
INTRODUCTION
Tobacco is a major cause of many diseases, such as cardiovascular diseases [1, 2], pulmonary diseases [3, 4], cancers [5, 6] and most notably lung cancer [6, 7]. Based on 2012 estimates, worldwide 21% of individuals aged 15 or greater smoked tobacco products [8]. Globally, 1.42 million cancer deaths in 2000 were attributable to cigarette smoke exposure, 60% of which were due to lung cancer [9]. Lung cancer alone is also responsible for 12.4% of all new cancer cases and 17.6% of all cancer mortality [10]. In the U.S., lung cancer is the second most common cancer in both men and women, and the 5-year survival rate of lung cancer patients is 15.6%, which is much lower than other common types of cancers such as breast cancer (5-year survival rate 89.7%) [11] and prostate cancer (99.2%) [12]. Given the high incidence and poor prognoses for lung cancer, the high prevalence of smoking, and the lack of early diagnostic testing methods, it is critical that we understand the mechanisms by which tobacco smoking might cause lung cancer [10].
DNA methylation, the addition of a methyl group to DNA, may mechanistically regulate gene function [13, 14]. Differential DNA methylation, hypermethylation and hypomethylation of promoter-specific genes within CpG islands of tumor suppressor and proto-oncogenes, has been correlated with human cancers [15–21]. The triggers leading to aberrant epigenetic changes are poorly understood in the cancer genome, but those in blood have been implicated in cigarette smoking [22, 23]. Cigarette smoking has also been shown to be associated with genomic instability, which leads to DNA copy number alterations in the lung cancer genome [24]. For example, polycyclic aromatic hydrocarbons (PAHs), among other carcinogens in tobacco smoke have been well documented in altering DNA [25, 26]. However, less is known about how carcinogens may alter epigenetic machinery in the cancer genome.
The effects of cigarette smoke on DNA methylation range from modification of tissue methylation patterns to the development of disease [27–29]. A number of these studies have previously examined the link between smoking and DNA methylation [1, 18, 30–33], and while many candidate methylation loci have been identified, most studies used blood samples from patients. However, DNA methylation profiles are tissue specific, and blood tissue is unlikely to accurately represent lung cancer etiology [34–36]. It is important to examine site-specific DNA methylation to best understand how these disease-associated patterns may manifest in vivo. In this study, we used disease-appropriate neoplastic tissue from patients with lung squamous cell carcinoma (LUSC) and lung adenocarcinoma (LUAD) to study the effects of smoke exposure on DNA methylation. Because we use lung tumor tissue, which is directly exposed to cigarette smoke, rather than relying on proxy specimens such as blood, our study may provide greater insight into the link between smoking and methylation in lung cancer.
RESULTS
In order to identify differentially methylated loci associated with smoking, we conducted an epigenome-wide analysis using a two-stage, discovery-validation approach in lung adenocarcinoma (LUAD) and lung squamous cell (LUSC) tissue samples of 511 subjects from The Cancer Genome Atlas (TCGA) database. Subjects were randomized into the discovery or validation analysis groups conditional on cell-type (LUAD or LUSC) in order to obtain a balanced distribution of each lung cancer tissue in the two subsets. Demographic characteristics for the 511 subjects by analytic group and smoking status (light or heavy smoking status, based on the median smoking pack-year (3.71)) are summarized in Table 1. None of the demographic and clinical characteristics were significantly different (Table 1) between the discovery (n = 326) and validation subsets (n = 185). Within-group distributions of sex, age and cell type of lung cancer were unbalanced between light and heavy smokers, i.e., heavy smokers were older, more likely to be male and to have squamous cell carcinoma (Table 1). Therefore, these potential confounders were adjusted either as covariates or stratifying factors in the epigenome-wide analyses.
Table 1: Demographic characters of sample by two-stage analysis and smoking status
Discovery Set |
Validation Set |
Discovery vs. Validation |
|||||||
|---|---|---|---|---|---|---|---|---|---|
Covariates |
Light† (n = 173) |
Heavy† (n = 153) |
P-value |
Light† (n = 97) |
Heavy† (n = 88) |
P-value |
Discovery (n = 326) |
Validation (n = 185) |
P-Value |
Age* |
66.36 (58, 74) |
70 (61, 73) |
0.04 |
66 (59, 72) |
67 (62, 72.25) |
0.048 |
67 (60, 73) |
66.36 (61, 72) |
0.81 |
% of Male |
91 (53%) |
103 (67%) |
0.007 |
54 (55.6%) |
61 (69.3%) |
0.055 |
194 (59.5%) |
115 (62.2%) |
0.56 |
Race‡ |
0.81 |
||||||||
Black |
10 (5.7%) |
4 (2.6%) |
0.37 |
6 (6.2%) |
3 (3.4%) |
0.68 |
14 (4.3%) |
9 (4.9%) |
|
White |
136 (78.6%) |
124 (81%) |
74 (76%) |
69 (78.4%) |
260 (79.8%) |
143 (77.3%) |
|||
Other |
27 (15.6%) |
25 (16.3%) |
17 (17.5%) |
16 (18.2%) |
52 (15.95%) |
33 (17.8%) |
|||
KRAS mutation |
5 (2.9%) |
2 (1.3%) |
0.55 |
4 (4.12%) |
1 (1.13%) |
0.43 |
7 (2.1%) |
5 (2.7%) |
0.92 |
EGFR mutation |
5 (2.9%) |
3 (2%) |
0.86 |
1 (1.03%) |
2 (2.27%) |
0.94 |
8 (2.5%) |
3 (1.6%) |
0.76 |
PackYears* |
3.367 (3.045, 3.58) |
4.11 (3.93, 4.39) |
2.20E-16 |
3.26 (3.05, 3.58) |
4.08 (3.93, 4.39) |
2.20E-16 |
3.71 (3.31, 4.11) |
3.714 (3.26, 4.04) |
0.71 |
% of ACA‡ |
105 (60.7%) |
59 (38.6%) |
1.00E-04 |
63 (65%) |
41 (46.6%) |
0.018 |
164 (50.3%) |
104 (56%) |
0.23 |
Smoking History‡ |
40 (23.1%) |
61 (39.9%) |
N/A |
23 (23.7%) |
36 (41%) |
0.02 |
101 (31%) |
59 (32.2%) |
0.16 |
*Median (1st, 3rd quartiles).
†Light and Heavy were determined by a median cutoff for the smoking packyears.
‡P-values were calculated using Chi-squre tests. All other p-values were calculated using Student’s t-test.
EWAS identification of differentially methylated sites associated with smoking
In the first stage, an epigenome-wide association scan was conducted in the discovery subset to test the relationship between smoking and DNA methylation at each CpG site, with adjustments for cell-type, EGFR and KRAS mutation status, age, sex, and race. We identified 263 out of 271,316 CpG sites, which were significant at FDR<0.05 in this stage. Of these, we identified 98 CpG loci which had 1) consistent directions of effect in both analytic stages, 2) p-value < 0.001 in the validation stage (Supplementary File S1) and were thus considered internally validated (Figure 1).
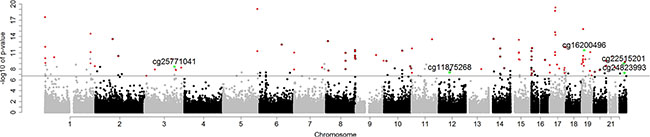
Figure 1: Manhattan plot of p-values for internally validated (No. of loci = 98) and externally validated (No. of loci = 5) CpG sites by chromosome. Bonferroni genome-wide significance (6.73) is represented as a horizontal solid line. Red dots are internally validated sites; green dots are internally and externally validated sites.
Externally validated candidate CpG loci
Further analysis of the 98 internally validated CpG sites in an external dataset, GSE56044 from the Gene Expression Omnibus database (demographic characteristics are summarized in Supplementary Table S1), identified five CpG sites with independently replicated signals for association between smoking and DNA methylation. As shown in Table 2, the five externally validated sites and their associated genes were: cg25771041 in WW domain containing transcription regulator 1 gene (WWTR1; pooled p-value = 3.63 × 10−9, external p-value = 0.046), cg11875268 in single-strand-selective monofunctional uracil DNA glycosylase gene (SMUG1; pooled p-value = 4.28 × 10−8, external p-value = 0.0017), cg16200496 in nuclear factor 1 x-type gene (NFIX; pooled p-value = 3.4 × 10−12, external p-value = 0.0344), cg22515201 in phospholipase A2 group VI gene (PLA2G6; pooled p-value = 1.04 × 10−9, external p-value (in LUAD) = 0.016), cg24823993 in NHP2-like protein 1 gene (NHP2L1; pooled p-value = 5.13 × 10−8, external p-value (in LUAD) = 0.047). We noted that the three most significant CpG sites that were validated internally: cg16579555 (pooled p = 4.2 × 10−20; located within RNF135 [ring finger protein 135]), cg00032419 (pooled p = 1.8 × 10−19; located within TP53I13 [tumor protein P53 inducible protein 13]) and cg16654732 (pooled p = 8.1 × 10−20; located within FGF18 [fibroblast growth factor 18]) seemed biologically interesting but were not validated by the external data.
Table 2: Results of the 5 externally validated CpG sites
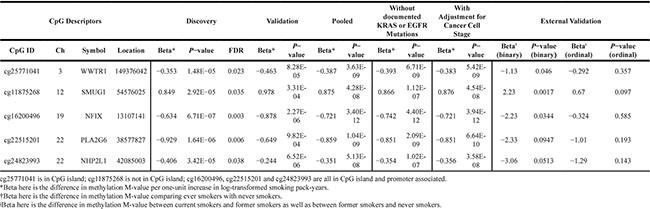
Higher smoking exposure was associated with decreased methylation at cg25771041, cg16200496, cg22515201, and cg24823993, and increased methylation at cg11875268. The direction of association for these loci was consistent across the internal discovery and validation subsets, as well as in the external validation analyses. Adjusting for cancer stage level (I-IV) also resulted in stronger statistical significance for all externally validated CpG loci (Table 2). Associations between CpG site methylation and RNA expression of their associated genes were also assessed for subjects who also had expression data available; only cg25771041 at NHP2L1 demonstrated a significant association between methylation and RNA expression (Supplementary Table S4).
DNA methylation signal profiles across candidate genes
DNA methylation profiles for all genes containing externally-validated CpG sites were mapped to explore whether any interesting methylation patterns could be discerned across the genes (Figure 2). In WWTR1, NHP2L1, and PLA2G6, evidence of significant methylation near the transcription start sites (TSS) was present based on the pooled TCGA analyses, with multiple significant CpG hits in the TSS neighborhoods in WWTR1 and PLA2G6. In NFIX, no significant methylation signals other than the externally validated finding at cg16200496 were present. Finally, in SMUG1, several significant methylation loci were present across the gene body but not in the TSS.
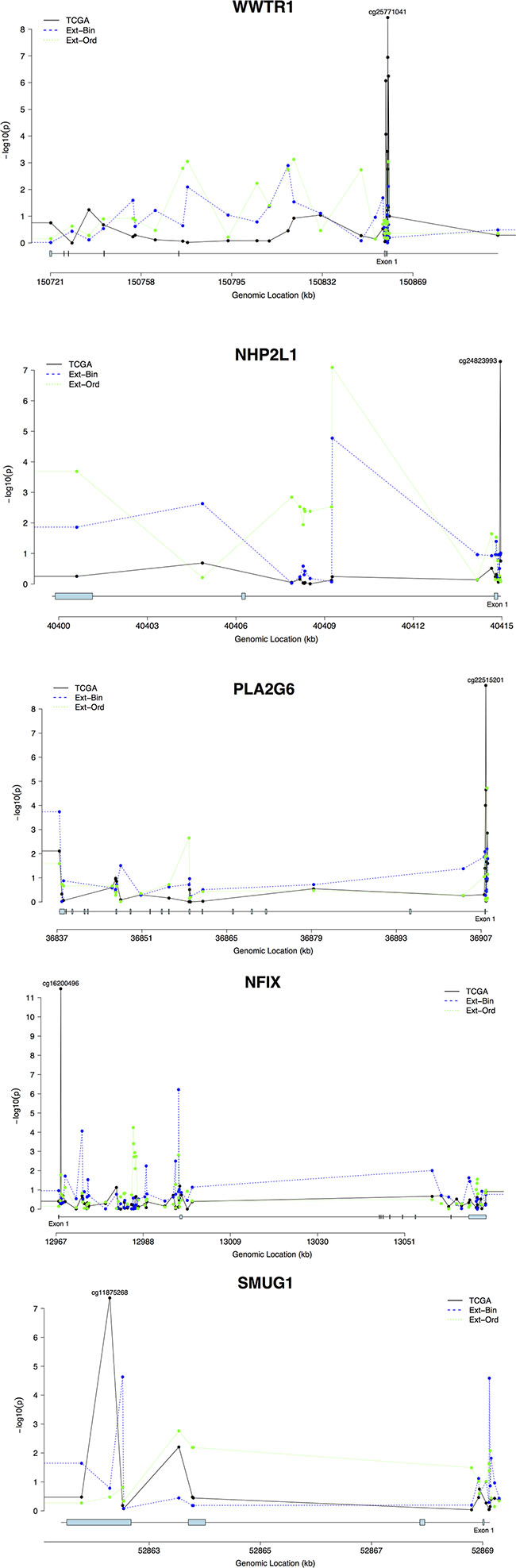
Figure 2: Methylation signal profiles for genes with externally validated methylation sites (A. WWTR1; B. NHP2L1; C. PLA2G6; D. NFIX; E. SMUG1). The analyses are plotted by genomic datasets. TCGA represents the TCGA dataset, and External-Binary (Ext-Bin) and External-Ordinal (Ext-Ord) represent the external validation analyses conducted in the GSE56044 dataset using binary and ordinal categorizations of smoking status. Signal strength is plotted via transformed p-values (-log10(p)) by genomic location (Mb) for each gene.
Several CpG loci in WWTR1 with significant association with smoking localize to a CpG island either within 200–1500 base pairs of the transcription start site (TSS), or in the 5′ untranslated region (UTR), depending on the isoform. The most significant association signal between DNA methylation and cigarette smoking in WWTR1 occurred at cg25771041 (Figure 2A). cg24823993 with the most significant signal between smoking and DNA methylation in NHP2L1, locates within 200 bp of the TSS (Figure 2B). The binary-smoking external validation analyses also identified cg24823993 as a marginally significant methylation site. cg22515201 in a CpG island within 200 bp of the TSS in PLA2G6 is the most significant CpG locus in the gene associated with cigarette smoking. The analyses also identified several highly significant methylation loci at the TSS and near exon 1 of PLA2G6 (Figure 2C). The analyses of CpG loci within NFIX identified highly significant association with smoking in exon 1 with the most significant signal in cg16200496, but no other regions demonstrate strong enrichment (Figure 2D). The association with smoking was more prominent in the fourth exon (cg11875268) of SMUG1 (Figure 2E).
Dose-response relationships between smoking and CpG methylation
To examine potential dose-response relationships between pack-years of cigarette smoking and methylation status, linear models with penalized spline (thin-plate regression spline) were constructed for the five validated CpG sites (Figure 3). There were statistically significant negative associations for all CpG sites as pack-years increased (cg25771041 [in WWTR1; p = 3.1 × 10−31, R2 = 0.079], cg16200496 [in NFIX; p = 4.8 × 10−39, R2 = 0.098], cg22515201 [in PLA2G6; p = 1.5 × 10−20, R2 = 0.072], cg24823993 [in NHP2L1; p = 0.008, R2 = 0.007]) except in cg11875268 (in SMUG1; p = 1 × 10−14, R2 = 0.068) (Figure 3A). Variation in cg22515201 (PLA2G6) was explained the most by smoking pack-years with a marginal R2 = 0.098. Marginal R2 values between smoking and methylation at the five loci were quite low (ranging from 0.007 to 0.098), suggesting that cigarette smoking alone does not fully explain changes in methylation status at these loci, and further implying the likely presence of other environmental and/or genetic determinants for epigenetic variations.
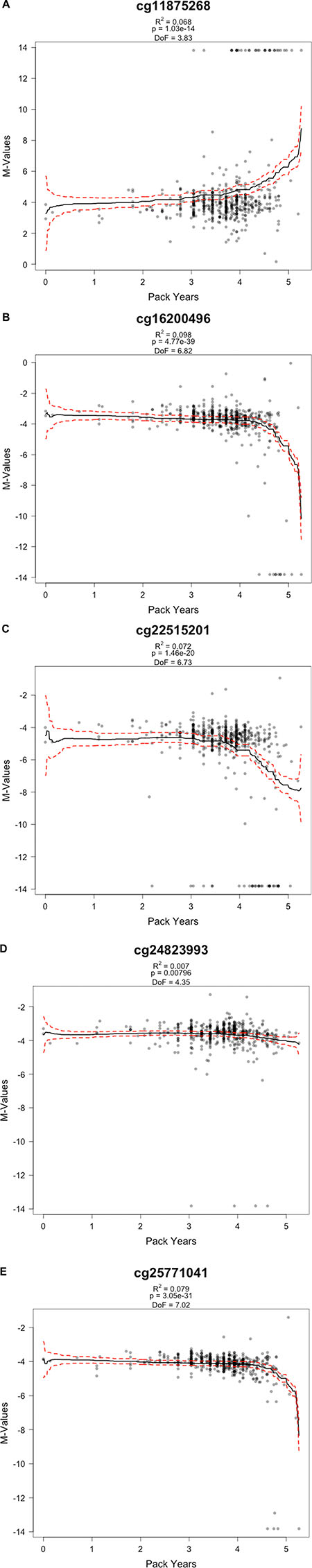
Figure 3: Dose-response relationships for externally validated CpG sites by M-values (logit transformed beta values) and smoking in pack years. The plots are based on generalized additive models with penalized spline using thin plate smoothing basis. Degree of Freedom (DoF) is provided for each plot. The solid black line represents the linear spline model of the change in M-value by pack years (smoking). The red, dotted line represents the upper and lower 95% confidence bounds. (A) cg11875268 in SMUG1; (B) cg16200496 in NFIX; (C) cg22515201 in PLA2G6; (D) cg24823993 in NHP2L1; (E) cg25771041 in WWTR1.
To check the robustness of the dose-response analyses, we also conducted sensitivity analyses which 1) removed extreme M-values and 2) substituted extreme M-values with less extreme values (the detectable largest/smallest values; Supplementary Figure S2). All validated CpG sites retained significant dose-response relationships with smoking in the analyses where extreme outliers were removed except cg11875268 (p = 0.052). All validated CpG sites retained significant dose-response relationships with smoking in the substitution analyses.
Cell-type restricted sub-analyses and sensitivity analyses
To identify cancer-specific methylation markers, sub-analyses were conducted by stratifying by cell-type and obtaining cell-type-specific estimates for the 98 internally validated CpG loci in the internal discovery, validation, and pooled samples respectively (Supplementary Files S2 and S3). The cell-type restricted subanalyses were then conducted in GSE56044 in an equivalent manner. In the internal subgroup analyses, the cell-type specific estimates retained directions of effect consistent with the effect estimates from the main analyses for all five externally validated loci. Interestingly, the strength of association was consistently more significant in the LUSC subgroup, with the strongest signal originating from cg16200946 in NFIX (β = −1.20, p = 6.6 × 10−10, R2 = 0.144) (Supplementary File S3 and Supplementary Figure S1D). The cell-type specific association between DNA methylation of the five externally validated loci (cg25771041, cg11875268, cg16200496, cg22515201 and cg24823993) and smoking was presented in Supplementary Tables S2 and S3, and their dose-response relationship was presented in Supplementary Figure S1. Further discussion of the cell-type-specific EWAS is available in the supplement.
Sensitivity analyses were conducted by reanalyzing the main, cell-specific, and external validation datasets after removing cases with KRAS or EGFR mutation from our analytical model. Each sensitivity analysis yielded consistently similar results to those from the main, cell-specific, and external validation analyses, respectively (Table 2), providing compelling support for the biological plausibility of our reported findings.
DISCUSSION
Our results identify five candidate methylation loci which may be influential in how smoking modifies DNA methylation, and thus also the development of lung cancer. Of our 98 internally validated CpG sites, five were externally validated in an independent data set: cg16200496 (NFIX), cg25771041 (WWTR1), cg11875268 (SMUG1), cg22515201 (PLA2G6), and cg24823993 (NHP2L1). Four of these loci localized near transcription start sites or within the first exons of their genes (WWTR1, NFIX, PLA2G6 and NHP2L1), and all four demonstrated negative associations between smoking and methylation status. We examined cancer stage as a confounder for smoking and DNA methylation. We utilized a missing indicator method to adjust for the available cancer stage information while keeping all subjects in the analyses. The analyses showed that our externally validated loci were more statistically significant than without adjustment. This suggests that smoking may alter neoplasm development with increased pack years being associated with higher cancer stage.
Among the five genes, a number have been previously implicated in smoking and lung cancer disease pathways. WWTR1 (also known as TAZ) is a well-described oncogenic transcriptional co-activator in many cancers including breast, liver, colon, thyroid, and lung [37, 38]. It is a part of the Hippo signaling pathway which is highly conserved in mammals and is thought to disrupt cell contact inhibition, an attribute commonly lost in cancer cells [39]. In one study, tumor-propagating cells were found to have gene expression signatures enriched for genes in the Hippo signaling pathway. Further experimentation with WWTR1/TAZ knockdowns resulted in decreased lung tumor progression, while constitutively active WWTR1/TAZ was found to be sufficient to drive lung tumor progression [40]. Additionally, higher TAZ expression levels in lung tumors have been shown to be predictive of worse prognoses [41, 42].
Another significant cancer-associated gene in our analyses was SMUG1, a glycosylase that removes damaged uracil in the base excision repair pathway [43]. The base excision repair pathway plays a critical role in removing oxidized and methylated bases from DNA, and has been implicated in a number of cancer subtypes including gastric, renal, lung, and colorectal cancers [44]. Importantly, SMUG1 has also been hypothesized to play a critical role in nucleic acid repair in lung fibroblasts suffering from cigarette-smoke induced oxidative stress [45]. This offers compelling biological implications for our finding of significant association between smoking and methylation status at cg11875268.
A third well-established cancer gene in our significant results was NFIX (Nuclear Factor I/X (CCAAT-binding transcription factor)). NFIX is a member of a family of transcription factors that are involved in regulating the transcriptional activity of genes [46, 47]; and has been involved in cancer progression in a number of cancers including breast and esophageal [46, 48]. In breast cancer, NFIX may interact with methyl-CpG binding protein 2 (MeCP2), an important epigenetic regulator, to suppress Z-DNA-mediated transcriptional suppression, thus enabling the overexpression of ADAM-12, a prominently up-regulated, metastasis-promoting protein in many cancer types.[49] In esophageal cancers, the down-regulation of NFIX allows for microRNA miR-1290 to promote tumor proliferation, migration and metastasis [46].
The roles of PLA2G6 and NHP2L1 are not well understood in LUSC and LUAD. One candidate gene pathway analysis identified PLA2G6, a member of the cell cycle pathway, as bearing a statistically significant single nucleotide polymorphism associated with lung cancer risk [50]. NHP2L1 is less understood, but is important in cell viability in yeast models and as an RNA-binding protein, specifically a small nucleolar RNA-protein complex, in eukaryotic models [51, 52]. Our study is the first to link these CpG sites to LUAD and LUSC in humans. Further research should examine the roles of these CpG sites in carcinogenesis given the strength of this finding.
In the internally validated-only CpG loci, cg16654732 was our strongest signal (pooled p-value = 8.1 × 10−20). This site localizes within 200bp of the TSS of gene FGF18, which was found to be down regulated in Italian LUAD cases compared to normal lung tissue [53]. In the present analyses, we found that methylation at cg16654732 was negatively associated with smoking, where higher pack years corresponded with lower methylation levels. In addition, cg13204512 and cg16579555 (within RNF135) were strongly, negatively associated with smoking pack-years (pooled p-value = 4.8 × 10−15 and 4.2 × 10−20, respectively). RNF135 gene has been well studied in malignant peripheral nerve sheath tumors and lymphoblastic leukemia [54, 55], but our study is the first to link these loci to smoking in non-small cell lung tumors. The TP53I13, TP53 inducible gene 13 also had two strong signals from the analyses of TCGA data (cg00032419, p = 1.8 × 10−19 and cg00265578, p = 1.6 × 10−15). TP53-inducible genes have been well documented to control many biological processes including cell cycle control, apoptosis, and DNA repair and may function to inhibit cancer progression [56]. The internal analysis showed these two CpG loci were negatively associated with smoking dosage, which may indicate these genes were active in the neoplastic tissue. Despite the strength of the association in our analyses, cg16654732, cg13204512, cg16579555, cg00032419 and cg00265578 were not found to be statistically significant in the external dataset suggesting this finding may have mechanistic heterogeneity and may not be generalizable to other studies.
For the five externally validated CpG sites, the binary effect estimates were larger than the ordinal effect estimates. It is difficult to distinguish whether there is a dose-response increase from never- to ever- and then current-smokers or a plateau effect that ever-/current-smokers share similar effects. While years since quitting smoking may help address the issue, such information was not collected in TCGA data. Research by van Osch et al. (2016) suggests that a plateau effect of smoking on bladder cancer risk and that heavy smokers are at high risk regardless of the timing of cessation for given packyears [57].
In considering the importance of tissue sample location, we utilized LUSC and LUAD neoplasms. Other studies have used whole blood samples in their EWAS, but few have used neoplasm site-specific analyses. In one site-specific analysis, Teschendorff et al. used buccal cells in their EWAS of epithelial cancers. However, we were unable to replicate their findings [58]. This lends credibility to the notion that the effect of smoking on differential methylation is site-specific. Despite smoking carcinogen presence in buccal cells, it seems there may be a different mechanism through which smoke may act on methylation profiles in different genes in different environments.
There were many strengths in our study analysis. We had a large sample size (n = 511), improving the power of our EWAS interrogation of 271,316 CpG sites. Furthermore, we obtained data collected from an appropriate target tissue—lung neoplasms. Many studies have performed EWAS using blood samples, but blood samples are not the ideal tissue to measure carcinogenesis in lung tissue and can only serve as a proxy for lung cancer rather than a direct sample of the cancer itself. Because our study used lung neoplasms, we were able to directly assess the methylation patterns of CpG sites within LUSC and LUAD. Cell-type specific analyses were also conducted to better understand differential methylation due to smoking in adenocarcinoma and squamous cell carcinoma separately. Not only were our results internally validated within the TCGA data, we were also able to replicate some of our findings in the external GSE56044 GEO dataset. The multi-step internal and external validation conducted in our analysis lowers the likelihood of obtaining false-positive CpG hits.
Although our study has strengths, there are some limitations. While our data suggest that smoking regulates methylation patterns in neoplasms, we are unable to directly link smoking and lung cancer. Our sample is based solely on patients with LUSC or LUAD neoplasms; we lack healthy participants and are therefore could not assess methylation patterns in the lung tissue of healthy smokers. Therefore, we were unable to directly assess lung cancer etiology. Furthermore, it is unclear whether the validated CpG sites had significant methylation profiles solely due to smoking or because of other unmeasured factors. Dichotomous cell-type adjustment of adenocarcinoma and squamous cell carcinoma may be coarse, but it is standard and widely available information in clinical practice. Without depending on more costly pathological profiling, the identified methylation biomarkers may have better translation utility. Alternatively, one may adjust for inferred cell mixture based on a bioinformatics algorithm [59] if the research interest is in the epigenetic association with smoking within a homogeneous cell population. Here we are more interested in such an association within a patient with non-small cell lung cancer, lung adenocarcinoma or lung squamous cell carcinoma.
We note that the five CpG hits in the external validation analyses were significant after adjustment of multiple comparisons. However, since 1) these sites were internally validated and 2) the external validation analyses did not stand alone, we were less concerned about the potential false positive due to multiple comparisons. Furthermore, the external validation data measured categorical smoking status rather than smoking pack-years, which may render the external validation less power and thus being non-ideal for conservative multiplicity adjustment.
Our data indicate that CpG sites in WWTR1, NFIX, PLA2G6, NHP2L1 and SMUG1 have differential methylation in LUAD and LUSC neoplasms. These internally and externally validated CpG sites may give insight into the mechanism by which smoking may cause lung cancer. Additional research should focus on how these CpG sites are mechanically altered after repeated smoke exposure and if there are hierarchical interactions with microRNA and proteins from other CpG sites.
MATERIALS AND METHODS
Study sample
Subject data (n = 820) were obtained from The Cancer Genome Atlas (TCGA) (https:// tcga-data.nci.nih.gov/tcga), a collaborative project between the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) that curates publicly available cancer datasets which have been comprehensively genotyped and assayed. Specific information on sample quality control has been previously reported [60]. We selected sample based on the availability of 1) epigenome-wide DNA methylation data from untreated neoplastic LUAD and LUSC tumor cells (classified as stages I–IV), and 2) clinical measures of smoke exposure. Subjects with missing methylation data (n = 139) and missing smoking measures (n = 170) were excluded, resulting in an analytical sample of 511 (268 LUAD and 243 LUSC) subjects.
Data processing
Key clinical and demographic variables of interest were re-categorized for analysis: smoking exposure in pack years, sex, age, race, KRAS mutation, EGFR mutation, and cell-type. Pack-years, defined as the packs of cigarette smoked multiplied by the duration of smoking, was log transformed due to skewness. Both KRAS and EGFR mutation types were re-categorized into binary variables based on whether any mutations were present: the presence of any mutation (e.g., exon 19 deletion, L858R, and others) or not. Race was re-categorized as a nominal, categorical variable with race designations “white”, “black”, and “other.” Missing information in age was imputed with the median values of age in the full sample.
Level 3 methylation data assayed on the Illumina Infinium Human Methylation 450K in LUSC and LUAD neoplasms were obtained from TCGA database. All CpG sites located in sex chromosomes were discarded, retaining only autosomal sites. We adjusted for batch effects using the ComBat method in the Surrogate Variable Analysis (sva) package from Bioconductor CpG sites with low variance were filtered out based on the first quartile of the variance for all autosomal CpG sites (σ = 0.147) [61]. After quality-control, 271,316 CpG sites were retained for EWAS analysis.
Statistical analysis
Internal discovery and validation analysis
Candidate methylation loci were identified and validated using a two-stage approach by randomizing all subjects (n = 511) into discovery (n = 326) and validation (n = 185) subsets. Randomized assignment was performed conditional on cell-type (LUAD or LUSC) in order to obtain a balanced distribution of each lung cancer tissue in the two subsets. Potential confounders were stratified or treated as covariates in regression analyses. In the first stage, an epigenome-wide association scan was conducted in the discovery subset using a linear model to test the relationship between DNA methylation and smoking at each CpG locus, with adjustments for cell-type, EGFR mutation status, KRAS mutation status, age, sex, and race. To adjust for multiple comparisons, we then applied a false discovery rate (FDR) < 0.05 threshold in the discovery analyses using the FDRtool R package [62]. CpG sites surviving the FDR < 0.05 threshold in the discovery stage were then re-analyzed in the validation subset using the same model specifications as in the discovery stage, and those loci with a validation p-value < 0.001 were retained for further cell-type-specific sub-analyses. All candidate loci surviving both the discovery and validation analyses were considered internally validated. Internally validated sites were then reassessed in the full study sample (i.e. the combined discovery and validation subsets) to obtain the final pooled p-values. Further adjustment for cancer stage and cancer stage missingness was conducted for the externally validated CpG sites using the missing indicator method [63].
External validation analysis
To validate our findings in an independent dataset, candidate CpG sites identified in the two-stage analyses were re-analyzed in the GSE56044 dataset (n = 124), which was obtained from the NCBI’s Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE56044). All covariates were operationalized as in the main analysis with one exception: in the GSE56044 dataset, smoking status was recorded as a categorical variable (never smoker, current smoker, and former smoker) rather than continuously in pack-years. To assess any underlying dose-response relationships, we recoded smoking as an ordinal variable (never smoker = 0, former smoker = 1, current smoker = 2) and as a binary variable (non-smoker = 0, ever-smoker = 1) in GSE56044. The model specification used for validation in the external dataset was identical to the model used in our main analyses. Finally, we also conducted sensitivity analyses to check the robustness of our dose-respond trends by re-analyzing the relationships after 1) removing extreme M-values and 2) substituting extreme M-values with less extreme values (the detectable largest/smallest values; Supplementary Figure S2).
Cell-Type specific sub-analyses and sensitivity analyses
To assess association between smoking and DNA methylation that may be specific to cell-type, we conducted sub-analyses restricting to cell-type. Cell-type-specific estimates for the 98 internally validated CpG loci were obtained in the pooled samples (Supplementary Files S2 and S3). The cell-type restricted subanalyses were then also conducted in GSE56044 in an equivalent manner. To examine the robustness of our findings, further sensitivity analyses were conducted by comparing estimates obtained by including vs excluding subjects with documented EGFR (n = 11) or KRAS (n = 12) mutations within the main and cell-type-specific analyses.
ACKNOWLEDGMENTS AND FUNDING
The work is supported by the Brown Junior Faculty Research Awards in Genetics and Population Studies (to J.R.F., S.C., T.H. and Y.T.H.). We also would like to thank the editor and the four anonymous referees for their thoughtful comments and suggestions.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Leone AA. Toxics of tobacco smoke and cardiovascular system: From functional to cellular damage. Curr Pharm Des. 2015.
2. Khan RJ, Stewart CP, Davis SK, Harvey DJ, Leistikow BN. The risk and burden of smoking related heart disease mortality among young people in the United States. Tob Induc Dis. 2015; 13:16.
3. Vancheri CC. IPF, comorbidities and management implications. Sarcoidosis Vasc Diffuse Lung Dis. 32:17.
4. Huang R, Wei Y, Hung RJ, Liu G, Su L, Zhang R, Zong X, Zhang ZF, Morgenstern H, Bruske I, Heinrich J, Hong YC, Kim JH, et al. Associated Links Among Smoking, Chronic Obstructive Pulmonary Disease, and Small Cell Lung Cancer: A Pooled Analysis in the International Lung Cancer Consortium. EBioMedicine. 2015; 2:1677–1685.
5. Wang Y, Duan H, Yang X, Guo J. Cigarette smoking and the risk of pancreatic cancer: a case-control study. Med Oncol. 2014; 31:184.
6. Liu M, Zhou C, Zheng J. Cigarette smoking impairs the response of EGFR-TKIs therapy in lung adenocarcinoma patients by promoting EGFR signaling and epithelial-mesenchymal transition. Am J Transl Res. 2015; 7:2026–2035.
7. World Health Organization. Tobacco. (http://www.who.int/mediacentre/factsheets/fs339/en/ WHO). 2015.
8. Organization WH. Prevalence of tobacco use. Global Health Observation (GHO) Data. (http://www.who.int/gho/tobacco/use/en/ WHO). 2016.
9. Ezzati M, Henley SJ, Lopez AD, Thun MJ. Role of smoking in global and regional cancer epidemiology: current patterns and data needs. Int J Cancer. 2005; 116:963–971.
10. Dela Cruz CS, Tanoue LT, Matthay RA. Lung cancer: epidemiology, etiology, and prevention. Clin Chest Med. 2011; 32:605–644.
11. Breast cancer survival rates, by stage. Breast Cancer 2016 5/4/2016 [cited 2016]; Available from: http://www.cancer.org/cancer/breastcancer/detailedguide/breast-cancer-survival-by-stage.
12. Prostate cancer stages. Prostate Cancer 3/11/2016. Available from: http://www.cancer.org/cancer/prostatecancer/detailedguide/prostate-cancer-survival-rates.
13. Miller OJ, Schnedl W, Allen J, Erlanger BF. 5-Methylcytosine localised in mammalian constitutive heterochromatin. Nature. 1974; 251:636–637.
14. Hou L, Zhang X, Wang D, Baccarelli A. Environmental chemical exposures and human epigenetics. Int J Epidemiol. 2012; 41:79–105.
15. Vaissiere T, Hung RJ, Zaridze D, Moukeria A, Cuenin C, Fasolo V, Ferro G, Paliwal A, Hainaut P, Brennan P, Tost J, Boffetta P, Herceg Z. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res. 2009; 69:243–252.
16. Vineis P, Perera F. Molecular epidemiology and biomarkers in etiologic cancer research: the new in light of the old. Cancer Epidemiol Biomarkers Prev. 2007; 16:1954–1965.
17. Villalba M, Diaz-Lagares A, Redrado M, de Aberasturi AL, Segura V, Bodegas ME, Pajares MJ, Pio R, Freire J, Gomez-Roman J, Montuenga LM, Esteller M, Sandoval J, et al. Epigenetic alterations leading to TMPRSS4 promoter hypomethylation and protein overexpression predict poor prognosis in squamous lung cancer patients. Oncotarget. 2016; 7:22752–22769. doi: 10.18632/oncotarget.8045.
18. Fasanelli F, Baglietto L, Ponzi E, Guida F, Campanella G, Johansson M, Grankvist K, Johansson M, Assumma MB, Naccarati A, Chadeau-Hyam M, Ala U, Faltus C, et al. Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat Commun. 2015; 6:10192.
19. Koh YW, Chun SM, Park YS, Song JS, Lee GK, Khang SK, Jang SJ. Association between the CpG island methylator phenotype and its prognostic significance in primary pulmonary adenocarcinoma. Tumour Biol. 2016.
20. Leduc N, Ahomadegbe C, Agossou M, Aline-Fardin A, Mahjoubi L, Dufrenot-Petitjean Roget L, Grossat N, Vinh-Hung V, Lamy A, Sabourin JC, Molinie V. Incidence of Lung Adenocarcinoma Biomarker in a Caribbean and African Caribbean Population. J Thorac Oncol. 2016; 11:769–773.
21. Jin Y, Xu P, Liu X, Zhang C, Tan C, Chen C, Sun X, Xu Y. Cigarette Smoking, BPDE-DNA Adducts, and Aberrant Promoter Methylations of Tumor Suppressor Genes (TSGs) in NSCLC from Chinese Population. Cancer Invest. 2016; 34:173–180.
22. Zeilinger SS. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PloS One. 2013; 8:e63812.
23. Sun YV. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Human Genet. 2013; 132:1027.
24. Huang YT, Lin X, Liu Y, Chirieac LR, McGovern R, Wain J, Heist R, Skaug V, Zienolddiny S, Haugen A, Su L, Fox EA, Wong KK, et al. Cigarette smoking increases copy number alterations in nonsmall-cell lung cancer. Proc Natl Acad Sci USA. 2011; 108:16345–16350.
25. Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol. 2002; 3:461–469.
26. Hecht SS, Stepanov I, Carmella SG. Exposure and Metabolic Activation Biomarkers of Carcinogenic Tobacco-Specific Nitrosamines. Acc Chem Res. 2016; 49:106–114.
27. Ligthart S, Steenaard RV, Peters MJ, van Meurs JB, Sijbrands EJ, Uitterlinden AG, Bonder MJ, consortium B, Hofman A, Franco OH, Dehghan A. Tobacco smoking is associated with DNA methylation of diabetes susceptibility genes. Diabetologia. 2016.
28. Dogan MV, Lei MK, Beach SR, Brody GH, Philibert RA. Alcohol and tobacco consumption alter hypothalamic pituitary adrenal axis DNA methylation. Psychoneuroendocrinology. 2016; 66:176–184.
29. Belinsky SA, Palmisano WA, Gilliland FD, Crooks LA, Divine KK, Winters SA, Grimes MJ, Harms HJ, Tellez CS, Smith TM, Moots PP, Lechner JF, Stidley CA, et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002; 62:2370–2377.
30. Ambatipudi S, Cuenin C, Hernandez-Vargas H, Ghantous A, Calvez-Kelm FL, Kaaks R, Barrdahl M, Boeing H, Aleksandrova K, Trichopoulou A, Lagiou P, Naska A, Palli D, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016.
31. Zhu X, Li J, Deng S, Yu K, Liu X, Deng Q, Sun H, Zhang X, He M, Guo H, Chen W, Yuan J, Zhang B, et al. Genome-Wide Analysis of DNA Methylation and Cigarette Smoking in Chinese. Environ Health Persp. 2016.
32. Cortessis VK. Environmental epigenetics: prospects for studying epigenetic mediation of exposure-response relationships. Human Genet. 2012; 131:1565.
33. Feil RR. Epigenetics and the environment: emerging patterns and implications. Nature Rev Genet. 2011; 13:97.
34. Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, Belvisi MG, Brown R, Vineis P, Flanagan JM. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 2013; 22:843–851.
35. Zaghlool SB. Association of DNA methylation with age, gender, and smoking in an Arab population. Clin Epigenetics. 2015; 7:6.
36. Besingi W, Johansson A. Smoke-related DNA methylation changes in the etiology of human disease. Hum Mol Gen. 2014; 23:2290–2297.
37. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013; 13:246–257.
38. Mo JS, Park HW, Guan KL. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014; 15:642–656.
39. Hong W, Guan KL. The YAP, TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. 2012; 23:785–793.
40. Lau AN, Curtis SJ, Fillmore CM, Rowbotham SP, Mohseni M, Wagner DE, Beede AM, Montoro DT, Sinkevicius KW, Walton ZE, Barrios J, Weiss DJ, Camargo FD, et al. Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J. 2014; 33:468–481.
41. Xie M, Zhang L, He CS, Hou JH, Lin SX, Hu ZH, Xu F, Zhao HY. Prognostic significance of TAZ expression in resected non-small cell lung cancer. J Thorac Oncol. 2012; 7:799–807.
42. Noguchi S, Saito A, Horie M, Mikami Y, Suzuki HI, Morishita Y, Ohshima M, Abiko Y, Mattsson JS, Konig H, Lohr M, Edlund K, Botling J, et al. An integrative analysis of the tumorigenic role of TAZ in human non-small cell lung cancer. Clin Cancer Res. 2014; 20:4660–4672.
43. Abdel-Fatah TM, Albarakati N, Bowell L, Agarwal D, Moseley P, Hawkes C, Ball G, Chan S, Ellis IO, Madhusudan S. Single-strand selective monofunctional uracil-DNA glycosylase (SMUG1) deficiency is linked to aggressive breast cancer and predicts response to adjuvant therapy. Breast Cancer Res Treat. 2013; 142:515–527.
44. Sweasy JB, Lang T, DiMaio D. Is base excision repair a tumor suppressor mechanism? Cell cycle. 2006; 5:250–259.
45. Deslee G, Adair-Kirk TL, Betsuyaku T, Woods JC, Moore CH, Gierada DS, Conradi SH, Atkinson JJ, Toennies HM, Battaile JT, Kobayashi DK, Patterson GA, Holtzman MJ, et al. Cigarette smoke induces nucleic-acid oxidation in lung fibroblasts. Am J Respir Cell Mol Biol. 2010; 43:576–584.
46. Mao YY. MiR-1290 promotes cancer progression by targeting nuclear factor I/X(NFIX) in esophageal squamous cell carcinoma (ESCC). Biomed Pharmacother. 2015; 76:82.
47. Nebl G, Cato AC. NFI/X proteins: a class of NFI family of transcription factors with positive and negative regulatory domains. Cell Mol Biol Res. 1995; 41:85–95.
48. Ordway JM, Budiman MA, Korshunova Y, Maloney RK, Bedell JA, Citek RW, Bacher B, Peterson S, Rohlfing T, Hall J, Brown R, Lakey N, Doerge RW, et al. Identification of novel high-frequency DNA methylation changes in breast cancer. PloS One. 2007; 2:e1314.
49. Ray BK, Dhar S, Henry C, Rich A, Ray A. Epigenetic regulation by Z-DNA silencer function controls cancer-associated ADAM-12 expression in breast cancer: cross-talk between MeCP2 and NF1 transcription factor family. Cancer Res. 2013; 73:736–744.
50. Hosgood HD, 3rd, Menashe I, Shen M, Yeager M, Yuenger J, Rajaraman P, He X, Chatterjee N, Caporaso NE, Zhu Y, Chanock SJ, Zheng T, et al. Pathway-based evaluation of 380 candidate genes and lung cancer susceptibility suggests the importance of the cell cycle pathway. Carcinogenesis. 2008; 29:1938–1943.
51. Saito H, Fujiwara T, Shin S, Okui K, Nakamura Y. Cloning and mapping of a human novel cDNA (NHP2L1) that encodes a protein highly homologous to yeast nuclear protein NHP2. Cytogenet Cell Genet. 1996; 72:191–193.
52. Schultz A, Nottrott S, Watkins NJ, Luhrmann R. Protein-protein and protein-RNA contacts both contribute to the 15.5K-mediated assembly of the U4/U6 snRNP and the box C/D snoRNPs. Mol Cell Biol. 2006; 26:5146–5154.
53. Falvella FS, Frullanti E, Galvan A, Spinola M, Noci S, De Cecco L, Nosotti M, Santambrogio L, Incarbone M, Alloisio M, Calabro E, Pastorino U, Skaug V, et al. FGFR4 Gly388Arg polymorphism may affect the clinical stage of patients with lung cancer by modulating the transcriptional profile of normal lung. Int J Cancer. 2009; 124:2880–2885.
54. Hicks C, Miele L, Koganti T, Young-Gaylor L, Rogers D, Vijayakumar V, Megason G. Analysis of Patterns of Gene Expression Variation within and between Ethnic Populations in Pediatric B-ALL. Cancer Inform. 2013; 12:155–173.
55. Pasmant E, Masliah-Planchon J, Levy P, Laurendeau I, Ortonne N, Parfait B, Valeyrie-Allanore L, Leroy K, Wolkenstein P, Vidaud M, Vidaud D, Bieche I. Identification of genes potentially involved in the increased risk of malignancy in NF1-microdeleted patients. Mol Med. 2011; 17:79–87.
56. Hata T, Ogawa T, Yokoyama TA, Fukushige S, Horii A, Furukawa T. DSCP1, a novel TP53-inducible gene, is upregulated by strong genotoxic stresses and its overexpression inhibits tumor cell growth in vitro. Int J Oncol. 2004; 24:513–520.
57. van Osch FH, Jochems SH, van Schooten FJ, Bryan RT, Zeegers MP. Significant Role of Lifetime Cigarette Smoking in Worsening Bladder Cancer and Upper Tract Urothelial Carcinoma Prognosis: A Meta-Analysis. J Urol. 2016; 195:872–879.
58. Teschendorff AE, Yang Z, Wong A, Pipinikas CP, Jiao Y, Jones A, Anjum S, Hardy R, Salvesen HB, Thirlwell C, Janes SM, Kuh D, Widschwendter M. Correlation of Smoking-Associated DNA Methylation Changes in Buccal Cells With DNA Methylation Changes in Epithelial Cancer. JAMA Oncol. 2015; 1:476–485.
59. Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014; 30:1431–1439.
60. International Cancer Genome C, Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, Guttmacher A, Guyer M, et al. International network of cancer genome projects. Nature. 2010; 464:993–998.
61. Leek JT JW, Parker HS, Fertig EJ, Jaffe AE, Storey JD. sva: Surrogate Variable Analysis. R package version 3. 2015; 19.0.
62. Strimmer K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 2008; 24:1461–1462.
63. White IR, Thompson SG. Adjusting for partially missing baseline measurements in randomized trials. Stat Med. 2005; 24:993–1007.