
Introduction
The most metastatic form of lung cancer is SCLC, a highly aggressive neuroendocrine cancer [1]. SCLC is typically metastatic at initial diagnosis, but little is known about the why this is so. The high frequency of metastasis at diagnosis raises the possibility that SCLC may acquire many of the genetic lesions needed for metastasis early, but there has been little work to compare primary SCLC to metastases using human samples. This is likely owing to the difficulty in accessing samples from a tumor type that rarely undergoes surgical resection. Thus, the timing of both genetic and non-genetic changes that promote SCLC progression and metastasis are very poorly understood.
Study of mouse models can overcome the limitations in accessing SCLC clinical samples. Human SCLCs almost universally exhibit RB and P53 inactivation [2, 3] and Rb/p53 deletion in a subset of cells in the murine lung leads to SCLC that arises with near complete penetrance [4, 5]. The resulting tumors resemble human SCLC, exhibiting strong neuroendocrine characteristics and frequent metastasis, especially to the liver. Tumors in the Rb/p53 mouse model undergo spontaneous secondary genetic alterations that also occur in human SCLC such as amplification of Mycl or deletion of Pten [6]. Moreover, overexpression of Mycl [7, 8] or deletion of Pten [6, 9] accelerates murine SCLC, showing that mouse models can be used to interrogate potential SCLC driver genes. We need a better understanding of which genetic changes occur very early in tumor initiation and which subsequent mutations contribute to tumor progression and metastasis.
Beyond Mycl amplification and Pten deletion, another genetic alteration that occurs in mouse models of SCLC is focal amplification of the Nfib gene [10]. These amplifications are high-level, and Nfib was found to be the only gene in the minimal region of amplification [10]. Nfib is one of four members of the Nfi family of transcription factors, a family that includes Nfia, Nfib, Nfic, and Nfix. Genetic inactivation of Nfib in mice revealed that Nfib is important both for brain and lung development [11, 12]. In human SCLC cells, NFIB can sometimes undergo high-level amplification [10]. An alternative mechanism through which NFIB can be highly expressed is through the presence of atypically large transcriptional enhancers or super-enhancers [13, 14]. Super-enhancers, marked by high levels and long stretches of histone H3K27 acetylation (and/or other features associated with active enhancers) have been proposed to promote the expression of genes important for cell lineage and oncogenesis [15-17]. Cell based studies support the notion that NFIB exhibits oncogenic activity [10], but in vivo analyses are required to rigorously assess Nfib oncogenic function. In this study, we overexpressed Nfib in a sensitized mouse model and also suppressed NFIB expression in SCLC cells; we report potent oncogenic activity of Nfib in promoting SCLC in vivo.
Results
Nfib amplifications occur more frequently in liver metastases over primary SCLC
We used next generation sequencing to compare genetic alterations between primary SCLC and liver metastases that arise in murine models of SCLC. These models are initiated by intra-tracheal delivery of either broadly expressing Ad-CMV-Cre or Ad-Cre driven by a neuroendocrine promoter (Ad-CGRP-Cre). We collected primary tumors and liver metastases across a series of 17 mice derived from three models: Rblox/lox;p53lox/lox [4] and Rblox/lox;p53lox/lox;Ptenlox/+ [9] mice infected with Ad-CMV-Cre, and an Rblox/lox;p53lox/lox;Ptenlox/lox model infected with Ad-CGRP-Cre [6, 7]. The use of neuroendocrine specific Ad-CGRP-Cre is necessary in the Rblox/lox;p53lox/lox;Ptenlox/lox model, as this dramatically reduces the incidence of adenocarcinoma that otherwise occurs with Ad-CMV-Cre infection in these mice [6, 7, 9]. We analyzed one lung tumor and one liver metastasis from each of 17 SCLC-bearing mice (Supplemental Table 1). We performed copy number variation (CNV) analyses using low-coverage whole genome sequencing across each lung tumor and liver metastasis. Matched normal tail DNA was used as a control. We were particularly interested in recurrent changes enriched in metastatic tumors. We employed Segseq [18] and CNVseq [19] to identify CNV changes. We did not find recurrent examples of metastasis-specific enrichment or newly acquired genetic amplifications in Mycl or deletions of Pten in the liver metastases (Supplemental Figure 1A, 2A and data not shown). However, we identified a region of focal amplification on chromosome 4 that was very frequently amplified in liver metastases over primary SCLC, this region harbored the Nfib gene (Figure 1A, Supplemental Figure 2). In the mouse models tested, multiple primary tumors can arise in the lung and so the liver metastasis was not always clonally related to the paired lung tumor. However, across some of the pairs, obvious clonal relationships could be discerned where it was clear that the Nfib amplification was present in the liver metastasis but undetectable in the bulk primary lung tumor. For example, Figure 1B shows a tumor from the Rb/p53 model, H6904, with very similar genome-wide CNV profiles between the lung and liver metastasis. However, multiple chromosome 4 amplifications, including Nfib (but not Mycl) were present in the liver metastasis but not the matched lung tumor. Similarly, across another Rb/p53 model pair that was clearly clonally related (H6210), Mycl amplification was present in the lung SCLC (which lacked Nfib amplification), and the liver metastasis exhibited Nfib amplification but lacked Mycl amplification (Supplemental Figure 1B). This example suggests different evolution of the lung tumor and liver metastasis by the time of sampling. We also compared Nfib expression across a series of primary SCLC vs. metastases from the Rb/p53 and Rb/p53/Ptenlox/+ models using real time PCR. We found significantly increased expression of Nfib in liver metastases over primary tumors (Figure 1C). We conclude that Nfib amplifications are enriched in liver metastases over primary tumors in mouse SCLC models. Our data suggest that positive selection of SCLC cells harboring Nfib amplification may contribute to SCLC progression.
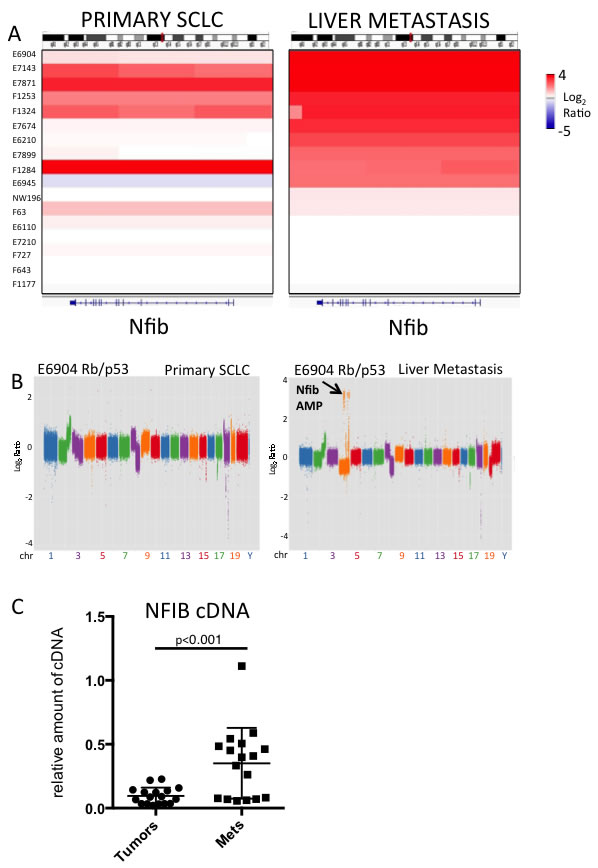
Figure 1: Nfib is amplified in liver metastases over primary tumors in mouse models of SCLC. A. Integrated genome viewer (IGV) plot showing Nfib copy number in a series of 17 primary murine SCLCs (left) and liver metastases (right). Log2 copy number ratios of tumor DNA relative to normal tail DNA is shown, with scale to the right. B. Copy number variation (CNV) analysis showing matched pair of primary SCLC and liver metastases, analyzed using CNVSeq. The majority of copy number alterations are similar between the primary and metastasis. Chromosome 4 harbors focal amplifications including Nfib amplification (arrow) exclusively in the metastasis. C. Real-time PCR analysis of Nfib expression (bottom) across a panel of primary SCLC and liver metastases. Data are normalized to Gapdh. Note the clear upregulation in Nfib expression in a large proportion of the liver metastases over matched primary lung tumors. p-value from Student’s T-test is indicated.
Nfib overexpression accelerates SCLC in a mouse model
We next used a novel TET-regulated Nfib transgenic strain (TRE-Nfib) to investigate the role of Nfib in SCLC initiation (see Methods for details on transgenic strain generation). By combining this allele with the Rosa26lox-stop-lox rtTA allele, rtTA, and consequently Nfib, is overexpressed upon Cre-mediated recombination (Figure 2A). Intra-tracheal AdenoCre delivery leads to Cre expression in lung epithelial cells. We used this inducible system to test oncogenic roles for Nfib in the sensitized Rblox/lox;p53lox/lox AdenoCre SCLC model [4]. We compared tumorigenesis in Rblox/lox;p53lox/lox;Rosa26LSL-rtTA/LSL-rtTA;TRE-Nfib (herein referred to as Rb/p53/TET-Nfib) vs. Rblox/lox;p53lox/lox;Rosa26LSL-rtTA/LSL-rtTA mice (herein referred to as Rb/p53). The expression of Nfib is activated by addition of doxycycline to the feed starting one week after Ad-CMV-Cre infection. We followed mice until respiratory changes from tumor burden necessitated euthanasia. Western blot analysis of lung tumors collected from Rb/p53/TET-Nfib mice showed increased expression of NFIB compared to lung tumors from Rb/p53 controls (Figure 2B). As shown in Figure 2C, we found that Nfib overexpression rapidly accelerated time to SCLC with a median time to morbidity of 256 days in the Rb/p53/TET-Nfib model vs. 324 days in the Rb/p53 controls (p = 0.04, log-rank test). Resulting tumors in the Nfib overexpressing model exhibited histology typical of the Rb/p53 model [20] and exhibited neuroendocrine features, as shown by Calcitonin Gene Related Peptide (CGRP) immunostaining (Figure 2D). While Rb/p53 deletion in this system leads to predominant SCLC, cooperating mutations can lead to a change in tumor spectrum, as in Cui et al [9], where Pten inactivation promoted lung adenocarcinoma. We found no instances of adenocarcinoma in the Rb/p53/TET-Nfib group. Given Nfib amplification enrichment in liver metastases (Figure 1), we were interested in whether Nfib-overexpressing SCLC exhibited higher rates of liver metastasis. Upon necropsy, gross liver metastases were found in 6/14 (43%) of lung tumor bearing mice from the Rb/p53/TET-Nfib compared to 7/15 (47%) of lung tumor bearing mice from the Rb/p53 group. Liver metastases were found at an average+/-s.d. of 299+/-53 days in the Rb/p53/TET-Nfib model and 318+/-29 days in the Rb/p53 model, a difference that was not statistically significant. Our data indicate that Nfib functions as a potent lung cancer oncogene in the Rb/p53 model and this ability of Nfib to function as an oncogene was not specific to SCLC metastasis.
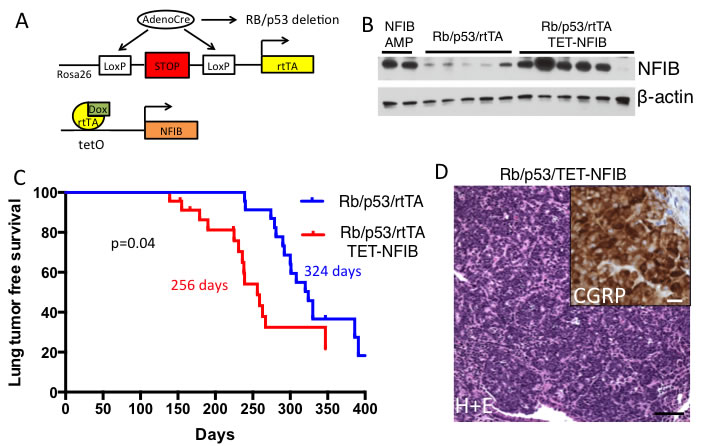
Figure 2: Nfib overexpression accelerates SCLC in vivo. A. Cartoon showing doxycycline inducible Nfib expression and Rb/p53 deletion driven by Adeno Cre expression, delivered using intratracheal delivery to the lung epithelium. B. Western blot showing NFIB overexpression in lung tumors from the Rb/p53/TET-Nfib model, as compared to Rb/p53 model controls. As a positive control, two liver metastases from tumors harboring spontaneous genomic amplification of Nfib in the Rb/p53 model are also shown (NFIB AMP). C. Kaplan-Meier curve showing acceleration of SCLC in Rb/p53/TET-Nfib compared to the Rb/p53 model. Median survival for each model is indicated and p-value from log-rank test is shown. D. Hematoxylin and eosin stain of representative SCLC arising in the Rb/p53/TET-Nfib model, black scale bar, 50mm. Inset: CGRP immunostaining of Rb/p53/TET-Nfib tumor, white scale bar, 10mm.
Suppressing NFIB levels reduces proliferation
We established cell lines from Rb/p53/TET-Nfib mice that had Nfib overexpressed in a doxycycline-dependent fashion. By washing out doxycycline from the media after cell line establishment, we could interrogate the cellular consequences of suppressing NFIB expression in tumors initiated with high levels of NFIB. Upon removal of doxycycline from the media, we found suppression of proliferation in two of three cell lines (Figure 3A) and sharply decreased NFIB protein levels (Figure 3B). In the responsive cell lines, removal of doxycycline led to increased apoptosis. We observed an increased sub-G1 population in FACS analysis for DNA content (Figure 3C, 3D and not shown). Increased cleaved caspase 3 was consistent with higher levels of apoptosis in responsive cells upon doxycycline removal (Figure 3B). We also found a decrease in the proportion of cells in S-phase upon NFIB suppression (Figure 3C, 3D, and data not shown), which correlated with increased expression of the cyclin dependent kinase inhibitor p27 in Western blot analysis (Figure 3B). In a human SCLC cell line, NCI-H446 that harbors a focal NFIB amplification [10] we used lentiviral CRISPR to generate NFIB-deleted single cell clones and confirmed complete loss of NFIB expression by Western blot analysis (Figure 3E). Deletion of NFIB across independent clones resulted in decreased numbers of viable cells (Figure 3F) and reduced cell migration, as assessed using a transwell migration assay (Figure 3G). Thus, NFIB regulates cell viability and migration in SCLC cells.
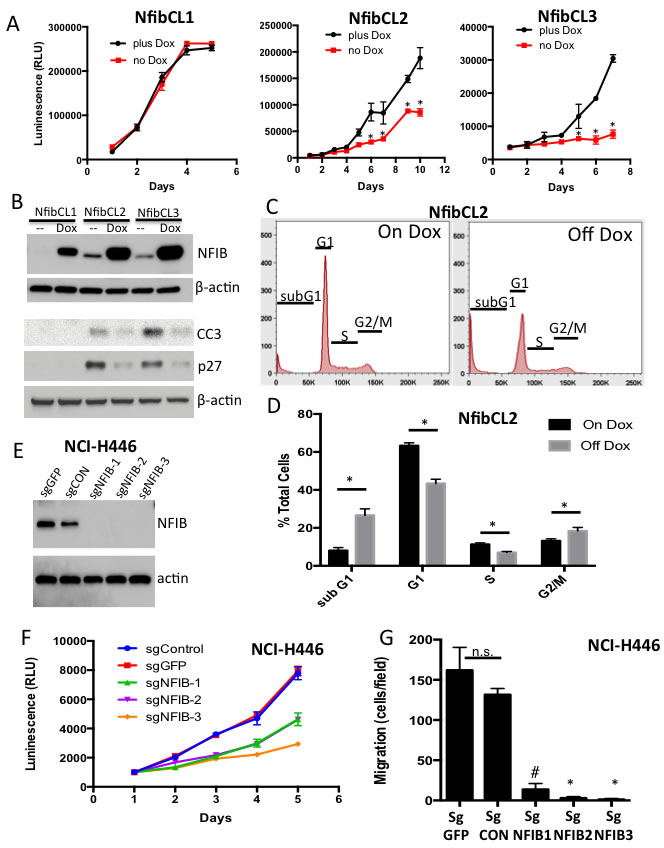
Figure 3: Suppression of NFIB expression reduces cell viability and migration. A. Cell TiterGlo assay showing proliferation in Rb/p53/TET-Nfib cell lines upon removal of doxycycline (DOX). Mean +/- s.d. of triplicate wells shown and data are representative of 3 independent experiments. *p < 0.001, Student’s T-test. B. Western blot showing NFIB, cleaved Caspase 3 (CC3) and p27 protein levels across three Rb/p53/TET-Nfib cell lines in the presence of DOX or 7 days post washout of DOX. Beta actin is used as a loading control. C. FACS analysis for DNA content shows increased subG1 population, indicative of apoptosis, and decreased G1 and S-phase populations upon doxycycline removal in NfibCL2 cell line from Rb/p53/TET-Nfib lung tumor. D. Quantification of FACS data. Data representative of 2 independent experiments, each done in triplicate. *p < 0.001, Student’s T-test. E. Western blot showing NFIB expression in NCI-H446 cell line that underwent lentiviral CRISPR-mediated NFIB deletion. Three NFIB-deleted sub-lines, derived from single cells, are shown along with two control sublines with guide RNAs targeting GFP, or a non-targeting control guide RNA. Two different sgRNA sequences were used, with one clone using sgRNA sequence NFIB#1 and two clones using sgRNA sequence NFIB#2 (see methods). F. CellTiter Glo experiment showing reduced proliferation in the NFIB-deleted H446 cells. G. Transwell migration assay in which NFIB-deleted and control H446 cells were transferred to transwells and allowed to migrate over 17 hours. Crystal violet stained migrated cells were counted. Each cell line was plated in triplicate transwells and results show mean +/- s.d. * p < 0.001, # p < 0.005, Student’s T-test.
NFIB transcriptional programs in SCLC
As a transcription factor that promotes SCLC, we were interested in identifying NFIB-regulated transcriptional programs in SCLC cells. The SCLC tumors and cells that differ in NFIB expression generated in this study provided ideal reagents to identify NFIB-regulated pathways. We performed RNAseq analyses on three complementary datasets. First, we compared gene expression in liver metastases from the Rb/p53 and Rb/p53/Ptenlox/+ models that exhibited high vs. low Nfib expression. Using RNAseq and EdgeR analysis, we identified 1174 genes to be differentially expressed (FDR <0.05) (Supplemental Table 2). Second, we compared SCLCs from the Rb/p53 (RTTA controls) vs. Rb/p53/TET-Nfib lung tumors that arose in the presence of doxycycline. Here, we identified 1324 genes that were differentially expressed in an NFIB dependent manner (Supplemental Table 3); 475 of these overlapped with the differentially expressed genes from the previous liver metastasis comparison (Figure 4A). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of this 475-gene set using DAVID (Database for Annotation, Visualization and Integrated Discovery) revealed significant enrichment in axon guidance, tight junctions, phosphatidylinositol signaling, and other pathways (Figure 4A). Third, we compared SCLC cell lines derived from Rb/p53/TET-Nfib tumors that were maintained in the presence of DOX or collected 7 days following DOX removal; here we identified 372 differentially expressed genes (Supplemental Table 4). This comparison was particularly important, as we expect that all of the cells are tumor cells, in contrast to the analyses of tumor tissues, where gene expression changes include a stromal cell contribution. KEGG analysis of this 372-gene set revealed highly significant NFIB-dependent pathways (focal adhesion p = 5*10-9, extracellular matrix-receptor interaction p = 5*10-8, axon guidance, p = 5*10-8 and pathways in cancer p = 5*10-4) shown in Figure 4B. Considering all three differential expression comparisons, i.e. liver metastases with high vs. low Nfib expression, DOX-inducible Rb/p53/TET-Nfib vs. Rb/p53 lung tumors, and TET-Nfib cell lines with or without DOX, we found 66 genes to be commonly changed depending on Nfib status (Figure 4C, Supplemental Table 5). 62 of these genes changed in the same direction, 43 were up-regulated when Nfib expression was increased and 19 down-regulated. KEGG pathway analysis of these consistently Nfib-regulated 62 genes, showed the highest enrichment in axon guidance genes (p = 0.00004, Benjamani-Hochberg FDR q = 0.001). Axon guidance related genes including Epha5, Epha8, Slit1 and Unc5d were upregulated and PlexinA1 and Robo1 downregulated upon Nfib overexpression (Figure 4D, Supplemental Figure 3).
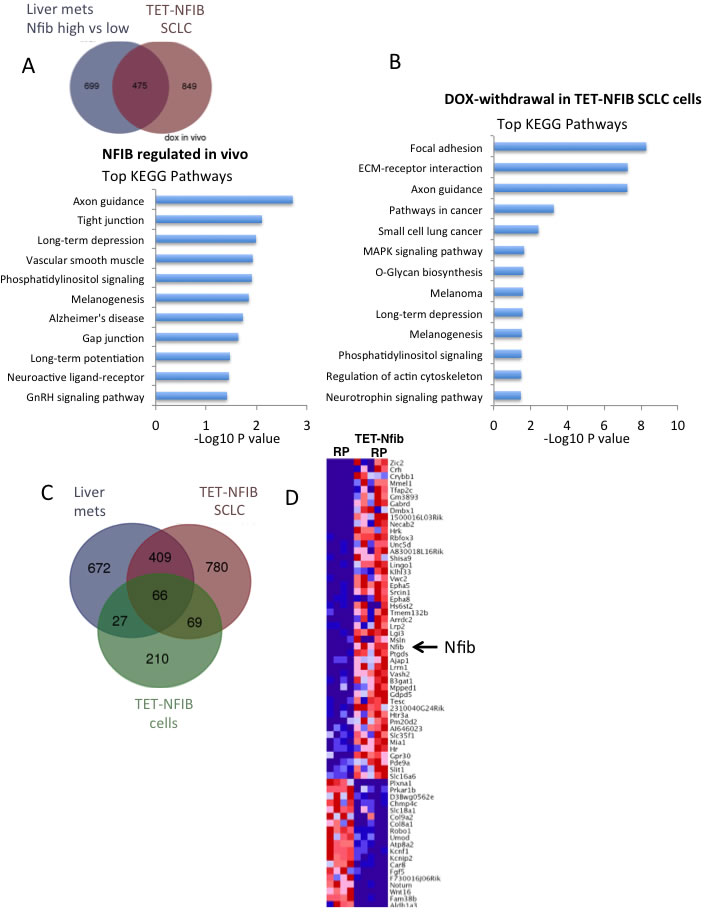
Figure 4: NFIB controlled transcriptional programs in SCLC. A. Top: Venn diagram showing overlapping differential expressed genes across two comparisons, including liver metastases with high vs. low Nfib expression and lung SCLCs from the Rb/p53 vs. Rb/p53/TET-Nfib models. 475 overlapping genes were differentially expressed in both liver metastasis and DOX-inducible lung tumor comparisons. Bottom: Top KEGG pathways enriched in this gene set are indicated with p-value shown on X-axis. B. KEGG pathways enriched in 372 genes differentially expressed in an RNAseq comparison of 4 Rb/p53/TET-Nfib SCLC cell lines, comparing cells on doxycycline vs. 7 days after doxycycline washout C. Venn diagram showing 66 genes were commonly differentially expressed across all three comparisons. D. Heat map showing the expression of the commonly altered 66 genes in the comparison of lung SCLCs from the Rb/p53 vs. Rb/p53/TET-Nfib models.
Discussion
The most metastatic form of lung cancer is small cell lung carcinoma (SCLC). Unfortunately, there is little known about the signaling pathways that control SCLC metastasis. Human metastatic SCLC is rarely resected, hindering our understanding of genetic determinants of SCLC metastasis. We compared genetic differences between primary SCLC and liver metastases from genetically engineered mouse models of SCLC. We identified Nfib as a gene that is frequently amplified in distant liver metastases (Figure 1). Nfib was previously found amplified in SCLC from the Rb/p53 model [10], but in vivo evidence that Nfib functions as an oncogene was needed. Thus, we interrogated Nfib oncogenic function using a sensitized mouse model of SCLC driven by inactivation of Rb and p53, genes inactivated in almost all human SCLCs. Our observation of acceleration of SCLC upon Nfib overexpression (Figure 2) provides definitive evidence that NFIB functions as an SCLC oncogene.
The mechanisms through which NFIB promotes SCLC are as yet unclear. We found that deletion of NFIB in a human SCLC cell line and NFIB suppression in cell lines derived from the Rb/p53/TET-Nfib SCLC mouse model led to reduced numbers of viable cells, with increased apoptosis and reduced proliferation (Figure 3). Our gene expression analysis showed that NFIB overexpression leads to activation of genes involved in axon guidance/neuronal function, which is consistent with roles for Nfib in brain development revealed through gene knockout studies [11]. Nfib also regulated genes involved in focal adhesions and extracellular matrix-receptor interactions (Figure 4). We also found reduced migration upon NFIB deletion in a human cell line harboring NFIB amplification. Our identification of NFIB-dependent gene expression differences in SCLC cells provides a starting point for functional interrogation of NFIB-regulated genes and pathways that promote SCLC.
By comparing primary SCLC to liver metastases from mouse SCLC models we found clear enrichment of Nfib amplifications in liver metastases (Figure 1). In matched pairs of lung tumors and liver metastases we observed Nfib amplifications in the metastasis but not in the bulk primary tumor. However, surprisingly, our mouse study did not reveal a higher rate of liver metastasis when NFIB was overexpressed at tumor initiation in the Rb/p53 mutant model. It may be that the acceleration of primary tumor development by Nfib overexpression limited our ability to detect a role for Nfib in driving liver metastasis, since the Nfib overexpressing mice died earlier with less time for metastasis to develop. Transplant experiments will help us address whether NFIB overexpressing tumor cells derived from the Rb/p53/TET-Nfib model exhibit increased propensity to metastasis. Also using the novel Rb/p53/TET-Nfib mouse model described, it will be interesting to activate Nfib expression not at tumor initiation but later in tumor progression. Following detection of lung tumor burden by magnetic resonance imaging, Nfib expression could be induced and the effect of Nfib overexpression in tumor progression discerned. For example, numbers of circulating tumor cells and influence of Nfib overexpression on rate of liver metastasis in this context could be assessed. It will also be interesting to remove doxycycline from Nfib-driven SCLC and assess the requirement for Nfib in the maintenance of lung tumors and liver metastases.
In human SCLC, NFIB expression can be deregulated through multiple mechanisms. It was previously shown that 16/46 human SCLC cell lines harbored NFIB copy number gains, including occasional focal high-level amplifications [10]. NFIB can also be highly expressed in SCLC cells through the presence of atypically large enhancers or super-enhancers [13, 14] and NFIB is a target gene of ASCL1, a master regulator of neuroendocrine cell fate [14]. As our mouse study shows that NFIB clearly exhibits oncogenic function in SCLC, it will be critical to compare human primary tumors and metastases to determine whether human SCLC also exhibits increased NFIB expression in metastases. Moreover, the extent to which genetic and non-genetic changes in human SCLC controls NFIB expression needs to be determined.
While this work was under review, two studies were published with data also supporting roles for NFIB in promoting SCLC and driving metastasis [21, 22]. Using a different transgenic allele, the Berns group found that NFIB overexpression also promoted small cell lung cancer in an Rb/p53-deleted background [22]. One difference compared to our work is the authors observed an increased rate of liver metastasis upon Nfib overexpression. It is possible that differences in the level of Nfib overexpression between the models could have an impact on metastasis rate. Overall, findings that Nfib amplifications were enriched in liver metastases over lung tumors (our study, Figure 1), along with the increased metastasis observed with NFIB overexpression in [22] and promotion of metastasis in transplant studies [21] all support NFIB as an oncogene in SCLC with roles in metastasis. NFIB was found to remodel chromatin and increase DNA accessibility in SCLC cells to promote metastasis [21]. It is now critical to identify the relevant NFIB target genes and mechanisms through which NFIB promotes advanced and metastatic SCLC.
Our data reveal that NFIB functions as a potent oncogene in SCLC. Patients with SCLC face a dismal clinical prognosis. It is essential that we make inroads to understand the pathways through which NFIB and other SCLC driver genes control the steps between tumor initiation and metastatic progression.
Materials and Methods
Mice
SCLC models bearing Rb/p53 and Rb/p53/Pten inactivation were previously described [4, 7, 9]. To test the effect of Nfib overexpression in vivo, the Rb/p53 floxed model was bred to mice with a Rosa26Lox-STOP-Lox reverse tetracycline controlled transactivator (rtTA) allele (Jackson Laboratories) and to a novel mouse strain that we generated harboring an inducible Tetracycline Responsive Element (TRE)-Nfib allele. Mice were maintained on a mixed genetic background. Adenoviral Cre driven by a CMV promoter was used to infect the lungs of mice via intratracheal delivery. A week after Cre delivery, mice were switched to doxycycline-containing chow (625mg/kg, Harlan) and were maintained on this feed through the duration of the experiment. Mice were euthanized when they exhibited signs of altered breathing patterns, which was typically caused by lung tumor burden.
Construction of TET-Nfib allele
To generate the doxycycline inducible Nfib transgene, the mouse Nfib cDNA containing an amino terminal hemagglutinin (HA) tag was placed under control of the (TetO)7CMV minimal promoter and the 3’ untranslated sequence and polyadenylation signal from the bovine growth hormone gene was added to generate a stable mRNA as described previously [23]. The HA-tagged Nfib cDNA was subcloned from pCHNFI-B2 [24] as a Not1- BamH1 (blunted with T4 polymerase) fragment into pCDNA3.1Teto-CMV-zeo [25] cut with Not1 and Xba1 (blunted with T4 polymerase). The final construct, pCMVtetoNFIB2, was verified by sequencing. This plasmid was digested with Spe1/Sph1 and the 2kb fragment containing (TetO)7CMVpromoter driven HA-tagged Nfib cDNA was purified and microinjected into F/VBN mouse oocytes by the transgenic core at Cincinnati Children’s Hospital Medical Center.
Next generation sequencing
RNAseq: Total RNA was isolated in Trizol (Invitrogen) and RNAseq libraries generated from oligo dT purified mRNA. We used the Truseq RNA kit (Illumina) for library generation. A HiSeq 2500 was used for sequencing, generating 50 base pair single end reads. After splicing aware alignment using Tophat [26] we used EdgeR [27] to identify differentially expressed transcripts. An FDR of 0.05 was used as a cutoff for identifying differentially expressed transcripts and the DAVID Bioinformatics Resources 6.7 online tool [28] (http://david.abcc.ncifcrf.gov/) was used to identify significant differentially expressed pathways, focusing on KEGG pathway analysis. We also generated FPKM values for each RNAseq comparison using Tophat/Cuffdiff (Supplemental Tables 6,7,8) [26].
CNV analysis: Genomic DNA was isolated from tumors and matched tail samples. Sequencing libraries were prepared using the NEBNext DNA library preparation kit (New England Biolabs). We used an Illumina Hiseq 2500, generating 50 base pair single end reads. The Burrows Wheeler Aligner (BWA) was used to align reads to the mm9 genome. Mapped reads were placed into 100kb bins and a read depth normalized ratio of tumor to normal was visualized using IGV. We also employed CNVseq to identify regions of copy number alteration [19].
Real-time RT-PCR
Total RNA from tumors were isolated using TRIzol (Invitrogen) following manufacturer’s instructions. cDNA was synthesized using SuperScript II reverse transcriptase (ThermoFisher Scientific). Quantitative PCR was performed with SYBR Green Real-Time PCR Master mix (ThermoFisher Scientific). Quantitative expression data were acquired and analyzed with a 7900 Real-time PCR System (Applied Biosystems). Primers were designed to detect endogenous Nifb and Gapdh (as endogenous control).
NFIB cDNA forward: GGGACTAAGCCCAAGAGACC
NFIB cDNA reverse: GTCCAGTCACAAATCCTCAGC
GAPDH (endogenous control) cDNA forward: AGCTTGTCATCAACGGGAAG
GAPDH (endogenous control) cDNA reverse: TTTGATGTTAGTGGGGTCTCG
CRISPR mediated NFIB deletion
The lentiCRISPR v2 CRISPR/Cas9 plasmid was obtained from Addgene (plasmid #52961 deposited by Feng Zhang). Guide RNA sequences were retrieved using the CRIPSR design tool at crispr.mit.edu. Sequences of primers for amplification of CRISPR-targeted genome follow:
Control forward: CACCG GTAGCGAACGTGTCCGGCGT
Control reverse: AAACACGCCGGACACGTTCGCTACC
GFP forward: CACCG GGGCGAGGAGCTGTTCACCG
GFP reverse: AAACCGGTGAACAGCTCCTCGCCCC
NFIB#1 forward: CACCGTCCGTGCAATTGCCTATACT
NFIB#1 reverse: AAACAGTATAGGCAATTGCACGGAC
NFIB#2 forward: CACCGGAGAATCGACTGCCTGCGAC
NFIB#2 reverse: AAACGTCGCAGGCAGTCGATTCTCC
CellTiter-Glo cell proliferation assay
Experiments were performed according to the manufacturer’s instructions (Promega). Briefly, 100μl of 5000 cells were seeded in white-walled 96-well plates. Plate was equilibrated at room temperature for 30 min before added CellTiter-Glo reagent at a 1:1 ratio to the cell culture volume. The plate was then mixed for 2 minutes on an orbital shaker and incubated at room temperature for another 10 minutes to stabilize the luminescent signal. Luminescence was recorded on plate reader (BioTek) at 1 second per well integration time.
FACS analysis
Cells were resuspended in 0.6 ml PBS and fixed with 1.4 ml ice-cold 100% ethanol. Cells were treated with 0.1mg/ml RNase and 25μg/ml propidium iodide and analyzed on a flow cytometer. FACS data was analyzed with FlowJo Software.
Cell migration assay
Briefly, 800μl of DMEM containing 10% FBS was placed in the lower chamber of 24-well 8 micron Transwells (Falcon). 200μl of 20,000 cells were placed in the upper chamber and allowed to migrate for 17 hours at 37 °C. At the end of the migration assay, the filter side of the upper chamber was cleaned with a cotton swab. The filters were then fixed in 100% methanol for 5 min, stained with crystal violet staining solution (0.05% crystal violet, 1% formaldehyde, 1% methanol in PBS) for 10 minutes and rinsed with water. The filters were air-dried and the cells that have migrated through filter pores were counted. For each migration condition, the number of cells that migrated across the filters in 4X high-power fields per insert was counted. Triplicate transwells were employed for each condition.
Histology and immunohistochemistry
We fixed lungs in formalin for 24 h before paraffin embedding. Paraffin blocks were sectioned at four-microns and stained with hematoxylin and eosin. Paraffin sections were cut and processed from xylene through a graded ethanol series (100%, 95% and 70%) to PBS. Unmasking was performed using microwave heating in sodium citrate buffer (0.01M at pH6.0). Endogenous peroxidases were blocked with 3.5% H2O2, and immunohistochemistry was performed with an overnight incubation with anti-CGRP antibody (Sigma C8198, 1:1000). Biotin-conjugated secondary antibodies (Vector Laboratories) were used at a dilution of 1/200 in blocking solution. After secondary antibody binding, detection was performed via a biotin-peroxidase complex (Vectastain ABC, Vector Laboratories) with DAB substrate (Vector Laboratories). Haematoxylin was used to counterstain.
Acknowledgments
The authors acknowledge the expert help of the Fred Hutch Genomics and Bioinformatics Shared Resource for carrying out next generation sequencing and providing assistance with data analysis. We thank Paul Shannon for help with the CNVseq analysis. We acknowledge the Fred Hutch Histology core for processing tissue and performing H+E staining. We thank Emily Eastwood for help with figure preparation.
Conflicts of Interests
We have no potential conflicts of interest to disclose.
Grant Support
DM acknowledges support from NIH grant R01CA197867. RMG acknowledges support from NYSTEM grants C026429 and C030133.
References
1. Jackman DM and Johnson BE. Small-cell lung cancer. Lancet. 2005; 366:1385-1396.
2. George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, Muller C, Dahmen I, Jahchan NS, Park KS, Yang D, Karnezis AN, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015; 524:47-53.
3. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, Bhatt D, Stinson J, Gnad F, Haverty PM, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nature genetics. 2012.
4. Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ and Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003; 4:181-189.
5. Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY and Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011; 19:754-764.
6. McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, Bhutkar A, McKenna A, Dooley A, Vernon A, Sougnez C, Malstrom S, Heimann M, Park J, Chen F, Farago AF, et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell. 2014; 156:1298-1311.
7. Kim DW, Wu N, Kim YC, Cheng PF, Basom R, Kim D, Dunn CT, Lee AY, Kim K, Lee CS, Singh A, Gazdar AF, Harris CR, Eisenman RN, Park KS and MacPherson D. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev. 2016; 30:1289-1299.
8. Huijbers IJ, Bin Ali R, Pritchard C, Cozijnsen M, Kwon MC, Proost N, Song JY, de Vries H, Badhai J, Sutherland K, Krimpenfort P, Michalak EM, Jonkers J and Berns A. Rapid target gene validation in complex cancer mouse models using re-derived embryonic stem cells. EMBO molecular medicine. 2014; 6:212-225.
9. Cui M, Augert A, Rongione M, Conkrite K, Parazzoli S, Nikitin AY, Ingolia N and Macpherson D. PTEN is a Potent Suppressor of Small Cell Lung Cancer. Mol Cancer Res. 2014.
10. Dooley AL, Winslow MM, Chiang DY, Banerji S, Stransky N, Dayton TL, Snyder EL, Senna S, Whittaker CA, Bronson RT, Crowley D, Barretina J, Garraway L, Meyerson M and Jacks T. Nuclear factor I/B is an oncogene in small cell lung cancer. Genes & development. 2011; 25:1470-1475.
11. Steele-Perkins G, Plachez C, Butz KG, Yang G, Bachurski CJ, Kinsman SL, Litwack ED, Richards LJ and Gronostajski RM. The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol Cell Biol. 2005; 25:685-698.
12. Grunder A, Ebel TT, Mallo M, Schwarzkopf G, Shimizu T, Sippel AE and Schrewe H. Nuclear factor I-B (Nfib) deficient mice have severe lung hypoplasia. Mech Dev. 2002; 112:69-77.
13. Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, Zhang J, Shimamura T, Capelletti M, Reibel JB, Cavanaugh JD, Gao P, et al. Targeting Transcriptional Addictions in Small Cell Lung Cancer with a Covalent CDK7 Inhibitor. Cancer Cell. 2014; 26:909-922.
14. Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, Girard L, Minna JD, Gazdar AF, Cobb MH and Johnson JE. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell reports. 2016.
15. Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, Reichert E, Kung AL, Rodig SJ, Young RA, Shipp MA and Bradner JE. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013; 24:777-790.
16. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA and Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934-947.
17. Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, Sanda T, Sim T, Kim ND, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014; 511:616-620.
18. Chiang DY, Getz G, Jaffe DB, O’Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M and Lander ES. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009; 6:99-103.
19. Xie C and Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009; 10:80.
20. Gazdar AF, Savage TK, Johnson JE, Berns A, Sage J, Linnoila RI, MacPherson D, McFadden DG, Farago A, Jacks T, Travis WD and Brambilla E. The comparative pathology of genetically engineered mouse models for neuroendocrine carcinomas of the lung. Journal of thoracic oncology. 2015; 10:553-564.
21. Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Gruner BM, Chiou SH, Schep AN, Baral J, Hamard C, Antoine M, Wislez M, Kong CS, Connolly AJ, Park KS, Sage J, et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell. 2016; 166:328-342.
22. Semenova EA, Kwon MC, Monkhorst K, Song JY, Bhaskaran R, Krijgsman O, Kuilman T, Peters D, Buikhuisen WA, Smit EF, Pritchard C, Cozijnsen M, van der Vliet J, Zevenhoven J, Lambooij JP, Proost N, et al. Transcription Factor NFIB Is a Driver of Small Cell Lung Cancer Progression in Mice and Marks Metastatic Disease in Patients. Cell reports. 2016; 16:631-643.
23. Bachurski CJ, Yang GH, Currier TA, Gronostajski RM and Hong D. Nuclear factor I/thyroid transcription factor 1 interactions modulate surfactant protein C transcription. Mol Cell Biol. 2003; 23:9014-9024.
24. Chaudhry AZ, Vitullo AD and Gronostajski RM. Nuclear factor I (NFI) isoforms differentially activate simple versus complex NFI-responsive promoters. J Biol Chem. 1998; 273:18538-18546.
25. Gossen M and Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992; 89:5547-5551.
26. Trapnell C, Pachter L and Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105-1111.
27. Robinson MD, McCarthy DJ and Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139-140.
28. Huang da W, Sherman BT and Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44-57.