
INTRODUCTION
Esophageal squamous cell carcinoma (ESCC) is one of the most prevalent cancers in African and Asian countries, accounting for >90% of all esophageal carcinomas in the worldwide [1]. Approximately 482,300 new cases of esophageal cancer are diagnosed annually, and the disease is responsible for approximately 406,800 deaths each year [2]. The disease is often advanced at presentation and has a poor prognosis despite the use of multidisciplinary therapy. The biological and pathological features of ESCC have not been well demonstrated, although multiple genetic and epigenetic changes have been detected in ESCC [3, 4]. Therefore, additional understanding of the molecular mechanisms of esophageal cancer is urgently needed to devise more effective treatment.
DNA methylation is one of the most common epigenetic modifications in mammalian genomes [5]. Aberrant methylation of gene promoter subsequently results in the inactivation of gene expression. In particular, hypermethylation of cytosine-guanine dinucleotide (CpG) islands in promoter regions has been strongly implicated in the onset and progression of cancer [6]. Aberrant methylation of the gene promoter has become widely recognized as a mechanism of gene inactivation in cancer [7].
The protein tyrosine phosphatase non-receptor type 22 (PTPN22) gene is located on chromosome 1p13.3–13.1 and participates in epithelial adhesion. In chronic lymphocytic leukemia (CLL), a study showed that overexpression of PTPN22 significantly inhibits antigen-induced apoptosis of primary CLL cells by blocking B-cell receptor (BCR) signaling pathways that negatively regulate lymphocyte survival. More importantly, this finding indicates that PTPN22 positively regulates antiapoptotic AKT kinase, providing a powerful survival signal to antigen-stimulated CLL cells [8]. Several large-scale comprehensive analyses, which were conducted using high-throughput sequencing technology, have revealed that the epidermal growth factor receptor (EGFR) signaling pathway and related downstream pathways, such as the PI3K/AKT signaling pathway, are considered to be involved in the development of ESCC [9-11]. Additionally, the TCGA database showed that there was a significant difference in the methylation level of PTPN22 between tumor tissues and normal tissues. However, despite the potential importance of the PTPN22 gene in carcinogenesis, there have been very few reports concerning ESCC. No study has been conducted regarding the expression and methylation of PTPN22 in ESCC. In the present study, we attempted to ascertain the methylation level and expression of PTPN22 in ESCC and the correlation between the PTPN22 methylation change and a series of clinicopathological parameters in a large sample of ESCC patients to elucidate the role of PTPN22 in the pathogenesis of ESCC and its potential role as a prognostic marker.
RESULTS
Methylation analysis of PTPN22
Using Vector NTI Advance 11 (Invitrogen), we compared the CpG density in different regions of the promoter and chose to analyze the region chr1:114,358,291-114,358,739 [GRCh37/hg19]. The selected region contains elevated CpG content and is easy to amplify. The amplicon was designed and included seven CpG sites (Figure 1). All seven CpG sites (CpG1, CpG2, CpG3, CpG4, CpG5, CpG6, and CpG7) were successfully detected using the quantitative Sequenom method (Table 1). The threshold value for methylation detection was 10%. The methylation level of the CpG sites in the selected region was subjected to paired analyses; three of the seven CpG sites (CpG2, CpG4, CpG5) exhibited significant differences between ANT and tumor tissue (80.2% vs. 88.3%, 91.4% vs. 94.9%, and 75.2% vs. 83.8%, respectively). All three of these CpG sites were significantly hypermethylated in ESCC tissues (Figure 2A). The mean methylation level of all CpG sites was 62.1% in ANT and 66.3% in tumor tissue. The difference was statistically significant (p=0.005, Table 1).
Figure 1: Schematic diagram of the CpG sites in the selected region. The CpG sites are depicted by lollipop markers. The binding sites for the forward and reverse primers are depicted as arrows below the diagram.
Table 1: Methylation level (%) of the detected sites in the selected region
CpGs |
Group |
Mean |
ΔMean |
P |
|---|---|---|---|---|
CpG1 |
Normal |
49.5 |
4.2 |
0.127 |
Tumor |
53.7 |
|||
CpG2 |
Normal |
80.2 |
8.1 |
<0.0001 |
Tumor |
88.3 |
|||
CpG3 |
Normal |
54.5 |
0.3 |
0.231 |
Tumor |
54.8 |
|||
CpG4 |
Normal |
91.4 |
3.5 |
0.007 |
Tumor |
94.9 |
|||
CpG5 |
Normal |
75.2 |
8.6 |
0.0002 |
Tumor |
83.8 |
|||
CpG6 |
Normal |
57.1 |
0.5 |
0.798 |
Tumor |
57.6 |
|||
CpG7 |
Normal |
43.9 |
0.7 |
0.836 |
Tumor |
44.6 |
|||
All Sites |
Normal |
62.1 |
4.2 |
0.005 |
Tumor |
66.3 |
ΔMean: the difference of mean methylation level between tumor and adjacent normal tissue
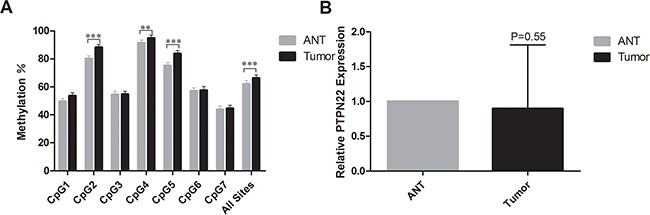
Figure 2: A. The mean methylation level for each CpG site in PTPN22. ***represents p<0.001, **represents p<0.01, and *represents p<0.05; B. The mRNA expression level of PTPN22. *represents p<0.05.
Relative PTPN22 expression level in ESCC samples by real-time PCR
The relative PTPN22 expression level was quantified in 43 paired tumor tissues and ANT. All samples were selected from the same cohort with methylation detected and run in triplicate to capture intra-assay variability. Then 31 paired samples were finally included to be analyzed. Lower expression was observed in 18 tumor samples, and higher expression was observed in 13 samples. The relative PTPN22 expression level was reduced in the tumor tissue compared with that in the corresponding ANT, but the difference was not statistically significant (ANT vs. tumor: 1 vs. 0.9, respectively; p=0.55) (Figure 2B). Linear regression analysis showed that lymph node metastasis and TNM stage were inversely associated with the expression level. Lower expression was more frequently observed in N1-3 and III stage patients, while higher expression was more likely to occur in N0 and I-II stage patients (Figure 3). The results were consistent with the reduced expression level in tumor tissue. Furthermore, overall survival was better in patients with lower PTPN22 expression than in patients with a higher expression level (Figure 4, p=0.04).
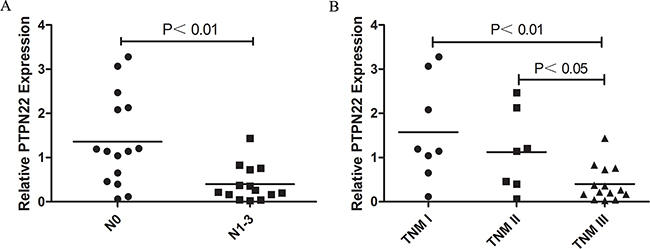
Figure 3: A. Correlation of PTPN22 expression and lymph node invasion; B. Correlation of PTPN22 expression and TNM stage.
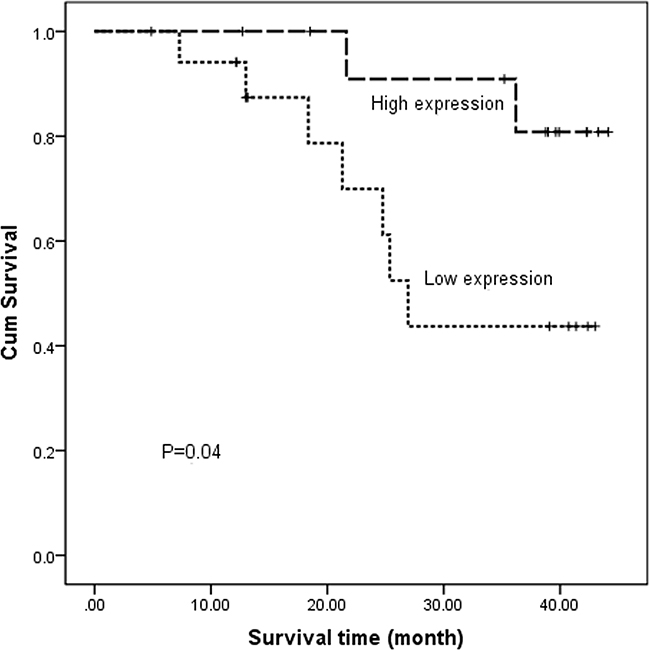
Figure 4: Kaplan–Meier estimates of overall survival for ESCC patients according to different PTPN22 expression levels.
Correlation of the PTPN22 methylation change and clinicopathological data
We analyzed the correlation between the methylation change (tumor/ANT) and multiple clinicopathological parameters. Smoking and N stage were found to be associated with the methylation change between the tumor and ANT samples (p<0.05) (Table 2). A distinct methylation change existed in non-smoking and N0 patients compared with smoking and N1-3 patients (8.3% vs. 2.1%, 7.5% vs. 1.4%, respectively). Additionally, the methylation level in the ANT of non-smokers was significantly lower than that in the ESCC tissue of smokers (59.1% vs. 65.8%, p=0.03); similarly, the methylation levels in ANT with no lymph node invasion were significant lower than those in tumor tissues with lymph node invasion (N0 57.6% vs. N1-3 67.5%, p=0.001). For other analyzed elements, including age, alcohol habit, nerve invasion, vessel invasion and TNM stage, there was no significant association with the methylation change in ESCC (Table 2).
Table 2: Association between methylation difference (%) and clinicopathological parameters
Parameters |
NO. |
Normal |
Tumor |
ΔMean |
P |
|---|---|---|---|---|---|
Gender |
0.66 |
||||
Male |
111 |
61.4 |
65.7 |
4.3 |
|
Female |
10 |
70.6 |
72.7 |
2.1 |
|
Age |
0.53 |
||||
<58 |
52 |
61.6 |
66.8 |
5.2 |
|
≥58 |
69 |
62.6 |
66.0 |
3.4 |
|
Alcohol habit |
0.43 |
||||
No |
99 |
59.8 |
65.1 |
5.3 |
|
Yes |
86 |
65.1 |
67.9 |
2.8 |
|
Smoking |
0.05 |
||||
No |
41 |
59.1 |
67.4 |
8.3 |
|
Yes |
80 |
63.7 |
65.8 |
2.1 |
|
Nerve invasion |
0.63 |
||||
No |
95 |
62.4 |
66.9 |
4.5 |
|
Yes |
26 |
61.2 |
64.2 |
3.0 |
|
Vessels invasion |
0.86 |
||||
No |
102 |
62.5 |
66.5 |
4.0 |
|
Yes |
19 |
60.4 |
65.5 |
5.1 |
|
T stage |
0.88 |
||||
T1-2 |
27 |
59.1 |
63.0 |
3.9 |
|
T3 |
94 |
63.0 |
67.3 |
4.3 |
|
N stage |
|||||
N0 |
57 |
57.6 |
65.1 |
7.5 |
0.04 |
N1-3 |
64 |
66.1 |
67.5 |
1.4 |
|
TNM stage |
0.13 |
||||
I-II |
61 |
58.6 |
65.1 |
6.5 |
|
III |
60 |
65.7 |
67.6 |
1.9 |
|
Differentiation |
0.46 |
||||
Well |
13 |
65.8 |
64.9 |
-0.9 |
|
Moderate |
68 |
61.1 |
66.5 |
5.4 |
|
Poor |
40 |
62.7 |
66.6 |
3.9 |
|
Lesion location |
0.15 |
||||
Upper thoracic |
44 |
61.7 |
67.9 |
6.2 |
|
Middle thoracic |
46 |
63.6 |
64.1 |
0.5 |
|
Low thoracic |
31 |
60.5 |
67.4 |
6.9 |
Association of the PTPN22 methylation level with overall survival and progression-free survival
To determine whether a correlation exists between PTPN22 methylation and patient survival, we analyzed five-year clinical follow-up data according to methylation levels. The PTPN22 methylation level was classified into two groups according to the cut-off value (70.3%), which was calculated as the mean of the normal samples (62.1%) to which we applied 0.5 SD (8.2%). A reduced methylation level in the tumor relative to the cut-off was defined as group 1. Similarly, an elevated methylation level in the tumor relative to the cut-off was defined as group 2. The 1-year, 3-year and 5-year rates of overall survival (OS) and progression-free survival (PFS) were analyzed using the Kaplan–Meier method (Table 3). No significant correlation was observed between methylation levels and prognosis (OS, p=0.554; PFS, p=0.614; respectively).
Table 3: Overall survival (OS) and progression-free survival (PFS) rates
OS rate (%) |
PFS rate (%) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
1-year |
3-year |
5-year |
P |
1-year |
3-year |
5-year |
P |
||
Region |
Group 1 |
83.5 |
58.1 |
42.2 |
0.554 |
73.6 |
46.5 |
31.6 |
0.614 |
Group 2 |
79.7 |
49.5 |
40.4 |
71.4 |
32.4 |
30.6 |
|||
Group 1: decreased methylation level of tumor relative to the cut-off (70.3%)
Group 2: elevated methylation level of tumor relative to the cut-off (70.3%)
DISCUSSION
In this study, we examined the methylation level of seven CpG sites in the selected region of PTPN22 (chr1:114,358,291-114,358,739 [GRCh37/hg19]) and observed that all seven sites exhibited hypermethylation in tumor tissue. The mean methylation level of all detected CpG sites was higher in ESCC. The selected region was not the conventional promoter region. The CpG site density in the promoter region was lower than that in the selected region in our analysis. There were only three or four CpG sites in the promoter region (Supplementary Figures 1, 3,000-5,200bp). According to the instructions of the SpectroCHIP® Arrays, quantification of at least four CpG sites was required for detection. In addition, the repeatability and consistency of the promoter region were poor because of rare CpG sites. Thus, combined with bioinformatics software, we selected the abovementioned region for analysis.
According to MATCHTM public version 1.0 software (BIOBASE BIOLOGICAL DATABASES, http://www.gene-regulation.com/index2.html), Paired box gene 4 (PAX4) was found to be bound in the selected region. PAX4 is a transcriptional modulator located on chromosome 7q32. Its expression is dysregulated in various human cancers. Patients with high PAX4 expression levels demonstrated lower 5-year survival rates in HNSCC, gastric cancer and breast cancer. Multiple studies have suggested that PAX4 may act as a driver gene in multiple tumors [12, 13]. Studies have indicated that miR-144 and miR-451(miR-144/451) upregulation inhibits cancer cell migration and invasion. However, PAX4 upregulation decreased miR-144/451 levels. PAX4 promoted migration and invasion in human epithelial cancers by decreasing the miR-144/451expression level [14]. We hypothesize that PAX4 may act as a regulator of PTPN22 in the development of ESCC. The analysis of the expression of PAX4 in ESCC is warranted in a future investigation. In vitro, the expression of PAX4 should be introduced to analyze the effect of the demethylation of PTPN22 on the expression of PAX4. Future experiments will focus on investigating the relationship between PTPN22 and PAX4.
Hypermethylation of CpG islands in gene promoter regions always results in the down-regulation of a gene [15, 16]. In the current study, the relative expression of the PTPN22 gene in tumor tissue was lower than that in ANT, although the difference was not significant. Combined with the hypermethylation in tumor tissue, the current results suggest that aberrant PTPN22 methylation may suppress the expression of PTPN22 mRNA in esophageal cancer. Additionally, lower expression more frequently occurred in N1-3 and III stage patients than in N0 and I-II stage patients. Combined with the higher methylation level in N1-3 and III stage patients, these phenomena were also consistent with the notion that gene hypermethylation down-regulates gene expression. PTPN22 expression was associated with prognosis. Overall survival was poor in patients with lower PTPN22 expression than in the cases with a higher expression level. PTPN22 expression may act as a predictor of prognosis in ESCC.
To assess the clinical significance of PTPN22 methylation, we examined the association between PTPN22 methylation changes and the clinicopathological characteristics of esophageal cancer. The mean methylation level was elevated in the tumors of patients with lymph node invasion and a history of smoking relative to normal tissues (Table 2). This finding is consistent with the opinion that various environmental and lifestyle exposures, such as those related to tobacco, alcohol, and radiation, are implicated in the development of human cancers by inducing DNA methylation changes [17]. Studies have demonstrated that tobacco smoking is associated with aberrant gene methylation in several cancers [18, 19]. Additionally, some studies have suggested a trend toward a higher risk of advanced T stage (p=0.05) or lymph node metastasis (p=0.02) when more adverse gene methylation profiles are present [20-23]. Our results confirmed that smoking and lymph node metastasis are risk factors for carcinogenesis. The results revealed that the difference in the methylation level of tumor tissue and ANT was larger in non-smoking and N0 patients than in smoking and N1-3 patients.
The methylation level in well-differentiated tumors was lower than that in normal tissue (ΔMean=-0.9%; Table 2). The methylation change was significantly different from that in tumors with moderate or poor differentiation (ΔMean=5.4% or 3.9%, respectively; Table 2). The functional consequences of DNA methylation changes in normal differentiation and cancer remain to be elucidated. With respect to the relationship between methylation and differentiation, studies have hypothesized that two potential mechanisms, loss of cell fate-determining transcription factors by methylation and functional inactivation of corresponding genomic-binding sites by DNA methylation, can promote cellular differentiation defects, thus enhancing the ability of tumor progenitor cells to transition toward ESCC [24]. Negative associations with other clinical factors may be due to the small sample size of this study. Another reason maybe that the analyzed region is not the key promoter region and do not affect PAX4 significantly. In the future, an intensive study with an increased number of patients included is needed to investigate the clinico-epigenetic combination.
No significant correlation has been observed between PTPN22 methylation and prognosis. In other malignancies, gene-specific DNA hypermethylation can predict PFS and OS and is always associated with unfavorable clinical outcomes, lower survival rates and aggressive behavior [25-27]. Our results were not as significant as those of previous studies. In previous studies, a qualitative research-methylation specific PCR (MSP) was used, according to which the methylated and unmethylated groups were divided [28, 29]. A significant correlation was more likely to be observed between methylation and clinicopathological features. However, in the current study, a quantitative research was used to investigate the clinical significance of aberrant methylation of PTPN22. The heterogeneity of cancer cells may impair the trend in the methylation change between ANT and tumor tissue [30]. The heterogeneity weakened the clinical significance of the methylation change in the present study. In addition, the included sample size may not be sufficient to observe a significant difference.
Additionally, there were some limitations to the study. No cell culture was conducted to further investigate the correlate on between PTPN22 and PAX4. This exploration is critical for some findings in the present study. What’s more, PTPN22 exhibits overexpression in CLL and positively regulates the antiapoptotic AKT kinase, which provides a powerful survival signal to antigen-stimulated CLL cells [8]. However, no significant difference in PTPN22 expression was shown in ESCC. The expression level of PTPN22 in ESCC merits further exploration in a larger sample size.
In conclusion, hypermethylation of the PTPN22 gene was observed in ESCC. Distinct methylation changes occurred more frequently in non-smoking and N0 stage patients. PTPN22 expression was inversely correlated with lymph node metastasis and TNM stage. Lower expression of PTPN22 was associated with poor overall survival. PTPN22 expression may act as a predictor of tumor grade.
MATERIALS AND METHODS
Patients and samples
Human primary ESCC and corresponding (5 cm from the tumor) adjacent normal tissue (ANT) were collected from 121 patients who were diagnosed and treated at Fudan University Cancer Hospital (Shanghai, China) from September 2007 to June 2011. The clinicopathological features of the patients are summarized in Table 4. The tissue samples were snap frozen in liquid nitrogen immediately after surgical resection and then were stored at -80°C until DNA and RNA were extracted. The pathological features were evaluated by independent pathologists according to the TNM staging system of the American Joint Committee on Cancer (AJCC 7th edition). Clinical, pathological, and follow-up data were stored in a database in accordance with hospital privacy rules.
Table 4: Clinicopathological characteristics of patients
Characters |
Type |
NO. |
|---|---|---|
Sex |
Female |
10 (8.3%) |
Male |
111 (91.7%) |
|
Alcohol habit (≥50g/day) |
No |
68 (56.2%) |
Yes |
53 (43.8%) |
|
Smoking |
No |
41 (33.9%) |
Yes |
80 (66.1%) |
|
Family History |
No |
110 (90.9%) |
Yes |
11 (9.1%) |
|
Age (mean ± SD) |
57.8±6.4 |
|
Diameter of tumor (mean ± SD) |
3.03±2.26 |
|
Tumor differentiation status |
Poor |
13 (10.6%) |
Moderate |
68 (55.3%) |
|
Well |
40 (32.5%) |
|
Lesion location |
Upper thoracic |
44 (36.4%) |
Middle thoracic |
46 (38.0%) |
|
Low thoracic |
31 (25.6%) |
|
T stage |
T1 |
7 (5.8%) |
T2 |
20 (16.5%) |
|
T3 |
94 (77.7%) |
|
N stage |
N0 |
57 (47.1%) |
N1 |
39 (32.2%) |
|
N2 |
16 (13.2%) |
|
N3 |
9 (7.4%) |
|
TNM stage |
I |
7 (5.8%) |
II |
54 (44.6%) |
|
III |
60 (49.6%) |
All patients were followed-up after primary treatment at intervals increasing from 3 months to 1 year until death or the end of the study. The follow-up period ranged from 2 months to 6.1 years (median: 1.7 years) for esophageal cancer patients. Routine examinations, including systemic review, tumor marker testing, endoscopic examination, chest X-ray, and computed tomography, were performed to evaluate the outcome of the disease, which was classified as disease-free, relapse, or death according to the WHO criteria for clinical response. The study protocol was approved by the ethics committee of Fudan University Cancer Center and informed consent was obtained from all participants.
DNA preparation and bisulfite conversion
Genomic DNA was isolated from ≥25 mg of tissue (tumor/normal) using the QIAamp DNA Mini Kit following the standard protocol (QIAGEN, Hilden, Germany). DNA concentrations were determined using the Thermo NanoDrop2000 system (Thermo, Wilmington, USA). The purity of DNA was verified by monitoring the ratio of absorbance at 260 nm to that at 280 nm, which was in the range of 1.8–2.0. Briefly, 400–500 ng of genomic DNA was chemically modified with sodium bisulfite using the EpiTect Bisulfite kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol.
MassARRAY quantitative methylation analysis
The sodium bisulfite-treated genomic DNA was amplified by PCR, and the PCR products were confirmed by gel electrophoresis. The samples were considered positive only if the selected band was comparable to 400- to 500-bp standards. The primer sets used for PCR were as follows: 5′-GGTTGATTAGTTTGAGTTTTTTTGG-3′ (forward primer) and 5′-ACAACAACTCTATCTCAATTCACTACAA-3′ (reverse primer). Only qualified samples were permitted for use in subsequent procedures, including the SAP cleanup, T Cleavage and Clean Resin steps.
The products were then transferred to SpectroCHIP® Arrays, and methylation of the selected region in PTPN22 was detected by MassARRAY spectrometry, an efficient EpiTYPER platform for high throughput analysis of DNA methylation patterns. The gene mass spectrogram was drawn using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS). The quantitative methylation level of each CpG site or aggregates of multiple CpG sites were analyzed using MassARRAY Analyzer 4 (Sequenom, USA).
RNA isolation and real-time PCR
Total RNA was isolated using Trizol reagent (Invitrogen, USA) according to the manufacturer’s instructions. Agarose gel (1%) electrophoresis and spectrophotometric analysis (OD260:280 nm ratio) were used to evaluate RNA quality. First-strand cDNA was synthesized according to the manufacturer’s instructions; 5 μg of total RNA was used to synthesize first-strand cDNA with random six-mer primers using a Superscript III-reverse transcriptase kit (Invitrogen, USA). Following first-strand synthesis, the reaction mixture was diluted to 100 μl with water. Subsequently, 2.5 μl of diluted cDNA mixture was used for PCR amplification in a final 20-μl reaction volume. FastStart Universal SYBR Green Master Mix Rox (Roche, Germany) was used as an amplification reaction mixture according to the manufacturer’s instructions. Real-time Quantitative PCR was performed in an ABI VIIATM 7 Thermal Cycler (Life Technologies, Carlsbad, California, USA). The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal control. The following oligonucleotide primers were used: PTPN22-FP, 5′-AGGCAGACAAAACCTATCCTACA-3′; PTPN22-RP, 5′-TGGGTGGCAATATAAGCCTTG-3′; GAPDH-FP, 5′-ACAACTTTGGTATCGTGGAAGG-3′; and GAPDH-RP, 5′-GCCATCACGCCACAGTTTC-3′. All samples were run in triplicate to capture intra-assay variability. The expression level of PTPN22 was analyzed using the 2-ΔΔCT method, where ΔCt = Ct (PTPN22)-Ct (GAPDH) and ΔΔCt = ΔCt (Tumor)−ΔCt (ANT). The expression level in control ANT was set as 1, and the 2-ΔΔCT value was the relative expression level in tumor tissue.
Statistical analysis
The total methylation differences or individual CpG site methylation differences between the tumor and ANT tissues were analyzed by paired two-tailed Student’s t-test. Associations between participant characteristics and methylation differences were assessed using an independent sample t-test and one-way ANOVA. The endpoints were death and disease progression. Overall survival (OS) was calculated from the date of surgery to the date of death or last contact on or before June 2011. Progression-free survival (PFS) was calculated from the date of surgery to the presentation of clinical or pathological evidence of disease recurrence or the last contact on or before June 2011. The Kaplan–Meier method was used for univariate survival analysis, and the log-rank test was used to assess the difference between survival curves. Cox’s proportional hazards analysis was used to estimate the prognostic effects of various variables. A P value <0.05 was considered to be statistically significant. These statistical calculations were performed using SPSS version 20.0.
ACKNOWLEDGMENTS
This study was financially supported by the National Natural Science Foundation of China Research (grant number 21172043), the National Natural Science Foundation of China (31200954 Beijing, PR China) and the China Postdoctoral Science Foundation Funded Project (2012M510110, 2013T60440 Beijing, PR China).
CONFLICTS OF INTEREST
The authors declare no conflicts of interests.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61: 69-90.
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65: 87-108.
3. Wang JD, Ma J, Wang FY, Peng LB, Wang X, Shi SS, Ma HH, Lu ZF, Lu GM, Zhou XJ. Amplification of the telomerase RNA component gene as a new genetic marker for disease progression and prognosis in esophageal squamous cell carcinoma. Dis Esophagus. 2013; 26: 737-45.
4. Dong Z, Guo W, Guo Y, Kuang G, Yang Z. Concordant promoter methylation of transforming growth factor-beta receptor types I and II occurs early in esophageal squamous cell carcinoma. Am J Med Sci. 2012; 343: 375-81.
5. Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004; 22: 4632-42.
6. Lewandowska J, Bartoszek A. DNA methylation in cancer development, diagnosis and therapy--multiple opportunities for genotoxic agents to act as methylome disruptors or remediators. Mutagenesis. 2011; 26: 475-87.
7. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004; 429: 457-63.
8. Negro R, Gobessi S, Longo PG, He Y, Zhang ZY, Laurenti L, Efremov DG. Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood. 2012; 119: 6278-87.
9. Song Y, Li L, Ou Y, Gao Z, Li E, Li X, Zhang W, Wang J, Xu L, Zhou Y, Ma X, Liu L, Zhao Z, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014; 509: 91-5.
10. Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, Sato Y, Okuno Y, Varela AM, Ding LW, Garg M, Liu LZ, Yang H, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014; 46: 467-73.
11. Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, Zhang F, Zhao ZR, Li ZT, Liu ZY, Zhao YD, Sun J, Zhou CC, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014; 46: 1097-102.
12. Zhang J, Qin X, Sun Q, Guo H, Wu X, Xie F, Xu Q, Yan M, Liu J, Han Z, Chen W. Transcriptional control of PAX4-regulated miR-144/451 modulates metastasis by suppressing ADAMs expression. Oncogene. 2015; 34: 3283-95.
13. Jiang HL, Sun HF, Gao SP, Li LD, Huang S, Hu X, Liu S, Wu J, Shao ZM, Jin W. SSBP1 Suppresses TGFbeta-Driven Epithelial-to-Mesenchymal Transition and Metastasis in Triple-Negative Breast Cancer by Regulating Mitochondrial Retrograde Signaling. Cancer Res. 2016; 76: 952-64.
14. Schaner ME, Ross DT, Ciaravino G, Sorlie T, Troyanskaya O, Diehn M, Wang YC, Duran GE, Sikic TL, Caldeira S, Skomedal H, Tu IP, Hernandez-Boussard T, et al. Gene expression patterns in ovarian carcinomas. Mol Biol Cell. 2003; 14: 4376-86.
15. Guo W, Zhang M, Shen S, Guo Y, Kuang G, Yang Z, Dong Z. Aberrant methylation and decreased expression of the TGF-beta/Smad target gene FBXO32 in esophageal squamous cell carcinoma. Cancer. 2014; 120: 2412-23.
16. Jia Y, Yang Y, Brock MV, Cao B, Zhan Q, Li Y, Yu Y, Herman JG, Guo M. Methylation of TFPI-2 is an early event of esophageal carcinogenesis. Epigenomics. 2012; 4: 135-46.
17. Herceg Z. Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors. Mutagenesis. 2007; 22: 91-103.
18. Beleford D, Liu Z, Rattan R, Quagliuolo L, Boccellino M, Baldi A, Maguire J, Staub J, Molina J, Shridhar V. Methylation induced gene silencing of HtrA3 in smoking-related lung cancer. Clin Cancer Res. 2010; 16: 398-409.
19. Yang J, Shen Y, Liu B, Tong Y. Promoter methylation of BRMS1 correlates with smoking history and poor survival in non-small cell lung cancer patients. Lung Cancer. 2011; 74: 305-9.
20. Li RN, Yu FJ, Wu CC, Chen YK, Yu CC, Chou SH, Lee JY, Cheng YJ, Wu MT, Wu IC. Methylation status of retinoic acid receptor beta2 promoter and global DNA in esophageal squamous cell carcinoma. J Surg Oncol. 2014; 109: 623-7.
21. Wu Y, Li G, He D, Yang F, He G, He L, Zhang H, Deng Y, Fan M, Shen L, Zhou D, Zhang Z. Telomerase reverse transcriptase methylation predicts lymph node metastasis and prognosis in patients with gastric cancer. Onco Targets Ther. 2016; 9: 279-86.
22. Kim MS, Lebron C, Nagpal JK, Chae YK, Chang X, Huang Y, Chuang T, Yamashita K, Trink B, Ratovitski EA, Califano JA, Sidransky D. Methylation of the DFNA5 increases risk of lymph node metastasis in human breast cancer. Biochem Biophys Res Commun. 2008; 370: 38-43.
23. Luo ZG, Li ZG, Gui SL, Chi BJ, Ma JG. Protocadherin-17 promoter methylation in serum-derived DNA is associated with poor prognosis of bladder cancer. J Int Med Res. 2014; 42: 35-41.
24. Kalari S, Jung M, Kernstine KH, Takahashi T, Pfeifer GP. The DNA methylation landscape of small cell lung cancer suggests a differentiation defect of neuroendocrine cells. Oncogene. 2013; 32: 3559-68.
25. Hu CY, Mohtat D, Yu Y, Ko YA, Shenoy N, Bhattacharya S, Izquierdo MC, Park AS, Giricz O, Vallumsetla N, Gundabolu K, Ware K, Bhagat TD, et al. Kidney cancer is characterized by aberrant methylation of tissue-specific enhancers that are prognostic for overall survival. Clin Cancer Res. 2014; 20: 4349-60.
26. Dai W, Teodoridis JM, Zeller C, Graham J, Hersey J, Flanagan JM, Stronach E, Millan DW, Siddiqui N, Paul J, Brown R. Systematic CpG islands methylation profiling of genes in the wnt pathway in epithelial ovarian cancer identifies biomarkers of progression-free survival. Clin Cancer Res. 2011; 17: 4052-62.
27. Jiang HL, Sun HF, Gao SP, Li LD, Hu X, Wu J, Jin W. Loss of RAB1B promotes triple-negative breast cancer metastasis by activating TGF-beta/SMAD signaling. Oncotarget. 2015; 6: 16352-65. doi: 10.18632/oncotarget.3877.
28. Lee HS, Yun JH, Jung J, Yang Y, Kim BJ, Lee SJ, Yoon JH, Moon Y, Kim JM, Kwon YI. Identification of differentially-expressed genes by DNA methylation in cervical cancer. Oncol Lett. 2015; 9: 1691-8.
29. Kumari A, Srinivasan R, Vasishta RK, Wig JD. Positive regulation of human telomerase reverse transcriptase gene expression and telomerase activity by DNA methylation in pancreatic cancer. Ann Surg Oncol. 2009; 16: 1051-9.
30. Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, Zhukova N, Walker EJ, Martin D, Merino D, Wasserman JD, Elizabeth C, Alon N, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol. 2013; 14: 534-42.