
INTRODUCTION
High-dose melphalan followed by autologous hematopoietic stem cell transplantation (HDM/AuHSCT) was the standard therapy for transplant-eligible multiple myeloma (MM) patients in the conventional chemotherapy era [1, 2], and it is likely to remain as such in the novel agents era [3]. Nonetheless, nearly all MM patients eventually progress after HDM/AuHSCT with a widely variable progression-free survival (PFS) [2]. Several factors are associated with short survival after HDM/AuHSCT, such as a high proportion of S-phase cells, advanced disease stage according to the International Staging System (ISS) III, high levels of lactate dehydrogenase (LDH), high-risk cytogenetic abnormalities (CAs) [e.g., t(4;14) and del 17p], and lack of complete response (CR) after HDM/AuHSCT [4-8], among others. All such factors are related to myeloma cells (MCs), but not to the surrounding microenvironment, which is well known to contribute to survival and the development of drug resistance in MCs [9].
Recently, immunomicroenvironments have been shown to play a crucial role in several cancers [10]. Both immunoscores and immunosignatures in tumor microenvironments—mainly containing CD8+ cytotoxic T lymphocytes (CTLs), programmed cell death 1 (PD-1) and its ligand 1 (PD-L1), and interferon (INF)-γ, in addition to other cytokines and antibodies—can predict treatment response and outcome in various cancer types [10, 11]. A member of the CD28 receptor family, PD-1, as well as its ligands PD-L1 and PD-L2, play a fundamental role in maintaining T-cell homeostasis by restricting T-cell activation and proliferation [12]. The interaction of PD-1+ T-cells with PD-L1-expressing cells inhibits T-cell responses [10, 12]. Indeed, the expression of PD-L1 in tumor cells promotes T-cell tolerance, suppressing the secretion of stimulatory cytokines by T-cells, and inhibiting tumor-reactive CTLs [13, 14]. Interestingly, primary MCs express higher levels of PD-L1 [15-22]. In addition, T-cells and nature killer (NK) cells from MM patients also exhibit increased PD-1 expression [12, 17, 18, 22-24]. These immune effector cells are usually surrounded by PD-L1+ MCs; therefore, MCs may escape antitumor immunity [20]. Use of a checkpoint blockade, like an anti-PD-L1 monoclonal antibody, enhances immune effector cell-mediated anti-MM response [18, 20, 23]. Notably, following HDM/AuHSCT, the expression of PD-1 in T-cells decreases or returns to normal levels, suggesting that HDM/AuHSCT can reset the immunomicroenvironment of bone marrow (BM) [17]. Furthermore, synergistic effects have been observed from combining HSCT with PD-L1 blockade or other immune checkpoints inhibitors in a 5T33 murine MM model [16, 25].
Previous studies suggests that suPD-L1 might contribute to hematological malignancies and that suPD-L1 levels might correlate with treatment response and outcome in patients with diffuse large B-cell lymphoma and newly diagnosed MM [26, 27]. Here, we analyzed the levels of suPD-L1 in BM plasma samples from 61 patients who had received uniform anti-MM treatment, to investigate whether suPD-L1 levels after HDM/AuHSCT can be used as a progression biomarker.
RESULTS
Patients and response after HDM/AuHSCT
The salient clinical characteristics of the 61 patients at diagnosis are shown in Table 1. These 61 patients had received median four cycles (range: 2–8 cycles) and one cycle (range: 1–3 cycles) for the anti-MM induction treatment and mobilization of PBSCs, respectively. The median time from diagnosis of MM to day 0 of HDM/AuHSCT was eight months (range: 3–17 months). The response evaluation performed at 100 days after HDM/AuHSCT revealed that 14 (23%), 15 (25%), 20 (33%), and 12 patients (19%) reached sCR, CR, VGPR, and PR, respectively (Supplementary Table S1).
Table 1: Salient clinical characteristics of the 61 MM patients at diagnosis and the comparison between those who had high (H) and normal-to-low (NL) suPD-L1 levels in the BM plasma at 100 days after receiving HDM/AuHSCT
suPD-L1 |
||||
|---|---|---|---|---|
Patients |
All |
H |
NL |
|
N |
61 |
18 |
43 |
P-value |
Sex (M/F) |
33/28 |
5/13 |
28/15 |
0.011 |
Age (yrs)* |
53.6±8.4 |
52.7±9.5 |
53.9±8.0 |
0.598 |
DSS [N (%)] |
0.097 |
|||
I/II |
30 (49) |
12 (67) |
18 (42) |
|
IIIa/b |
31 (51) |
6 (33) |
25 (58) |
|
ISS [N (%)] |
0.381 |
|||
I/II |
42 (69) |
14 (78) |
28 (65) |
|
III |
19 (31) |
4 (22) |
15 (35) |
|
Isotype [N (%)] |
0.273# |
|||
IgG |
38 (62) |
9 (50) |
29 (67) |
|
IgA |
9 (15) |
2 (11) |
7 (16) |
|
IgD |
5 (8) |
3 (17) |
2 (5) |
|
Light-chain |
9 (15) |
4 (22) |
5 (12) |
|
Kappa:Lambda ratio |
2.2:1 |
3.5:1 |
1.9:1 |
0.381 |
Hemoglobin (gm/dL)* |
9.5±2.7 |
9.3±2.5 |
9.6±2.8 |
0.728 |
White blood cell (x109/L)* |
6.7±4.4 |
6.9±5.5 |
6.6±3.9 |
0.809 |
Platelet (x1011/L)* |
2.1±0.9 |
2.1±0.9 |
2.0±0.9 |
0.882 |
Creatinine (mg/dL)* |
1.8±2.2 |
1.9±3.1 |
1.7±1.7 |
0.727 |
Calcium (μmol/L)* |
2.3±0.4 |
2.2±0.2 |
2.3±0.4 |
0.441 |
LDH (IU/L)* |
329±319 |
425±496 |
289±202 |
0.290 |
ALP (IU/L)* |
183±199 |
169±144 |
189±217 |
0.742 |
CRP (mg/dL)* |
1.1±1.8 |
0.8±1.1 |
1.2±2.1 |
0.509 |
Albumin (gm/dL)* |
3.6±0.8 |
3.9±0.6 |
3.5±0.9 |
0.110 |
β2M (mg/L)* |
5.8±5.6 |
6.2±7.5 |
5.6±4.7 |
0.747 |
Plasma cell in BM (%)* |
57.9±30.6 |
63.6±32.8 |
55.8±29.9 |
0.386 |
High risk CAs [N (%)] |
15 (25) |
6 (33) |
9 (21) |
0.340# |
CAs detected by CG |
8 (13) |
3 (17) |
5 (12) |
0.669# |
FISH_t(4;14) |
5 (8) |
1 (6) |
4 (9) |
0.517# |
FISH_t(14;16) |
2 (3) |
1 (6) |
1 (2) |
1.000# |
FISH_del 17p |
8 (13) |
4 (22) |
4 (9) |
0.795# |
EMD [N (%)] |
13 (21) |
5 (28) |
8 (19) |
0.499# |
*. mean±SD; #. Fisher’s exact test
Abbreviations: ALP, alkaline phosphatase; BM, bone marrow; CAs, cytogenetic abnormalities; CG, conventional G-banding; CRP, C-reactive protein; DSS, Durie-Salmon staging; EMD, extramedullary disease; F, female; HDM/AuHSCT, high dose melphalan followed by autologous hematopoietic stem cell transplantation; ISS, International staging system; LDH, lactate dehydrogenase; M, male; MM, multiple myeloma; suPD-L1, soluble PD-L1;β2M, beta2-microglobulin
Levels of suPD-L1
All plasma samples were analyzed at least in triplicate, and the mean intra-assay coefficient of variability was 4.2% and 7.6% in the normal controls and MM patients, respectively. The mean ± SD level of suPD-L1 in the 28 normal BM donors was 2.81 ± 0.77 ng/mL (range: 1.71–4.53 ng/mL). For the 61 MM patients, the mean ± SD level of suPD-L1 was 4.15 ± 2.01 ng/mL (range: 1.0–14.24 ng/mL). A comparison between the suPD-L1 levels of the normal controls and MM patients is presented in Figure 1; the difference was statistically significant (P < .001). In the normal controls, we observed no significant correlation between age and the level of suPD-L1 (Pearson’s correlation: 0.158, P = .421; Spearman’s correlation: 0.168, P = .394). The levels of suPD-L1 for the patients who had PR were significantly lower than those for patients who had VGPR or better (mean ± SD: 3.03 ± 1.70 ng/mL vs 4.42 ± 1.99 ng/mL, respectively; P = .030) (Figure 2). By applying the cutoff of 4.54 ng/mL, selected from the uppermost level of the normal controls, we determined that 18 (30%) patients had suPD-L1 levels higher than 4.54 ng/mL; these patients constituted the high-suPD-L1 group (H group). The remaining 43 (70%) patients constituted the normal-to-low group (NL group) (≤4.54 ng/mL). The comparison between the clinical characteristics at diagnosis of the H and NL groups is shown in Table 1; no differences were found between these groups, except that the H group had a higher proportion of female patients compared with the NL group (P = 0.011). The response distribution after HDM/AuHSCT between the H and NL groups is shown in Supplementary Table S1; the difference was not statistically significant.
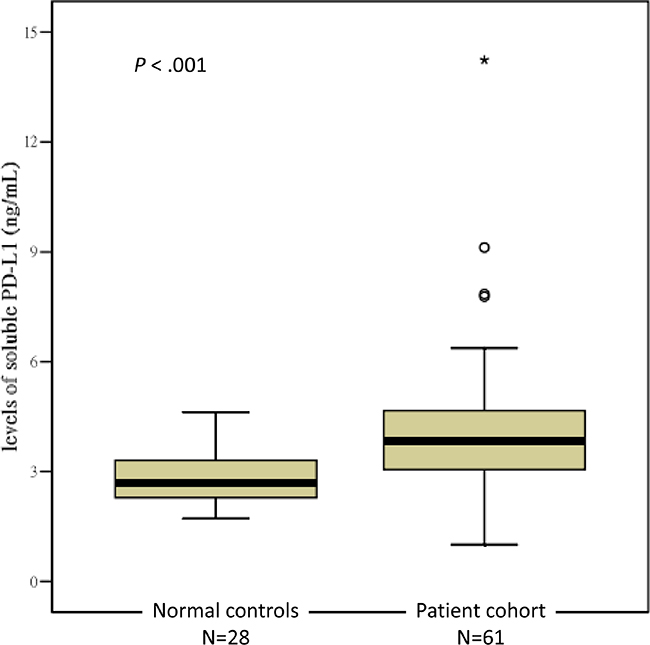
Figure 1: Levels of suPD-L1 between the normal controls and the experimental patients.
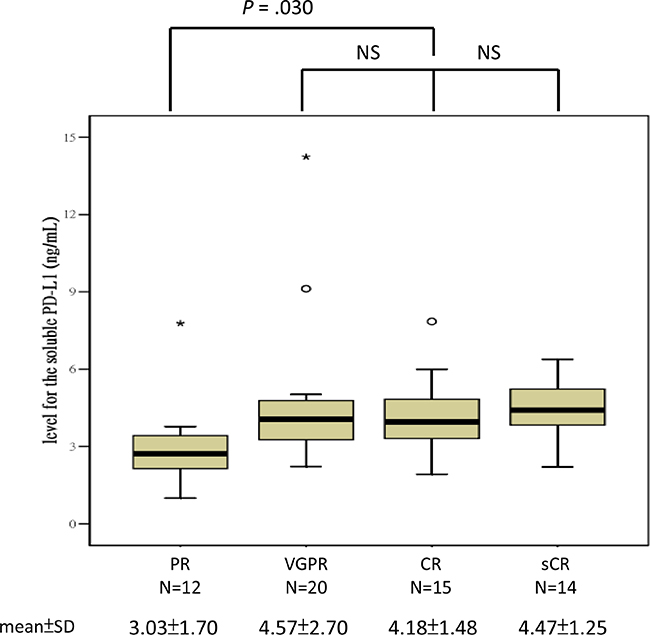
Figure 2: Levels of suPD-L1 among the various treatment response groups after HDM/AuHSCT.
Association of high suPD-L1 level and shorter response period in patients with VGPR or better after HDM/AuHSCT
The response period for HDM/AuHSCT (RpSCT), defined from day 0 of HDM/AuHSCT to the date of documented progressive disease (PD), for patients who only had PR after single HDM/AuHSCT was shorter than that for those who had VGPR or better [median: 11 months (95% CI: 0–26.28) vs 50 months (95% CI: 35.8–64.2); P = .0001]. Among the 49 patients who had reached VGPR or better, patients in the H group had a shorter RpSCT than those in the NL group [median: 17 months (95% CI: 14.3–19.8) vs not reached (NR); P = .0006] (Figure 3A). The PFS of the front-line anti-MM treatment was also shorter for patients in the H group than for those in the NL group [median: 28.5 months (95% CI: 23.6–33.4 months) vs NR; P = .0002] (Figure 3B). After a median follow-up period of 50 months (95% CI: 47.7–52.4 months), the median OS was found to be shorter in the H group than in the NL group (61 months vs NR; P = .0015) (Figure 3C).
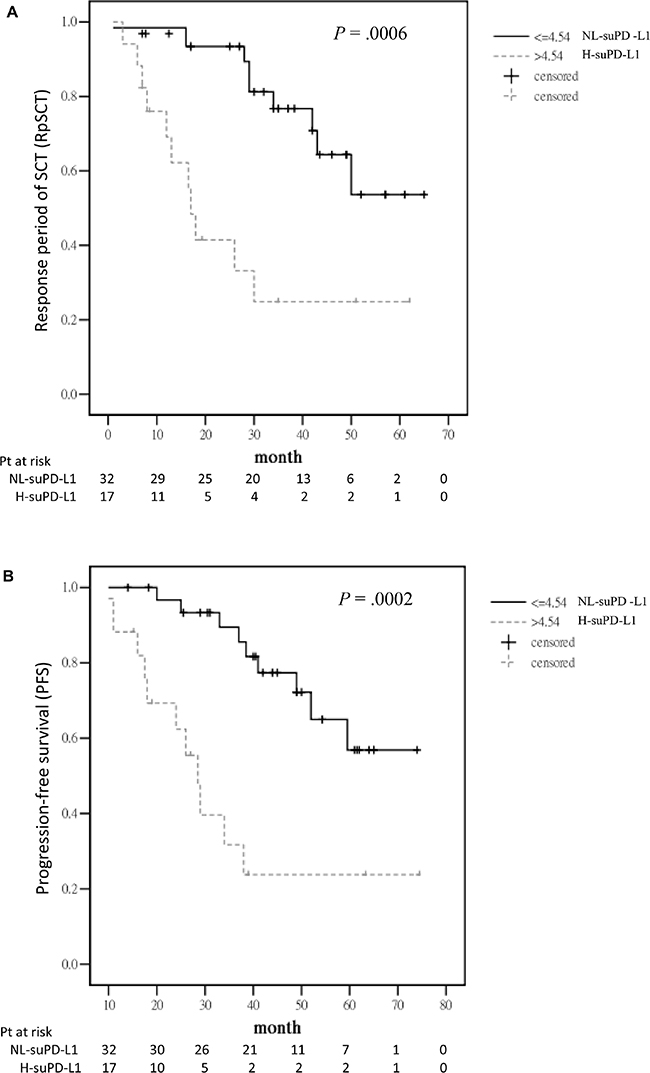
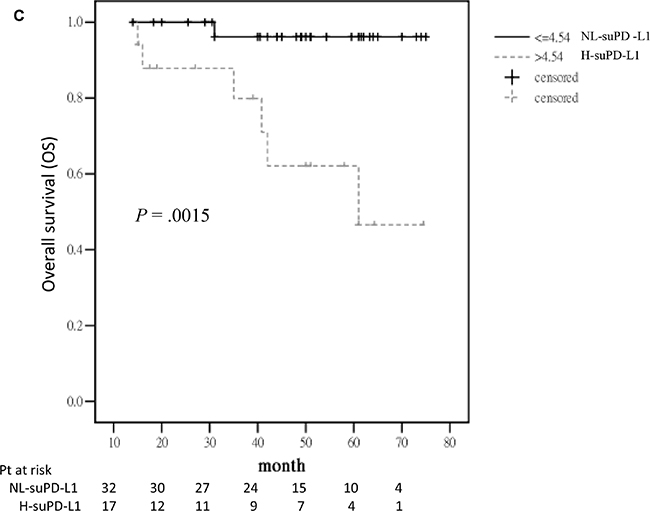
Figure 3: Outcome for the 49 patients with VGPR or better after HDM/AuHSCT. A. RpSCT between the H and NL groups measured by suPD-L1 levels. B. PFS for front-line anti-MM treatment between the H and NL groups. C. OS between the H and NL groups.
Multivariate analysis confirmation of high suPD-L1 as a negative independent predictor for the RpSCT and OS
Among the 49 patients who had VGPR or better after HDM/AuHSCT, several factors associated with the RpSCT and OS detected by Cox regression univariate analysis are presented in Tables 2 and 3, respectively. After a multivariate analysis, the high suPD-L1 (>4.54 ng/mL) was confirmed to be a negative independent factor for RpSCT (Hazard ratio: 4.322; 95% CI: 1.708–10.936; P = .002) and OS (Hazard ratio: 9.181; 95% CI: 1.069–78.820; P = .043). The other significant factor, high-risk CAs, remained independent for both the RpSCT and OS (Tables 2 and 3). The results of comprehensive uni- and multivariate Cox regression analyses for PFS and OS are presented in Supplementary Tables S2 and S3.
Table 2: Cox regression analysis among levels of suPD-L1 and salient clinical features at diagnosis associated with progression of HDM/AuHSCT in the 49 MM patients with VGPR or better after HDM/AuHSCT
Univariate analysis |
Multivariate analysis |
|
|---|---|---|
Item |
Hazard Ratio (95% CI) |
|
suPD-L1 (ng/mL) |
||
<= 4.54 |
ref |
ref |
> 4.54 |
4.295 (1.746-10.565)** |
4.322 (1.708-10.936)** |
Hb >= 10 gm/dL |
0.352 (0.127-0.981)* |
- |
High risk CAs |
4.243 (1.605-11.218)** |
4.268 (1.547-11.771)** |
*. Statistical significance, p<0.05; **. p<0.01
Abbreviations: CAs, cytogenetic abnormalities; CI, confidence interval; Hb, hemoglobin; HDM/AuHSCT, high dose melphalan followed by autologous hematopoietic stem cell transplantation; suPD-L1, soluble PD-L1; VGPR, very good partial response
Table 3: Cox regression analysis among levels of suPD-L1 and salient clinical features at diagnosis associated with overall survival in the 49 MM patients with VGPR or better after HDM/AuHSCT.
Univariate analysis |
Multivariate analysis |
|
|---|---|---|
Item |
Hazard Ratio (95% CI) |
|
suPD-L1 (ng/mL) |
||
<= 4.54 |
ref |
ref |
> 4.54 |
13.796 (1.658-114.818)* |
9.181 (1.069-78.820)* |
LDH > ULN |
7.453 (1.636-33.959)** |
- |
PC > 50% |
9.477 (1.110-80.955)* |
- |
High risk CAs |
18.759 (2.183-161.225)** |
10.406 (1.186-91.330)* |
*. Statistical significance, p<0.05; **. p<0.01
Abbreviations: CAs, cytogenetic abnormalities; CI, confidence interval; HDM/AuHSCT, high dose melphalan followed by autologous hematopoietic stem cell transplantation; LDH, lactate dehydrogenase; MM, multiple myeloma; PC, plasma cells; ref, reference; suPD-L1, soluble PD-L1; ULN, upper limit of normal range
Optimal cutoff for suPD-L1 derived from the ROC curve
For the RpSCT, the optimal cutoff for suPD-L1 derived from the ROC curve was 4.55 ng/mL (with an AUC of 0.5672), and this provided positive and negative predictive values of 55% and 79%, respectively (Supplementary Figure S1). For OS, the optimal cutoff for suPD-L1 was 4.52 ng/mL (with an AUC of 0.8061), and this provided positive and negative predictive values of 86% and 71%, respectively (Supplementary Figure S2).
Correlation between suPD-L1 and the other immunological parameters
Among the 61 MM patients, we evaluated several immunological parameters such as incidence of total recovery of immunoparesis and absolute counts of neutrophils, lymphocytes, and monocytes in the PB taken at the time of BM examination; however, we determined no correlations among these factors and the level of suPD-L1 (Supplementary Table S4). Among the 49 patients who had VGPR or better after HDM/AuHSCT, we observed no differences concerning these immunological parameters between the H and NL groups (Supplementary Table S5).
Cell density of CD8+ CTLs
Among the 61 patients, 56 BM biopsied samples were adequate for measurement of the cell density of CD8+ CTLs, which was 7.22 ± 5.69% (mean ± SD) with a range from 1.50% to 38.33%. The findings between independent observers showed good correlation (Pearson’s correlation, 0.81; P < .001). The representative CD8+ CTLs within BM identified by the IHC staining were shown in Supplementary Figure S3A and there were no differences among the mean cell density of CD8+ CTLs between patients with various responses (PR vs VGPR vs CR vs sCR, 7.60% vs 7.22% vs 5.31% vs 9.22%, respectively; P = .387). Among the different cell density scores of CD8+ CTLs, there were no differences on RpSCT (Supplementary Figure S3B) or other immunological parameters, including the levels of suPD-L1 (Supplementary Table S6).
DISCUSSION
Our study demonstrates for the first time that the levels of suPD-L1 in BM plasma at 100 days after HDM/AuHSCT in MM patients with VGPR or better are significantly and independently associated with patient outcome. By applying a cutoff (>4.54 ng/mL) determined by the upper limit of our normal controls, we observed that approximately 30% of our patients had higher suPD-L1 levels (i.e., the H group). These patients had a shorter PFS and OS compared with those who had lower suPD-L1 levels (i.e., NL group). Notably, the optimal cutoff generated from our ROC curve analysis was nearly the same level as that determined by the normal controls, which showed moderate AUC, providing fair-to-good model fitness as well as positive and negative predictive values for both PFS and OS (Supplementary Figures S1 and S2). SuPD-L1 might be able to inhibit the function of CTLs by binding to PD-1 on T-cells, similar to its membrane counterpart (mPD-L1) [10, 33], thereby preventing host immunity from eradicating the minimal residual disease (MRD) or preventing immediate resurgence of MCs. Supporting this notion, a previous study in vitro indicated that suPD-L1 could suppress the proliferation of T-cells and induce a Th2 immune response [34], suggesting that suPD-L1 can act as a fully functional molecule. This study also supports the hypothesis that suPD-L1 could be used as a biomarker to predict the outcome of MM patients after HDM/AuHSCT. In a recent study, a subpopulation of T-cells with an exhaustion/senescence phenotype identified at 3 months after HDM/AuHSCT was determined to be an early distinguishing feature ahead of clinical relapse [35]. Taken together, these data indicate that the immunomicroenvironment in BM after HDM/AuHSCT in MM patients, particularly at a low tumor mass burden, plays a role in determining the outcome. Our multivariate analyses confirmed that high suPD-L1 was an independent prognostic factor. Another independent factor was high-risk CAs, which is a well-known prognostic factor in MM patients [4]. Furthermore, suPD-L1 remained an independent factor, even in the presence of other prognostic factors including age over 60 years, high level of LDH and ISS III (data not shown). However, the female predominance in the H group is inexplicable.
Our observations also raise the question as to how suPD-L1s are produced. Previous studies have shown that suPD-Ll might be produced when matrix metalloproteinases (MMPs) cleave the extracellular fraction of mPD-L1 [33, 36]. A similar process was observed for soluble B7-H3, another co-stimulatory molecule on antigen presenting cells (APCs) that could be released from cell membranes through MMPs cleavage [37]. In support of this notion, suPD-L1 was detectable in supernatants from mPD-L1+ cells but not in those from mPD-L1− cell lines [33]. However, whether such cleavage occurs randomly or is regulated by specific mechanisms remains to be determined. Moreover, whether suPD-L1 can be produced by other mechanisms, such as alternative splicing, is unclear [38, 39].
No association has been observed between suPD-L1 levels and tumor PD-L1 expression in patients with DLBCL and renal cell carcinoma [26, 40]; therefore, nonmalignant cells in the tumor microenvironment may produce suPD-L1 as well. Whether non-MCs in BM produce suPD-L1 remains unknown, but there may be several candidates that can express mPD-L1 and produce suPD-L1 through MMPs cleavage, including myeloid-derived suppressor cells (MDSCs), tissue histiocytes, toll-like receptor APCs, and plasmacytoid DC [41-44]. Among these, MDSCs in the BM of MM patients remain as PD-L1+ even at remission because of persistent hypoxia in BM [18, 20, 45]. Furthermore, the frequency of MDSCs in the BM of MM patients appears to be higher at remission than at diagnosis or relapse [20]. Notably, ibrutinib, a bruton tyrosin kinase inhibitor, was found to deplete MDSCs in tumor models and to synergize with anti-PD-L1 therapy [46]. Accordingly, measuring suPD-L1 in BM plasma, as conducted in this study, may be more useful than measuring mPD-L1 on MCs alone. Moreover, the plasma obtained from BM is likely to be more relevant than that from PB because it is closer to the immunomicroenvironment surrounding MCs [19, 20].
Similar to the findings of other studies [6, 8], approximately 20% of our MM patients had PR only after HDM/AuHSCT, and their outcome was poor compared with that of patients who had VGPR or better. However, the reason suPD-L1 was significantly lower in patients with PR after HDM/AuHSCT than in those who had VGPR or better is unclear (Figure 2). Considering that syndecan-1/CD138 shedding induced by apoptosis of MCs after chemotherapy [47], we hypothesize that killing fewer MCs would induce less mPD-L1 shedding, thereby resulting in lower levels of suPD-L1 within the matrix.
The various mechanisms that might contribute to relapse/progression of MM after HDM/AuSCT can be divided into 1) those related to tumor biology, e.g. tumor resistance and aggressiveness, and 2) those related to the status of the BM microenvironment, e.g. immune competence within the BM microenvironment. In some of our patients who had PR already before HDM/AuSCT, the response could not be further intensified after HDM/AuSCT, suggesting treatment resistance and high-risk tumor biology. On the other hand, in patients with VGPR either before or after the HDM/AuSCT, the tumor immunomicroenvironment may play a more predominant role than tumor biology in determining relapse/progression.
Our study has several limitations. First, this study was a retrospective analysis with a heterogeneous background and a limited sample size. However, the patients were treated in a single institute under a homogenous protocol, which might have minimized the variation in treatment effects among individual patients. Second, this study lacked a sensitive MRD detection approach for comparison either by next-generation flow cytometry or sequencing-based techniques, which are powerful prognostic factors [48]. Nevertheless, we employed four-color flow cytometry for any aberrant immunophenotypic MCs according to the updated recommendation by IMWG [29]. Third, no sex- or age-matched controls were employed in this study. Notably, similar to another study [27], the distribution of suPD-L1 in our normal controls did not correlate with age. However, a correlation between suPD-L1 and age in healthy donors was reported in a different study [49]. Forth, our study lacked a longitudinal follow-up for suPD-L1 at various clinical status levels. Finally, statuses were not identified for other checkpoints such as TIM-3, LAG-3, and CTLA-4, all of which might play a synergistic role with PD-L1 in immune inhibition [10, 25]. Although analyzing more samples with longer follow-up times could further substantiate our findings, our data indicate that suPD-L1 can serve as a biomarker to predict the outcome of MM patients with VGPR or better after HDM/AuHSCT.
MATERIALS AND METHODS
Patients and BM plasma
During July 2009–January 2015, a total of 61 patients with MM who had responded, terms of partial response (PR) or better, to uniform anti-MM induction treatment at our institute followed by single autologous transplantation were enrolled. EDTA anticoagulated BM plasma samples (10–20 mL) obtained upon routine BM evaluation, including BM needle aspiration and biopsy, performed at 100 days after the transplantation, were collected and processed as described previously [28]. In addition, BM plasma samples collected from 28 healthy BM donors (comprising 13 men and 15 women with a median age of 48 years: range, 13–67 years), during 2014–2015 were used as normal controls. Among these aspirated BM blood samples, the first drawn sample, if available, was the preferred one to use. This study and its consent procedure were approved by the National Taiwan University Hospital Research Ethics Committee (NTUHREC: 201505162RINC). Written informed consent was obtained from all study participants in accordance with the Declaration of Helsinki.
Induction regimen, treatment response and outcome
The uniform anti-MM induction treatment for transplant-eligible patients at our institute was as previously described [28]. Namely BTD+Cy: bortezomib (B) [1.3 mg/m2 (sc or iv) at days 1, 4, 8, and 11] plus thalidomide (T) (100–200 mg/d) and oral dexamethasone (D) (20–40 mg/d at days 1–4) and oral cyclophosphamide (Cy) (100 mg at days 1–4) in a 21-day cycle. Upon reaching PR or better after induction, autologous PB stem cells (PBSCs) were mobilized with high-dose Cy (2 gm/m2) plus G-CSF (5–10 μg/Kg) and collected. HDM (140–200 mg/m2) was employed to condition AuHSCT. T 50–100 mg/d was provided for post-HDM/AuHSCT maintenance. The treatment outcomes, comprising CR, stringent CR (sCR), PR, very good partial response (VGPR), relapse, progressive disease (PD), PFS, and overall survival (OS) measured from diagnosis, were reevaluated in each patient on the basis of the IMWG consensus criteria [29]. The response period for HDM/AuHSCT (RpSCT) was defined from day 0 of HDM/AuHSCT to the date of documented PD.
Enzyme-linked immunosorbent assay (ELISA) for suPD-L1
The suPD-L1 levels in the BM plasma samples were measured using an enzyme-linked immunosorbent assay (ELISA) (PDCD1LG1 ELISA kit; USCN Life Science, Wuhan, China),according to manufacturer instructions. In brief, 100 μL plasma sample or standard protein was added to each well and incubated at 37 ºC for 2 h. Detection reagent A (100 μL) was added and incubated at 37 ºC for 1 h. Next, the wells were incubated with detection reagent B at 37 ºC for 30 min, and subsequently with 90 μL substrate solution at room temperature for 20 min and protected from light. The wells were finally incubated with 50 μL stopping solution. The wells were then placed on an ELISA plate reader (Perkin Elmer, CA, USA), and the absorbance of each well was recorded at 450 nm. Standard PD-L1 protein with serial dilutions (0–10 ng/μL) was used as a standard in each experiment. Each sample and standard protein was analyzed in triplicate; the minimum detection level of suPD-L1 was 0.156 ng/mL. The suPD-L1 levels (ng/mL) were calculated by linear regression.
Immunohistochemical stain for CD8+ CTLs
The procedures for immunohistochemical (IHC) staining in our laboratory were executed as described in a previous study [30], but optimized for the current study. Briefly, BM biopsied samples were fixed in 10% neutral buffered formaldehyde for at least 24 h, decalcified with Shandon TBO-2 decalcifier (Thermo Scientific, US) for 2 h, and embedded in paraffin. Paraffin-embedded BM tissue sections measuring 4–5 μm were deparaffinized in xylene, rehydrated with ethanol, and rinsed in PBS. After deparaffinization and rehydration, the slides were placed in the target retrieval solution (S1700, Dako, Denmark) and heated (90–99 ºC) for 40 min. Endogenous peroxidase was then blocked with 3% hydrogen peroxide for 10 min (Dako, Denmark); nonspecific protein binding was blocked with 3% BSA and 10% FBS (Corning NY, USA) and was then incubated at room temperature for 60 min. After blocking, the slides were incubated overnight with the primary antibody at 4 ºC. The primary antibody was used to identify the CD8 (1:100) (clone IF6; Novocastra, Newcastle upon Tyne, UK). For the maximal cell density of the CD8+ CTLs, we first identified three locations with the maximum number of CD8+ CTLs under a low-power field (100x), then switched to a high-power field (HPF, 600x), counting the CD8+ CTLs in each field (number of CD8+ CTLs/total amount of nucleated cells). The cell density of the CD8+ CTLs was averaged from the three counted areas. Scoring cutoffs for the averaged maximal density of CD8+ CTLs was set as follows: score 0, 0% to <= 10% positive cells; score 1, > 10% to <= 20% positive cells; score 2, > 20% positive cells.
Flow cytometry
In our laboratory experiment, four-color flow cytometry was employed to detect the clonotypic plasma cells in the BM. The panels of four-color fluorescent monoclonal antibodies (Becton Dickinson, BD Bioscience, San Jose, CA) are listed as follows: CD45-PerCP/CD38-PE/CD138-APC/cytoplasmic Kappa-FITC; CD45-PerCP/CD38-PE/CD138-APC/cytoplasmic Lambda-FITC; and CD45-PerCP/CD117-PE/CD138-APC/CD20-FITC. Additionally, three-color panels were used, listed as follows: CD45-PerCP/CD38-PE/CD56-FITC; CD45-PerCP/CD19-PE/surface Kappa-FITC; and CD45-PerCP/CD19-PE/surface Lambda-FITC. The experimental procedures were performed on cells isolated from BM by lysing and washing, as described previously [31]. Briefly, for each analysis, at least 100,000 events with the appropriate ratio of forward scatter to side scatter were collected and analyzed on a FACS Canto II flow cytometer (BD Bioscience, San Jose, CA) by using WinList software. Optimally titrated antibodies were added to 100 μL BM aspirates and incubated for 20 min at room temperature in the dark. After incubation, red blood cells were lysed by ammonium chloride, and the remnant targeted cells were fixed to stabilize cell membranes and prevent dissociation of the fluorescent monoclonal antibodies. Subsequently, all cells were permeabilized using a commercial kit (Cytofix/CytopermTM Fixation/Permeabilization; BD Bioscience, San Jose, CA) for intracellular staining.
Statistics
Chi squared or Fisher’s exact tests were used for intergroup comparisons of the discrete variables. A two-sample t test was employed for intergroup comparisons of the means. Pearson’s or Spearman’s correlation tests were used to determine the correlation between continuous variables. Kaplan–Meier survival curves were constructed to estimate the PFS, OS, and RpSCT, and the intergroup differences were compared using a log-rank test. In the analyses, identified salient variables for the clinical and laboratory data were categorized as previously described [28] and are listed as follows: age ≥ 60 years; stage ≥ Durie–Salmon Stage III; stage ≥ International Staging System III; nonIgG isotype; Hb < 10 g/dL; WBC < 4.0 × 109/L; PLA < 1.5 × 1011/L; LDH ≥ upper limit of normal range (ULN); ALP ≥ ULN; Ca ≥ 2.4 μmol/L; Cr ≥ 2.0 mg/dL; and C-reactive protein (CRP) ≥ ULN (0.8 mg/dL). High-risk CAs represented clonal changes detected by the conventional G-banding technique, and/or t (4;14), t (14;16), del (17p) detected by fluorescence in situ hybridization, performed as previously described [32]. Factors that provided statistically significant predictive power in the univariate analysis were further subjected to multivariate regression analysis of the linear, logistic, or Cox type with backward elimination and stepwise entering. Regarding the RpSCT and OS, the optimal cutoff value of suPD-L1 was selected with Youden’s index by using a receiver-operating characteristic (ROC) curve; the area under the ROC curve (AUC) was then calculated. All directional P values were two-tailed, and P ≤ 0.05 was considered significant for all tests. All analyses were performed using SPSS Version 19.0 software (Chicago, IL, USA).
ACKNOWLEDGMENTS
The authors acknowledge statistical assistance provided by the Taiwan Clinical Trial Statistical Center, Training Center, and Pharmacogenomics Laboratory (Which is founded by National Research Program for Biopharmaceuticals (NRPB) at the Ministry of Science and Technology of Taiwan; MOST 104-2325-B-002-032). And the Department of Medical Research in National Taiwan University Hospital. The authors would also like to thank the 3rd core facility at NTUH for technical assistance and facility support.
CONFLICTS Of INTEREST
No conflicts or interests to disclose.
FUNDING
This study was supported by grants from the National Science Council (NSC97-2314-B-002-036-MY3; NSC101-2314-B-002-086; NSC102-2628-B-002-052-MY3) and National Taiwan University Hospital (NTUH 99-S1031, 100-S1659, 101-S1801, 102-S2161, 103-S2435, 104-S2720).
REFERENCES
1. Cavo M, Rajkumar SV, Palumbo A, Moreau P, Orlowski R, Bladé J, Sezer O, Ludwig H, Dimopoulos MA, Attal M, Sonneveld P, Boccadoro M, Anderson KC, et al; International Myeloma Working Group. International Myeloma Working Group consensus approach to the treatment of multiple myeloma patients who are candidates for autologous stem cell transplantation. Blood. 2011; 117: 6063-6073.
2. Lendvai N, Cohen AD, Cho HJ. Beyond consolidation: auto-SCT and immunotherapy for plasma cell myeloma. Bone Marrow Transplantation. 2015; 50: 770–780.
3. Attal M, Lauwers-Cances V, Hulin C, Facon T, Caillot D, Escoffre M, Arnulf B, MACRO M, Belhadj K, Garderet L, Roussel M, Mathiot C, Avet-Loiseau H, et al. Autologous transplantation for multiple myeloma in the era of new drugs: a phase III study of the Intergroupe Francophone Du Myelome (IFM/DFCI 2009 Trial). Blood. 2015; 126: 391.
4. Gutiérrez NC, Castellanos MV, Martín ML, Mateos MV, Hernández JM, Fernández M, Carrera D, Rosiñol L, Ribera JM, Ojanguren JM, Palomera L, Gardella S, Escoda L, et al; GEM/PETHEMA Spanish Group. Prognostic and biological implications of genetic abnormalities in multiple myeloma undergoing autologous stem cell transplantation: t(4;14) is the most relevant adverse prognostic factor, whereas RB deletion as a unique abnormality is not associated with adverse prognosis. Leukemia. 2007; 21: 143-150.
5. van de Velde HJ, Liu X, Chen G, Cakana A, Deraedt W, Bayssas M. Complete response correlates with long-term survival and progression-free survival in high-dose therapy in multiple myeloma. Haematologica. 2007; 92: 1399-1406.
6. Kumar L, Cyriac SL, Tejomurtula TVSVGK, Bahl A, Biswas B, Sahoo RK, Mukherjee A, Sharma O. Autologous stem cell transplantation for multiple myeloma: identification of prognostic factors. Clinical Lymphoma, Myeloma & Leukemia. 2013; 13: 32-41.
7. Chim CS, Sim J, Tam S, Tse E, Lie AKW, Kwong YL. LDH is an adverse prognostic factor independent of ISS in transplant-eligible myeloma patients receiving bortezomib-based induction regimens. European Journal of Haematology. 2014; 94: 330–335.
8. Martino M, Postorino M, Gallo GA, Messina G, Neri S, Piro E, Gentile M, Moscato T, Monteleone R, Fedele R, Mazzone C, Console G, Penna G, et al. Long-term results in multiple myeloma after high-dose melphalan and autologous transplantation according to response categories in the era of old drugs. Clinical Lymphoma, Myeloma & Leukemia. 2014; 14:148-154.
9. Kawano Y, Moschetta M, Manier S, Glavey S, Görgün GT, Roccaro AM, Anderson KC, Ghobrial IM. Targeting the bone marrow microenvironment in multiple myeloma. Immunological Reviews. 2015; 263: 160-172.
10. Perez-Gracia JL, Labiano S, Rodriguez-Ruiz ME, Sanmamed MF, Melero I. Orchestrating immune check-point blockade for cancer immunotherapy in combinations. Current Opinion in Immunology. 2014; 27: 89-97.
11. Becht E, Giraldo NA, Germain C, de Reyniès A, Laurent-Puig P, Zucman-Rossi J, Dieu-Nosjean MC, Sautès-Fridman C, Fridman WH. Immune contexture, immunoscore, and malignant cell molecular subgroups for prognostic and theranostic classifications of cancers. Advances in Immunology. 2016; 130: 95-190.
12. Atanackovic D, Luetkens T, Kroger N. Coinhibitory molecule PD-1 as a potential target for the immunotherapy of multiple myeloma. Leukemia. 2014; 28: 993–1000.
13. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature Medicine. 2002; 8: 793–800.
14. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99: 12293–12297.
15. Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, Saudemont A, Quesnel B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-gamma and TLR ligands via a MyD88-,TRAF6-, and MEK-dependent pathway. Blood. 2007; 110: 296–304.
16. Hallett WH, Jing W, Drobyski WR, Johnson BD. Immunosuppressive effects of multiple myeloma are overcome by PD-L1 blockade. Biology of Blood and Marrow Transplantation. 2011; 17: 1133–1145.
17. Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, Joyce RM, Wellenstein K, Keefe W, Schickler M, Rotem-Yehudar R, Kufe D, Avigan D. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. Journal of Immunotherapy. 2011; 34: 409-418.
18. Gorgun G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, White RE, Singh A, Ohguchi H, Suzuki R, Kikuchi S, Harada T, Hideshima T, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clinical Cancer Research. 2015; 21: 4607-4618.
19. Sponaas AM, Moharrami NN, Feyzi E, Standal T, Holth Rustad E, Waage A, Sundan A. PDL1 expression on plasma and dendritic cells in myeloma bone marrow suggests benefit of targeted anti PD1-PDL1 therapy. PLoS One. 2015; 10: e0139867.
20. Castella B, Foglietta M, Sciancalepore P, Rigoni M, Coscia M, Griggio V, Vitale C, Ferracini R, Saraci E, Omedé P, Riganti C, Palumbo A,Boccadoro M, Massaia M. Anergic bone marrow Vγ9Vδ2 T cells as early and long-lasting markers of PD-1-targetable microenvironment-induced immune suppression in human myeloma. Oncoimmunology. 2015; 4: e1047580.
21. Paiva B, Azpilikueta A, Puig N, Ocio EM, Sharma R, Oyajobi BO, Labiano S, San-Segundo L, Rodriguez A, Aires-Mejia I, Rodriguez I, Escalante F, de Coca AG, et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia. 2015; 29: 2110-2113.
22. Ray A, Das DS, Song Y, Richardson P, Munshi NC, Chauhan D, Anderson KC. Targeting PD1-PDL1 immune checkpoint in plasmacytoid dendritic cell interactions with T cells, natural killer cells and multiple myeloma cells. Leukemia. 2015; 29: 1441-1444.
23. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, Baiocchi RA, Zhang J, Yu J, Smith MK, Greenfield CN, Porcu P, Devine SM, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010; 116: 2286-2294.
24. Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A, Hyodo H, Shinya E, Takahashi H, Dong H, Tamada K, Chen L, Dan K, Ogata K. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia. 2013; 27: 464-472.
25. Jing W, Gershan JA, Weber J, Tlomak D, McOlash L, Sabatos-Peyton C, Johnson BD. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. Journal for Immunotherapy of Cancer. 2015; 3: 2.
26. Rossille D, Gressier M, Damotte D, Maucort-Boulch D, Pangault C, Semana G, Le Gouill S, Haioun C, Tarte K, Lamy T, Milpied N, Fest T. High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-Cell lymphoma: results from a French multicenter clinical trial. Leukemia. 2014; 28: 2367-2375.
27. Wang L, Wang H, Chen H, Wang WD, Chen XQ, Geng QR, Xia ZJ, Lu Y. Serum levels of soluble programmed death ligand 1 predict treatment response and progression free survival in multiple myeloma. Oncotarget. 2015; 6: 41228-41236. doi: 10.18632/oncotarget.5682.
28. Huang SY, Lin HH, Yao M, Tang JL, Wu SJ, Hou HA, Chou WC, Chou SC, Hsu SC, Ko BS, Lu HY, Tsay W, Tien HF. Higher decorin levels in bone marrow plasma are associated with superior treatment response to novel agent-based induction in patients with newly diagnosed myeloma- a retrospective study. PLoS One. 2015; 10: e0137552.
29. Rajkumar SV, Harousseau JL, Durie B, Anderson KC, Dimopoulos M, Kyle R, Blade J, Richardson P, Orlowski R, Siegel D, Jagannath S, Facon T, Avet-Loiseau H, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011; 117: 4691-4695.
30. Huang SY, Lin CW, Lin HH, Yao M, Tang JL, Wu SJ, Chen YC, Lu HY, Hou HA, Chen CY, Chou WC, Tsay W, Chou SJ, Tien HF. Expression of cereblon protein assessed by immunohistochemical staining in myeloma cells is associated with superior response of thalidomide- and lenalidomide-based treatment, but not bortezomib-based treatment, in patients with multiple myeloma. Annals of Hematology. 2014 ; 93: 1371–1380.
31. Terstappen LWMM, Johnsen S, Segers-Nolten I, Loken MR. Identification and characterization of plasma cells in normal human bone marrow by high resolution flow cytometry. Blood. 1990; 76: 1739-1747.
32. Huang SY, Yao M, Tang JL, Tsay W, Lee FY, Liu MC, Wang CH, Chen YC, Shen MC, Tien HF. Clinical significance of cytogenetics and interphase fluorescence in situ hybridization analysis in newly diagnosed multiple myeloma in Taiwan. Annals of Oncology. 2005; 16: 1530-1538.
33. Chen Y, Wang Q, Shi B, Xu P, Hu Z, Bai L, Zhang X. Development of a sandwich ELISA for evaluating soluble PD-L1 (CD274) in human sera of different ages as well as supernatants of PD-L1+ cell lines. Cytokine. 2011; 56: 231–238.
34. Li Y, Xiao Y, Su M, Zhang R, Ding J, Hao X, Ma Y. Role of soluble programmed death-1 (sPD-1) and sPD-ligand 1 in patients with cystic echinococcosis. Experimental and Therapeutic Medicine. 2016;11: 251-256.
35. Chung DJ, Pronschinske KB, Shyer JA, Sharma S, Leung S, Curran SA, Lesokhin AM, Devlin SM, Giralt SA, Young JW. T-cell exhaustion in multiple myeloma relapse after autotransplant: optimal timing of immunotherapy. Cancer Immunology Research. 2016; 4: 61-71.
36. Dai S, Jia R, Zhang X, Fang Q, Huang L. The PD-1/PD-Ls pathway and autoimmune diseases. Cellular Immunology. 2014; 290: 72-79.
37. Zhang G, Hou J, Shi J, Yu G, Lu B, Zhang X. Soluble CD276 (B7-H3) is released from monocytes, dendritic cells and activated T cells and is detectable in normal human serum. Immunology. 2008; 123: 538–546.
38. Oaks MK, Hallett KM. Cutting edge: a soluble form of CTLA-4 in patients with autoimmune thyroid disease. Journal of Immunology. 2000; 164: 5015–5018.
39. Liu C, Jiang J, Gao L, Wang X, Hu X, Wu M, Wu J, Xu T, Shi Q, Zhang X. Soluble PD-1 aggravates progression of collagen-induced arthritis through Th1 and Th17 pathways. Arthritis Research & Therapy. 2015; 17: 340.
40. Ruf M, Moch H, Schraml P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. International Journal of Cancer. 2016; Mar 4. doi: 10.1002/ijc.30077. [Epub ahead of print]
41. Dhodapkar MV, Sexton R, Das R, Dhodapkar KM, Zhang L, Sundaram R, Soni S, Crowley JJ, Orlowski RZ, Barlogie B. Prospective analysis of antigen-specific immunity, stem-cell antigens, and immune checkpoints in monoclonal gammopathy. Blood. 2015; 126: 2475-2478.
42. Meier A, Bagchi A, Sidhu HK, Alter G, Suscovich TJ, Kavanagh DG, Streeck H, Brockman MA, LeGall S, Hellman J, Altfeld M. Upregulation of PD-L1 on monocytes and dendritic cells by HIV–1 derived TLR ligands. AIDS. 2008; 22: 655–658.
43. Myklebust JH, Irish JM, Brody J, Czerwinski DK, Houot R, Kohrt HE, Timmerman J, Said J, Green MR, Delabie J, Kolstad A, Alizadeh AA, Levy R. High PD-1 expression and suppressed cytokine signaling distinguish T cells infiltrating follicular lymphoma tumors from peripheral T cells. Blood. 2013; 121: 1367-1376.
44. Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nature Reviews Immunology. 2008; 8: 594–606.
45. Martin SK, Diamond P, Gronthos S, Peet DJ, Zannettino AC. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia. 2011; 25: 1533-1542.
46. Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S, McMichael EL, Duggan M, Campbell A, Keller K, Landi I, Zhong Y, Dubovsky J, et al. Myeloid-derived suppressor cells express Bruton’s tyrosine kinase and can be depleted in tumor bearing hosts by ibrutinib treatment. Cancer Research. 2016; Feb 15. pii: canres.1490.2015. [Epub ahead of print]
47. Ramani VC, Sanderson RD. Chemotherapy stimulates syndecan-1 shedding: a potentially negative effect of treatment that may promote tumor relapse. Matrix Biology. 2014; 35: 215-222.
48. Landgren O, Owen RG. Better therapy requires better response evaluation: paving the way for minimal residual disease testing for every myeloma patient. Cytometry Part B. 2016; 90B: 14–20.
49. Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, Oh S, Shin JG, Yao S, Chen L, Choi IH. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7–H1 (CD274). FEBS Letters. 2006; 580: 755–762.