16. Springer S, Wang Y, Molin MD, Masica DL, Jiao Y, Kinde I, Blackford A, Raman SP, Wolfgang CL, Tomita T, Niknafs N, Douville C, Ptak J, et al. A Combination of Molecular Markers and Clinical Features Improve the Classification of Pancreatic Cysts. Gastroenterology. 2015.
17. Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, Rene-Corail F, Elarouci N, Sbiera S, Kroiss M, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet. 2014; 46:607-12.
18. Juhlin CC, Goh G, Healy JM, Fonseca AL, Scholl UI, Stenman A, Kunstman JW, Brown TC, Overton JD, Mane SM, Nelson-Williams C, Backdahl M, Suttorp AC, et al. Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. J Clin Endocrinol Metab. 2015; 100:E493-502.
19. Nord KH, Nilsson J, Arbajian E, Vult von Steyern F, Brosjo O, Cleton-Jansen AM, Szuhai K, Hogendoorn PC. Recurrent chromosome 22 deletions in osteoblastoma affect inhibitors of the Wnt/beta-catenin signaling pathway. PLoS One. 2013; 8:e80725.
20. Jiang X, Hao HX, Growney JD, Woolfenden S, Bottiglio C, Ng N, Lu B, Hsieh MH, Bagdasarian L, Meyer R, Smith TR, Avello M, Charlat O, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A. 2013; 110:12649-54.
21. Bond CE, Umapathy A, Ramsnes I, Greco SA, Zhen Zhao Z, Mallitt KA, Buttenshaw RL, Montgomery GW, Leggett BA, Whitehall VL. p53 mutation is common in microsatellite stable, BRAF mutant colorectal cancers. Int J Cancer. 2012; 130:1567-76.
22. Bond CE, Nancarrow DJ, Wockner LF, Wallace L, Montgomery GW, Leggett BA, Whitehall VL. Microsatellite stable colorectal cancers stratified by the BRAF V600E mutation show distinct patterns of chromosomal instability. PLoS One. 2014; 9:e91739.
23. Bond CE, Bettington ML, Pearson SA, McKeone DM, Leggett BA, Whitehall VL. Methylation and expression of the tumour suppressor, PRDM5, in colorectal cancer and polyp subgroups. BMC cancer. 2015; 15:20.
24. Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, Arango D, Strausberg RL, Buchanan D, Wormald S, O'Connor L, Wilding JL, Bicknell D, et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer research. 2014; 74:3238-47.
25. Suter CM, Norrie M, Ku SL, Cheong KF, Tomlinson I, Ward RL. CpG island methylation is a common finding in colorectal cancer cell lines. British journal of cancer. 2003; 88:413-9.
26. Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK, Nieuwenhuis EE, Beekman JM, Clevers H. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell stem cell. 2013; 13:653-8.
27. Yan HH, Lai JC, Ho SL, Leung WK, Law WL, Lee JF, Chan AK, Tsui WY, Chan AS, Lee BC, Yue SS, Man AH, Clevers H, et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut. 2016.
28. Bettington ML, Walker NI, Rosty C, Brown IS, Clouston AD, McKeone DM, Pearson SA, Klein K, Leggett BA, Whitehall VL. A clinicopathological and molecular analysis of 200 traditional serrated adenomas. Modern pathology. 2015; 28:414-27.
29. Bettington M, Walker N, Rosty C, Brown I, Clouston A, McKeone D, Pearson SA, Leggett B, Whitehall V. Clinicopathological and molecular features of sessile serrated adenomas with dysplasia or carcinoma. Gut. 2015.
30. Sekine S, Yamashita S, Tanabe T, Hashimoto T, Yoshida H, Taniguchi H, Kojima M, Shinmura K, Saito Y, Hiraoka N, Ushijima T, Ochiai A. Frequent PTPRK-RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. The Journal of pathology. 2016.
31. Xi S, Zhang X, Chen H, Zhong Z, Lu J, Hu W, Wu Q, Zeng J. Downregulation of ring-finger protein 43 in glioma associates with poor prognosis. Int J Clin Exp Pathol. 2015; 8:490-6.
32. Niu L, Qin HZ, Xi HQ, Wei B, Xia SY, Chen L. RNF43 Inhibits Cancer Cell Proliferation and Could be a Potential Prognostic Factor for Human Gastric Carcinoma. Cell Physiol Biochem. 2015; 36:1835-46.
33. Sugiura T, Yamaguchi A, Miyamoto K. A cancer-associated RING finger protein, RNF43, is a ubiquitin ligase that interacts with a nuclear protein, HAP95. Exp Cell Res. 2008; 314:1519-28.
34. Loregger A, Grandl M, Mejias-Luque R, Allgauer M, Degenhart K, Haselmann V, Oikonomou C, Hatzis P, Janssen KP, Nitsche U, Gradl D, van den Broek O, Destree O, et al. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated beta-catenin by sequestering TCF4 to the nuclear membrane. Sci Signal. 2015; 8:ra90.
35. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, McLaren-Douglas A, Blokker J, Jaksani S, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015; 161:933-45.
36. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer research. 1998; 58:5248-57.
37. Nagasaka T, Koi M, Kloor M, Gebert J, Vilkin A, Nishida N, Shin SK, Sasamoto H, Tanaka N, Matsubara N, Boland CR, Goel A. Mutations in both KRAS and BRAF may contribute to the methylator phenotype in colon cancer. Gastroenterology. 2008; 134:1950-60, 60 e1.
38. Spring KJ, Zhao ZZ, Karamatic R, Walsh MD, Whitehall VL, Pike T, Simms LA, Young J, James M, Montgomery GW, Appleyard M, Hewett D, Togashi K, et al. High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology. 2006; 131:1400-7.
39. Zhao ZZ, Nyholt DR, Le L, Martin NG, James MR, Treloar SA, Montgomery GW. KRAS variation and risk of endometriosis. Mol Hum Reprod. 2006; 12:671-6.
40. Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006; 38:787-93.
41. Srihari S, Kalimutho M, Lal S, Singla J, Patel D, Simpson PT, Khanna KK, Ragan MA. Understanding the functional impact of copy number alterations in breast cancer using a network modeling approach. Molecular bioSystems. 2016; 12:963-72.