
INTRODUCTION
Pancreatic cancer is a fatal disease in humans [1, 2] and is often referred to as being a silent killer because in general, there are no symptoms until late tumor stages, at which point the tumor cells have metastasized and multiple lesions are formed [3]. Consequently, only 20% of the tumors are resectable [4], which limits translational research using cancer specimens. Currently, a few chemotherapeutic options are available for pancreatic cancer, such as Gemcitabine or fluorouracil (5-FU). However, these are not effective (extending the survival by only a few months) and produce substantial side effects [1, 5]. Therefore, there is an urgent clinical need for the development of novel diagnostic and therapeutic options.
The establishment of a preclinical model for pancreatic cancer is a prerequisite for developing new treatments. A genetically engineered mouse model is currently available for pancreatic cancer, in which activated Kras and/or Trp53 mutant proteins are specifically induced in the pancreatic ductal epithelial cells [6, 7]. However, this model cannot fully reflect human pancreatic cancer, which is genetically heterogeneous. Consequently, the use of patient-derived xenograft (PDX) models is becoming an attractive option because the tumor specimens are directly transplanted into immuno-compromised mice, providing a faithful representation of individual tumors [8]. However, establishing PDX can also be a challenge, with the success rate varying according to several factors, including the type of tumor, recipient mouse, transplant technique, and time gap between surgery and transplantation [9].
Recent studies have successfully generated PDXs for pancreatic cancer. For example, Helene et al. described 12 PDAC PDXs and PDX-derived cell lines that showed sonic hedgehog (SHH) signaling activation [10]. Delitto et al. also successfully generated 15 PDXs from 25 specimens and demonstrated that they had a conserved histology with the primary tumors [11]. Furthermore, they also found that mouse stromal cells infiltrated the human cancer cells, suggesting active tumor-stromal interactions in pancreatic cancer. Regarding the molecular analysis of PDAC PDXs, Matti et. al. reported KRAS and PIK3CA mutation analysis up to eight passages and found a similar mutation frequency in PDXs [9]. Therefore, it seems that use of the PDX model is very successful for pancreatic cancer, suggesting that it would be a good preclinical model for understanding this complex disease.
Here, we describe 20 pancreatic PDXs originating from PDAC patients who underwent surgery at the Asan Medical Center, Seoul, Korea. Clinical information and analysis of the molecular data revealed that these pancreatic PDXs have novel and heterologous characteristics.
RESULTS
Generation of pancreatic patient-derived xenografts (PDXs) and primary cells
In total, we obtained 29 freshly dissected specimens from surgery, in which we carefully selected the region that exhibited enriched tumor cells. Approximately1 cm3 of tumor tissue was obtained and cut into small pieces (1–2mm3on average). Three or four of these pieces were then subcutaneously transferred into NOD/SCID mice under anaesthetized conditions. From here, it usually takes 1~2 months for the tumor to grow. Using this method, we successfully produced 20 PDXs, representing a 72.4% success rate. Representative pictures of these PDXs are shown in Figure 1A and 1B, and Supplementary Figure S1. We also obtained six primary cancer cell lines from the PDXs (Supplementary Figure S2; also see Supplementary Table S1 for clinical information) and utilized some of these cells in our genomic analysis, along with human pancreatic ductal epithelial (HPDE) cells and PDAC (Panc1) cells.
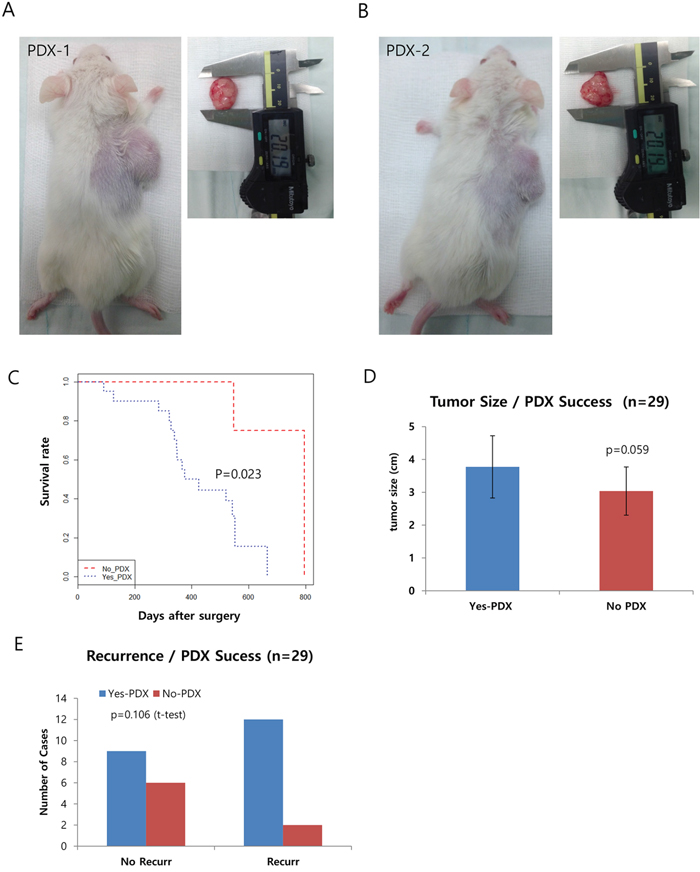
Figure 1: Generation of pancreatic patient-derived xenografts (PDXs) and the clinical features affecting their success. A-B. Representative pictures of PDXs in NOD/SCID mice. The right panels show the dissected tumors being measured with calipers. C. Kaplan-Meyer curve of the two groups of successful xenograft (Yes PDX in Blue) or failed xenograft (No PDX in Red). D and E. Graphs showing a positive correlation between the success of the xenograft and other clinical factors, including survival (D) and recurrence (E).
Analysis of clinical data reveals several criteria affecting the success of PDX
Next, we checked the clinical information to determine which factor(s) affected the success of PDX (see Table 1 for a summary). Due to the highly metastatic properties of PDAC, most of our PDX samples fell into Stage IIA or IIB, exhibiting lymph node metastasis but not distance metastasis. Here, we specifically focused on tumor size at surgery, recurrence, gender, and survival/death of the patient. Other factors such as lymphatic/vascular invasion, histological type, and distant metastasis were not considered due to the limited number of cases for each. Among the clinical characteristics analyzed, we found survival/death of the patient was significantly associated with the success rate of PDX (P=0.023 by Cox proportional hazard regression analyses, Figure 1C). In addition, tumor size (P=0.059) and recurrence showed a positive correlation but was not significant (Figure 1D and 1E). Multivariate analysis of the recurrence and tumor size, however, revealed the tumor size is a significant factor for the success of PDX (p=0.048, Table 2).
Table 1: Clinical characteristics of the parental tumors of the patient-derived xenografts (PDXs)
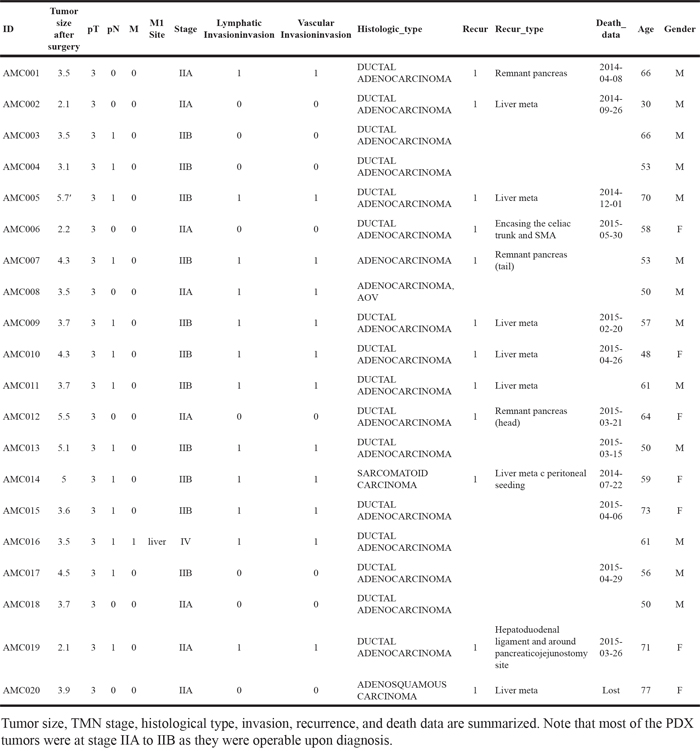
Table 2: Summary of the multivariate analysis affecting the success rate of PDX
|
Coefficient |
Std. error |
p-value |
|---|---|---|---|
Tumor_size |
1.8052 |
0.9135 |
0.0481 |
Recur |
2.6846 |
1.4763 |
0.069 |
(intercept) |
−6.0514 |
3.2551 |
0.063 |
For the tumor size and recurrence, a logistic regression method was used to determine the effect of multiple clinical factors on the success of PDX.
Comparison of the histological features of the PDX and its original primary tumor
To confirm that the gross histology of the primary tumor was conserved in the PDXs, we performed hematoxylin and eosin (H&E) staining and immunostaining with anti-P53 and SMAD4 antibodies. These data are summarized in Table 3. Overall, we observed similarities between the gross histology of the primary tumors and PDX tumors (Figure 2A, 2D, and 2G, and Supplementary Figure S3). In addition, P53 and SMAD4 staining showed a comparable reactivity in most cases (13 out of 16) of PDX-primary tumor pairs (Figure 2B, 2C, 2E, 2F, 2H, and 2I, and Supplementary Figure S3). These results show that the PDAC PDXs generated in this study recapitulated the primary tumors histologically.
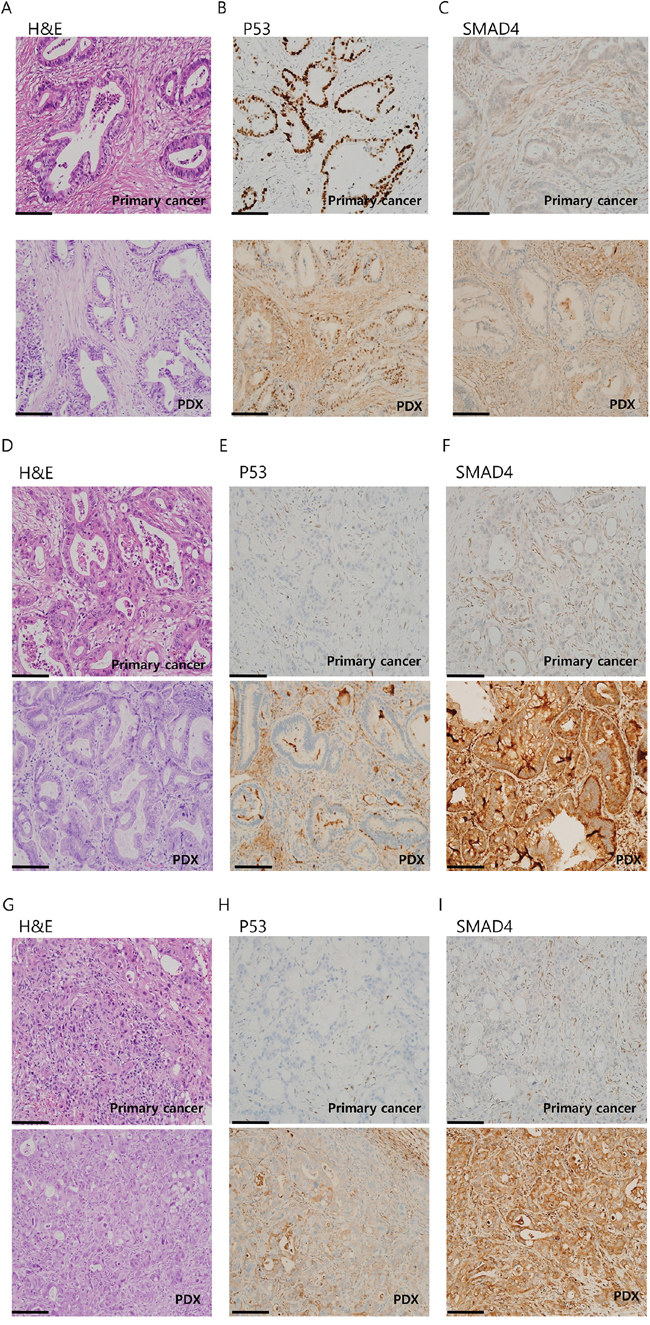
Figure 2: Immunohistochemistry (IHC) analysis of patient-derived xenograft (PDX)-primary tumor pairs reveals a conserved histology. IHC images of three PDX-primary tumor pairs: AMC001 A-C. AMC002 B-D. and AMC003 E-G. stained with hematoxylin and eosin (H&E) (A, D, G), P53 (B, E, H), or SMAD4 (C, F, I). Scale bar:100μm.
Table 3: List of genes containing deleterious variants that are frequently found in pancreatic patient-derived xenografts (PDXs)
An exome single nucleotide polymorphism (SNP) array enables grouping of the PDXs and identifies putatively functional SNPs
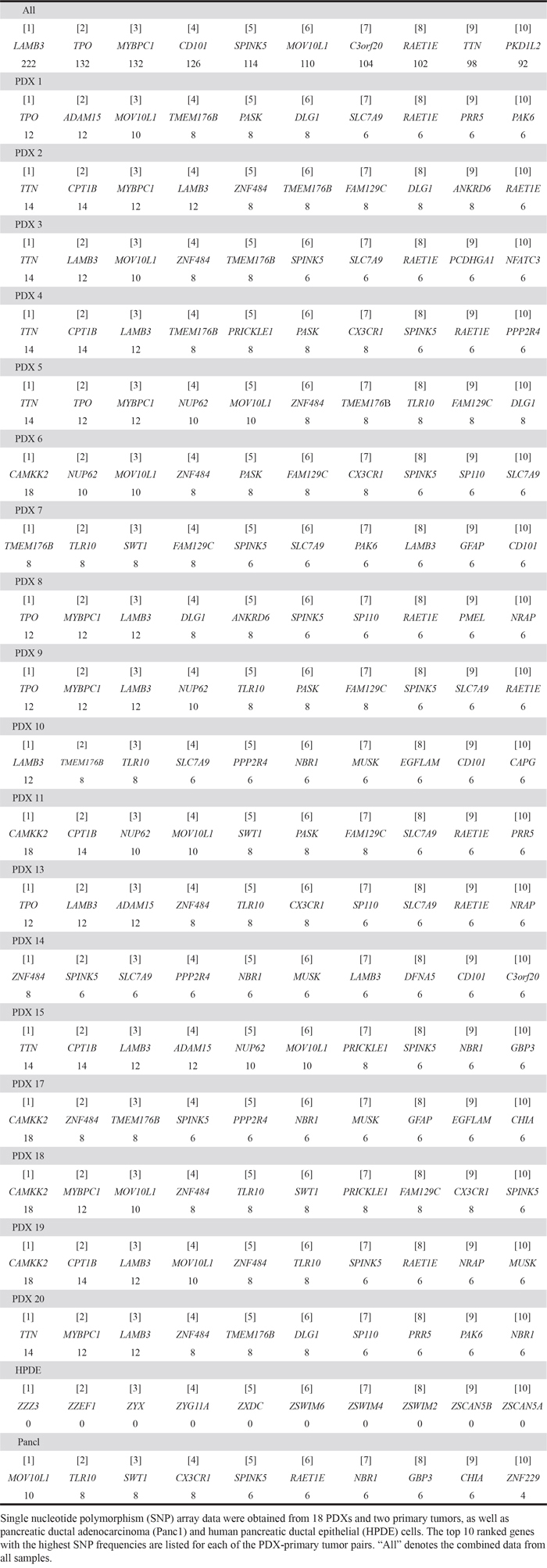
To characterize the PDXs at the molecular level, we performed an exome SNP array. Among the 20 PDXs, we excluded #12 and 16 due to the poor data quality. Instead, we included primary cancer cell line (59390), HPDE cells and Panc1 cells as cancer and normal cell controls. We aimed to compare the SNP profile of each sample so that we could subcategorize PDXs, and discover putatively functional SNPs. These functional SNPs could help us to better understand the molecular mechanisms of tumorigenesis as well as tumor heterogeneity.
We first selected 24,000 non-rare variants from 244,770 variants using plink (option – maf 0.1). We then found 1,385 deleterious variants, as predicted by Polyphen2 and Sorting Intolerant from Tolerant (SIFT) (see methods). Following this, we removed variants whose risk alleles were present in HPDE to obtain only cancer-specific variants, which left us with 623 variants (Supplementary Table S2). Table 3 summarizes the top 10 genes for each PDX that showed a high number of deleterious variants. We found that there was little overlap between these variants among the PDXs, with the sum of the top 10 variants for each PDX tumor comprising only a minor portion, ranging from 62 (10.9%) to 102 (16.4%), which implied that pancreatic PDXs are heterogeneous. However, a phylogenetic tree analysis of the functional SNPs yielded three groups of clusters (Figure 3A). An Information-Based Similarity (IBS) matrix analysis of the deleterious SNPs (Figure 3B) and a multidimensional scaling (MDS) plot analysis (Figure 3C) showed 70~80% similarity (with the exception of #8), confirming the diversity of genetic variants among the pancreatic PDXs.
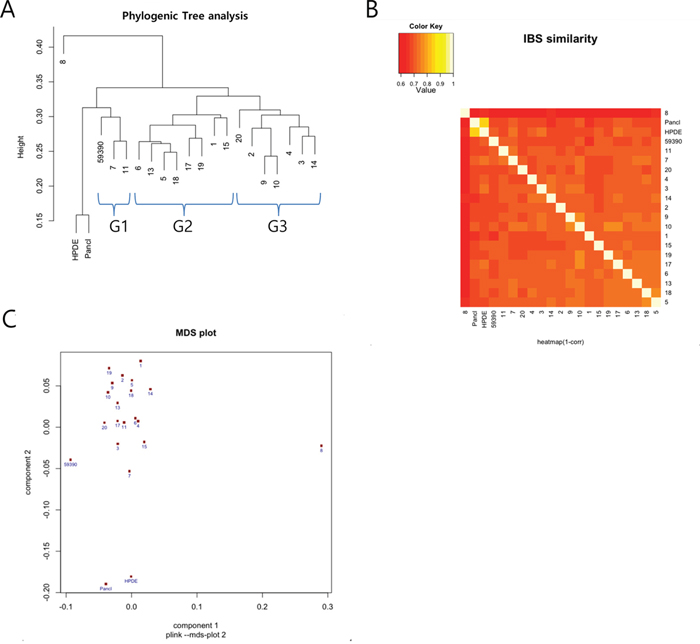
Figure 3: Summary of a single nucleotide polymorphism (SNP) array analysis from 18 patient-derived xenografts (PDXs), a primary tumor cell line (59390), pancreatic ductal adenocarcinoma (Panc1) cells, and human pancreatic ductal epithelial (HPDE) cells. The results were obtained from 623 deleterious cancer-specific SNPs. A. Phylogenetic tree showing three main clusters of the variants (marked G1, G2, and G3) occurring among the PDXs. B. Information-Based Similarity (IBS) matrix based on the SNP variants among the PDXs. C. Multidimensional scaling (MDS) plot showing a clustering pattern.
Comprehensive Cancer Panel reveals unknown genetic alterations specific to pancreatic cancer
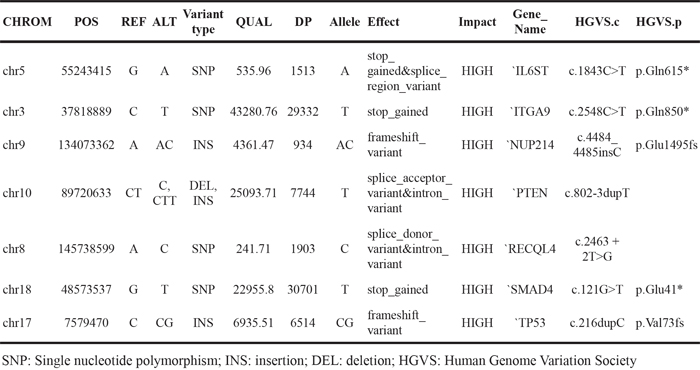
Although the data shown in Figure 3 and Table 3 generated by the exome SNP array provided useful information to classify the18 PDXs along with the primary cancer cell lines, they were insufficient for determining the molecular characteristics of the PDX-primary tumor pairs in terms of cancer-related genes. Therefore, to examine how the cancer-related mutations were conserved between the PDXs and primary tumors, we conducted an analysis of eight PDX-primary tumor pairs using Ion Ampliseq Comprehensive Cancer Panel, which covers 409 cancer-related genes (Supplementary Figure S4 for general data; for the gene list, see Thermofisher.com). The total number of variants was 40,827, of which 10,031 were novel (Supplementary Table S3). There were up to 1,804 variants in the coding region and untranslated region (UTR), and 13 of the genes with these variants were predicted to be highly affected by them. Table 4 shows examples of the variants that had a large impact. Notably, we found that PTEN, SMAD4, and TP53 were in this list, confirming previous findings [12, 13] (for raw data, see Supplementary Table S4).
Table 4: Examples of variants with a high impact, as identified from the Comprehensive Cancer Panel analysis
Clustering analysis (Figure 4A) showed that there was a high similarity between each PDX and primary tumor, with the exception of PDX #20. Furthermore, in the similarity matrix (Figure 4B), we could clearly see the conservation of most cancer gene variants between each pair of PDX-primary tumors (ranging from 90.2% to 97.4%). Interestingly, however, all other combinations among the 18 primary tumors showed much less similarity (from 59% to 67.7%), suggesting heterogeneity of the PDX tumors. The numbers of variants found in the tumors were very close to each other (around 700; Figure 4C and Supplementary Table S5, column F), implying that there was comparable genetic alteration among the tumors. This was further confirmed by counting the number of novel variants (Supplementary Table S6).
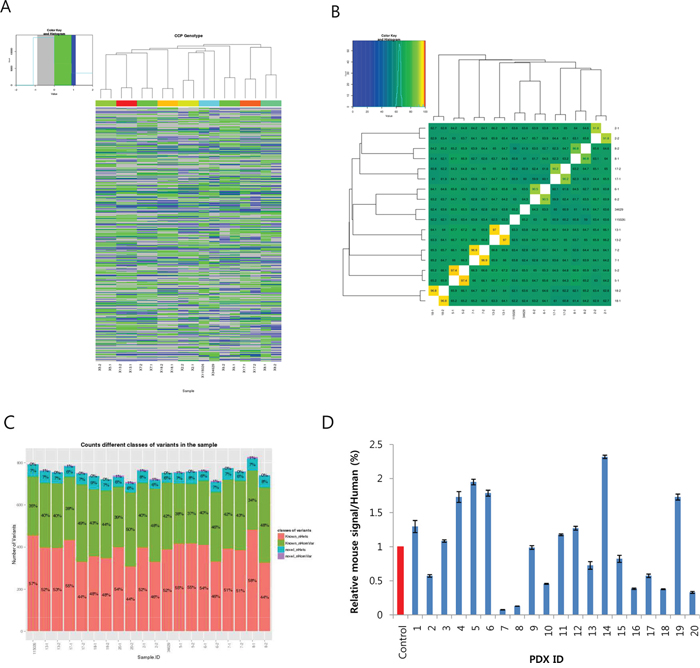
Figure 4: Summary of the Ion-Ampliseq Comprehensive Cancer Panel analysis for eight patient-derived xenograft (PDX)-primary tumor pairs. A. Cluster analysis of 18 samples (8 pairs and two cell lines) based on the variants found in 402 cancer genes. B. Similarity matrix showing conservation of the variants between PDX and primary tumors ranging from 90.5% to 97.4%. C. Number of variants found in each PDX and primary tumor sample. The numbers in the bar denote the proportion of known/novel and homologous/heterologous variants, respectively. D. Estimation of the proportion of infiltrated mouse cells in the PDXs calculated by dividing mouse RP13a expression by humanRP13a expression. The control was 5% mouse cells mixed with human pancreatic ductal epithelial (HPDE) cells.
Lastly, we measured the degree of mouse cell infiltration by measuring the relative mouse RPL13a expression to human RPL13a in the PDXs. We also included a control comprised of 95% HPDE mixed with 5% mouse fibroblast cells. This showed that there was 1–12% mouse RPL13a expression (Figure 4D), suggesting variable mouse cell infiltration in the PDXs.
Western blot analysis of PDXs for the major growth signaling/cell cycle regulatory proteins reveals their heterologous nature
In addition to the genetic analyses described above, which used an SNP array and Comprehensive Cancer Panel, we performed a series of western blot analyses to check the levels of the major growth signaling and cell cycle regulatory proteins that have previously been implicated in pancreatic cancer [14–17]. Accordingly, we found heterogeneous expression levels of these proteins (Figure 5). In particular, we observed the frequent loss of TP53 expression (by approximately 50%), as well as the minimal expression of P16. In contrast, we detected various levels of p-BRAF and p-MEK, which are major downstream effectors of K-Ras [18]. Interestingly, some of the PDXs (#4, 9, and 15) showed discordant p-BRAF and p-MEK levels, suggesting that some alternative pathway activates p-MEK in these tumors. We detected a relatively consistent level of p-AKT and SMAD4, whereas the levels of p-ERK and MTAP varied greatly. Therefore, our protein analysis revealed that the PDXs have a heterologous molecular nature that resembles the known heterologous character of primary tumors [19], supporting the strategy of using PDX as a preclinical model in pancreatic cancer.
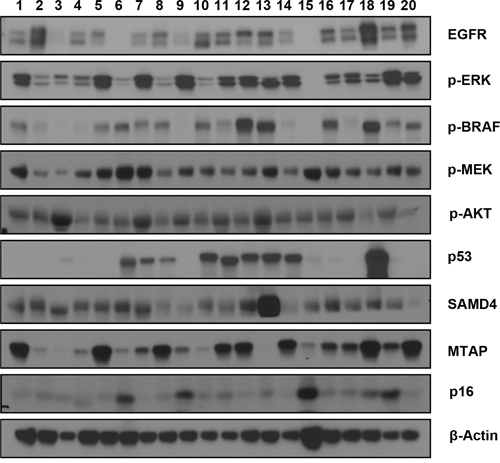
Figure 5: Western blots showing the expression levels of various growth signaling and cell cycle regulatory proteins. In total, 20 pancreatic patient-derived xenografts (PDXs) were analyzed. The name of each protein is marked on the right. The beta-actin antibody was used to ensure equal loading.
DISCUSSION
Xenograft transplantation of human PDAC cells or tissues was first performed in the late 1990s [20], with subsequent studies reporting a high degree of similarity between the PDXs and primary cancer cells, and passage-dependent genetic changes [9]. A recent study using 96 PDAC patient samples estimated the frequency of mutations in a panel of 22 cancer predisposition genes, which led to the identification of 14 pathogenic mutations in 13 patients (13.5%) [21]. Other studies on pancreatic cancer xenografts have analyzed gene expression and/or copy number variations, but have discovered only small numbers of genetic variants [9, 22, 23]. Therefore, our report provides more information about the potentially deleterious variants to pancreatic cancer research field.
In our SNP analysis, we initially found 762 deleterious (as predicted by SIFT and Polyphen2) variants in HPDE. Since we included HPDE as a non-cancer cell control, we subtracted these variants from the total variants to obtain the number of cancer-specific deleterious variants. However, we cannot exclude the possibility that this subtraction might have missed some variants that is functional in cancer cells. The list of top-ranked genes (which shows frequent SNPs in multiple PDXs) included a number of promising candidates for functional analysis. For example, LAMB3, the top-ranked gene, produces lamininb3, which is one of the major components of the extracellular matrix (ECM) of pancreatic cancer [24], and these variants generate diverse types of missense mutations, whose function needs to be further analyzed. By contrast, CD101, which has been reported as a potential risk-associated variant for PDAC [25], plays a role as an inhibitor of CD3-induced T-cell proliferation [26], and so the variants of this gene may have the immuno-modulatory effect on cancer cells. Further molecular study will reveal the exact function of these variants in pancreatic cancer.
Because different number of samples were analyzed in SNP array and CCP (18 PDXs in SNP, 8 primary tumor-PDX pairs in CCP), a direct comparison of the clustering result from the two analysis was not possible. However, we were able to compare several samples analyzed in both platforms. For example, we could see the #5/#18 and #6/13 pairs are closely related in the Group 2 of SNP data (Figure 3A) but only #5/#18 pair is closely related in the CCP analysis (Figure 4A). Therefore, we think the results of the two techniques are only partly matching. The possible reason of this result might be the difference of analytical platform. Specifically, the SNP array covers about 20,000 exome SNPs throughout the genome but the CCP covers only around 400 cancer genes.
Taken together, our findings indicate that the PDX model can provide a faithful representation of patient tumors. Furthermore, these PDXs retained the heterologous nature of pancreatic cancer cells, enabling us to use this model for preclinical research, as well as the basic study of this disease.
MATERIALS AND METHODS
Tumor implantation into mice
The animal care protocol for this study was approved by the International Animal Care and Use Committee (IACUC) of the Laboratory of Animal Research at the Asan Medical Center, Seoul, Korea. Five-week-old male NOD/SCID mice were used for tumor engraftment and were grown in a specific pathogen-free facility. The surgical specimens were obtained under permission from the institutional review board (IRB) of the Asan Medical Center (No. S2013-0744-0009).
Fresh tumor tissues were obtained from pancreatic cancer patients who underwent surgery and were immediately placed in RPMI medium (10% FBS, 1% penicillin/streptomycin) at 4°Cin the refrigerator. As soon as possible after this, the samples were spliced into one to two 2-mm3 fragments and implanted into the interscapular fat pad of the mice subcutaneously. All of the animals were anesthetized with 15 mg/kg of Zoletil® (Virbac, USA) and 2.5 mg/kg of Rompun® (Bayer Korea, Korea) by intraperitoneal injection for tumor implantation. Following implantation, the mice were monitored twice per week for at least 12 months. Once the xenograft tumor had attained a size of 300–500 mm2, the tumor was excised and the mice were euthanized following the protocol of the Laboratory of Animal Research at the Asan Medical Center. Part of the tumor that had been excised from the mouse was then engrafted into another NOD/SCID mouse for expansion, while the residual part of the tumor was placed in a freezing medium with dimethyl sulfoxide (DMSO) and kept in a deep freezer.
Immunohistochemical staining
Tumors were fixed in 10% formalin for at least 24hand then embedded in paraffin. Both human and mouse tumor tissues were sectioned at a 5μm thickness and stained with H&E. Immunohistochemistry(IHC) was performed to examine the expression of p53 and DPC4 in the primary human tumors, as previously described [27], following the protocol of the Department of Diagnostic Pathology at the Asan Medical Center. Briefly, after deparaffinization and antigenic retrieval, the slides were labeled with a monoclonal antibody against p53 (cloneDO-7, 1:3,000; DAKO, Glostrup, Denmark) and DPC4 (clone EP618Y, 1:100; GeneTex, Irvine, CA, USA). Labeling was detected using the avidin-biotin complex staining method. 3, 3′-diaminobenzidine (DAB) was used as the chromogen for p53and 3-amino-9-ethylcarbazole was used for DPC4. A pathologist who was experienced in pancreatic cancer reviewed the slides to compare the tumor architecture and desmoplastic appearance.
Collection of exonic variants
Genetic variant data for the PDX samples were gathered using the InfiniumHumanExomee12 v1.2 BeadChip. This platform targets putative functional exonic variants selected from over 12,000 individual exome and whole-genome sequences. The output data contain both SNP and single base insertion or deletion information. The data also include the GeneCall score for each variant of the samples, which is a quality control measure that was scaled between 0 and 1.
Quality control for genetic data
For each sample, we counted the number of variants that completely failed in genotype calling (GeneCall score = 0) (Supplementary Table S7). This resulted in the exclusion of two samples(#12 and 16) that had an exceedingly large number of failed genotypes (>10,000). We then chose 217,793 variants (from a total of 244,770) that had a positive GeneCall score in all remaining 21 samples, and used these variants in the subsequent genetic analysis.
Genetic similarity and MDS analysis
We used plink v1.07to perform a similarity analysis using the genetic data. We calculated the identify-by-state (IBS) pairwise similarity between samples using the--cluster-distance-matrix options in plink. We then generated a heatmap and dendrogram using R. We also generated an MDS plot using the--cluster--mds-plot options in plink and the R package heatmap v3.
Prediction and selection of deleterious variants
We used SIFT [28] and Polyphen2 [29] to predict and select putatively important variants that may cause protein damage. Polyphen2 predicted which variants were possibly damaging, probably damaging, or benign, while SIFT predicted which variants were damaging or tolerated based on the Rapid Stain Identification Series (RSID) of each variant. We defined a variant as deleterious if the Polyphen2 prediction was possibly/probably damaging or if the SIFT prediction was damaging. Among 244,770 variants (i.e., all variants before applying the quality controls), 13,613 were predicted as being deleterious.
Defining the gene disruption variable
To analyze the data at the gene level, we newly defined a genetic variable that indicated whether the gene was disrupted or not. We defined a gene as being disrupted if any variant that was predicted as being deleterious within the gene carried the risk allele. Since Polyphen2 and SIFT did not provide information about the risk allele, we obtained this information from Illumina, and confirmed this by comparing the data to the predictions from Ensemble.
Western blot analysis
Western blot analysis was performed as previously described [30]. Briefly, cells were lysed in lysis buffer (150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 50 mMTris-HCl [pH 7.5], 2 mM ethylenediaminetetraacetic acid [EDTA;pH 8.0], and 0.1% SDS). Following this, 10~50 μg of protein were separated on SDS PAGE, transferred to a nitrocellulose membrane, and probed with anti-EGFR, p-ERK, p-BRAF, p-MEK, p-AKT, P53, SMAD4, MTAP, and p16 (1:1,000; Cell Signaling Technology). The membranes were then stripped and re-probed with anti β-actin antibody (1:1,000; Santa Cruz Biotechnology, CA, USA) to ensure equal loading.
Statistics
For the analysis of clinical factors affecting successful xenograft, we applied a univariate and multivariate statistical models. For univariate statistical analysis, the statistical significance was measured by a t-test or a chi-square test. For multivariate analysis, a logistic regression method was used to determine the effect of multiple clinical factors. The survival curve was plotted using Kaplan-Meier method and the significance of the differences between the two curves was calculated by a log-rank test. Cox proportional hazards regression model was also used both for individual variable and for the multivariate analysis. All the statistical analysis was also carried out by Microsoft Excel or the R package (ver.3.3).
ACKNOWLEDGMENTS
This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (Grant Number: HI14C2640).
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Ko AH. Progress in the treatment of metastatic pancreatic cancer and the search for next opportunities. Journal of clinical oncology. 2015; 33:1779-1786.
2. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016.
3. Heestand GM, Murphy JD, Lowy AM. Approach to patients with pancreatic cancer without detectable metastases. Journal of clinical oncology. 2015; 33:1770-1778.
4. Gillen S, Schuster T, Meyer Zum Buschenfelde C, Friess H, Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS medicine. 2010; 7:e1000267.
5. Spadi R, Brusa F, Ponzetti A, Chiappino I, Birocco N, Ciuffreda L, Satolli MA. Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians. World journal of clinical oncology. 2016; 7:27-43.
6. Cook N, Olive KP, Frese K, Tuveson DA. K-Ras-driven pancreatic cancer mouse model for anticancer inhibitor analyses. Methods in enzymology. 2008; 439:73-85.
7. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer cell. 2003; 4:437-450.
8. Colvin EK, Scarlett CJ. A historical perspective of pancreatic cancer mouse models. Seminars in cell & developmental biology. 2014; 27:96-105.
9. Mattie M, Christensen A, Chang MS, Yeh W, Said S, Shostak Y, Capo L, Verlinsky A, An Z, Joseph I, Zhang Y, Kumar-Ganesan S, Morrison K, Stover D, Challita-Eid P. Molecular characterization of patient-derived human pancreatic tumor xenograft models for preclinical and translational development of cancer therapeutics. Neoplasia. 2013; 15:1138-1150.
10. Damhofer H, Ebbing EA, Steins A, Welling L, Tol JA, Krishnadath KK, van Leusden T, van de Vijver MJ, Besselink MG, Busch OR, van Berge Henegouwen MI, van Delden O, Meijer SL, Dijk F, Medema JP, van Laarhoven HW, et al. Establishment of patient-derived xenograft models and cell lines for malignancies of the upper gastrointestinal tract. Journal of translational medicine. 2015; 13:115.
11. Delitto D, Pham K, Vlada AC, Sarosi GA, Thomas RM, Behrns KE, Liu C, Hughes SJ, Wallet SM, Trevino JG. Patient-derived xenograft models for pancreatic adenocarcinoma demonstrate retention of tumor morphology through incorporation of murine stromal elements. The American journal of pathology. 2015; 185:1297-1303.
12. Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Eshleman JR, Goggins M, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Cameron JL, Olino K, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clinical cancer research. 2009; 15:4674-4679.
13. Moore PS, Orlandini S, Zamboni G, Capelli P, Rigaud G, Falconi M, Bassi C, Lemoine NR, Scarpa A. Pancreatic tumours: molecular pathways implicated in ductal cancer are involved in ampullary but not in exocrine nonductal or endocrine tumorigenesis. British journal of cancer. 2001; 84:253-262.
14. Heestand GM, Kurzrock R. Molecular landscape of pancreatic cancer: implications for current clinical trials. Oncotarget. 2015; 6:4553-4561. doi: 10.18632/oncotarget.2972.
15. Nedaeinia R, Avan A, Manian M, Salehi R, Ghayour-Mobarhan M. EGFR as a potential target for the treatment of pancreatic cancer: dilemma and controversies. Current drug targets. 2014; 15:1293-1301.
16. Matthaios D, Zarogoulidis P, Balgouranidou I, Chatzaki E, Kakolyris S. Molecular pathogenesis of pancreatic cancer and clinical perspectives. Oncology. 2011; 81:259-272.
17. Li M, Becnel LS, Li W, Fisher WE, Chen C, Yao Q. Signal transduction in human pancreatic cancer: roles of transforming growth factor beta, somatostatin receptors, and other signal intermediates. Archivum immunologiae et therapiae experimentalis. 2005; 53:381-387.
18. Gysin S, Lee SH, Dean NM, McMahon M. Pharmacologic inhibition of RAF-->MEK-->ERK signaling elicits pancreatic cancer cell cycle arrest through induced expression of p27Kip1. Cancer research. 2005; 65:4870-4880.
19. Bartsch DK, Gress TM, Langer P. Familial pancreatic cancer--current knowledge. Nature reviews Gastroenterology & hepatology. 2012; 9:445-453.
20. Mohammad RM, Dugan MC, Mohamed AN, Almatchy VP, Flake TM, Dergham ST, Shields AF, Al-Katib AA, Vaitkevicius VK, Sarkar FH. Establishment of a human pancreatic tumor xenograft model: potential application for preclinical evaluation of novel therapeutic agents. Pancreas. 1998; 16:19-25.
21. Hu C, Hart SN, Bamlet WR, Moore RM, Nandakumar K, Eckloff BW, Lee YK, Petersen GM, McWilliams RR, Couch FJ. Prevalence of Pathogenic Mutations in Cancer Predisposition Genes among Pancreatic Cancer Patients. Cancer epidemiology, biomarkers & prevention. 2016; 25:207-211.
22. Wennerstrom AB, Lothe IM, Sandhu V, Kure EH, Myklebost O, Munthe E. Generation and characterisation of novel pancreatic adenocarcinoma xenograft models and corresponding primary cell lines. PloS one. 2014; 9:e103873.
23. Walters DM, Stokes JB, Adair SJ, Stelow EB, Borgman CA, Lowrey BT, Xin W, Blais EM, Lee JK, Papin JA, Parsons JT, Bauer TW. Clinical, molecular and genetic validation of a murine orthotopic xenograft model of pancreatic adenocarcinoma using fresh human specimens. PloS one. 2013; 8:e77065.
24. Zboralski D, Bockmann M, Zapatka M, Hoppe S, Schoneck A, Hahn SA, Schmiegel W, Schwarte-Waldhoff I. Divergent mechanisms underlie Smad4-mediated positive regulation of the three genes encoding the basement membrane component laminin-332 (laminin-5). BMC cancer. 2008; 8:215.
25. Reid-Lombardo KM, Fridley BL, Bamlet WR, Cunningham JM, Sarr MG, Petersen GM. Inflammation-related gene variants as risk factors for pancreatic cancer. Cancer epidemiology, biomarkers & prevention. 2011; 20:1251-1254.
26. Soares LR, Tsavaler L, Rivas A, Engleman EG. V7 (CD101) ligation inhibits TCR/CD3-induced IL-2 production by blocking Ca2+ flux and nuclear factor of activated T cell nuclear translocation. Journal of immunology. 1998; 161:209-217.
27. Shin SH, Kim SC, Hong SM, Kim YH, Song KB, Park KM, Lee YJ. Genetic alterations of K-ras, p53, c-erbB-2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas. 2013; 42:216-222.
28. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic acids research. 2012; 40:W452-457.
29. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics. 2013; Chapter 7:Unit7 20.
30. Chang S, Wang RH, Akagi K, Kim KA, Martin BK, Cavallone L, Kathleen Cuningham Foundation Consortium for Research into Familial Breast C, Haines DC, Basik M, Mai P, Poggi E, Isaacs C, Looi LM, Mun KS, Greene MH, Byers SW, et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nature medicine. 2011; 17:1275-1282.