
INTRODUCTION
Epigenetic mechanisms, mainly including DNA methylation such as covalent modification of cytosine bases leading to 5-methylcytosine (5mC) and histone modifications, are fundamental to regulate many biological and pathological processes in gene expression, cellular differentiation, embryogenesis, neural development and carcinogenesis. TET1 protein was initially described as a fusion partner of the mixed lineage leukemia (MLL) gene in acute myeloid leukemia (AML) as well as pediatric leukemias bearing the t(10:11)(q22;q23) translocation [1-3]. A recent study discovered that human TET1 protein could catalyze the conversion of 5mC of DNA to 5-hydroxymethylcytosine (5hmC), indicating that DNA demethylation may be a TET1-mediated process [4]. Its family members, TET2 and TET3 also have the capacity to catalyze a similar reaction [4-8]. Currently, an accumulating body of evidence mainly involves the enzymatic functions of TET proteins. The catalytic mechanism of TET family proteins is that they not only catalyze the conversion of 5mC to 5hmC, but also further catalyze the conversion of 5hmC to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), which are subsequently recognized and cleaved by thymine DNA glycosylase (TDG) and the base-excision repair (BER) process in an active DNA demethylation manner [4-8] (Figure 1). Alternatively, 5hmC may be further deaminated to produce 5-hydroxymethyluridine (5hmU) by activation-induced deaminase/apolioprotein B mRNA-editing enzyme catalytic polypeptide (AID/APOBEC) deaminases [9, 10] (Figure 1). Extensive studies were focused on the catalytic enzymatic roles of TET family proteins; while some investigations illustrated a non-catalytic activity of TET proteins. Several studies have reported that TET proteins interact with other epigenetic modifiers and some transcriptional regulators, such as the histone deacetylase 2 (Hdac 2), the O-glcNAC transferase (OGT), the Sin 3A complex and hypoxiainducible factors (HIFs), independent of their enzymatic activity [12-16]. Furthermore, Cartron et al. observed that TET1 interacts with MeCP2, HDAC1/6/7, EZH2, mSin3A, PCNA, and LSD1 to control its DNA-demethylating function [11]. Thus, TET family proteins act as transcriptional activation or repression of target genes through their enzymatic and non-enzymatic activity in lots of cellular processes. This review outlines recent advances with respect to the catalytic and non-catalytic roles of TET proteins in human tumors and neural development. The elucidation of dual roles of TET proteins will facilitate the better understanding of the process of neural development, tumorigenesis and tumor progression.
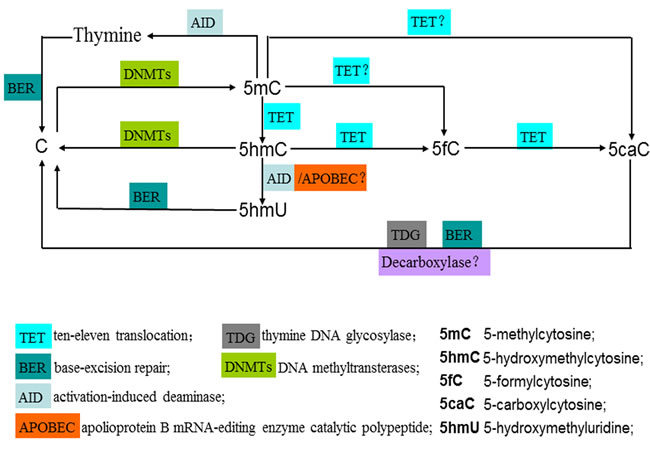
Figure 1: Enzymatic activity of TET family proteins: achieving the dynamic conversion cycle of C, 5mC and 5hmC. 5mC is catalyzed by DNA methyltransterases (DNMTs) via the addition of a methylgroup at the 5-position of cytosine. TET family proteins (TET1/2/3) catalyze the stepwise oxidation of 5mC to yield 5hmC, 5fC and 5caC. 5fC and 5caC are able to be recognized and excised by thymine DNA glycosylase (TDG) and replaced with unmethylated cytosines through the base excision repair (BER) machinery, leading to the active demethylation. An alternative active demethylation pathway may be achieved via deamination of 5hmC to 5-hydroxymethyluridine (5hmU) by AID/APOBEC enzymes, followed by BER. However, this deamination mechanism is controversial. During DNA replication, the downregulation or loss of the maintenance activity of DNMT1 dilutes the level of 5mC after each cell cycle, which results in passive demethylation. DNMT3A and DNMT3B could display a reduction-oxidation state-dependent DNA dehydroxymethylase activity, opening a simple pathway for the direct conversion of 5hmC to C. The stem-cell nuclear extracts could be capable of achieving the decarboxylation of 5caC, indicating the stem-cell nuclear extracts may be a decarboxylase. In a proportion of gliomas and breast cancers, a possible mechanism that leads to decreased levels of 5hmC by Eleftheriou et al. is that TET proteins are preactivated in certain cancer-related environment resulting in the preferential catalytic conversion of 5mC to 5caC and 5fC, instead of 5hmC.
5hmC levels in tumors and neural development
It is well known that TET proteins catalyze the oxidation of 5mC to generate 5hmC and thereby 5hmC levels reflect diverse functions of TET proteins. Some studies have revealed that 5hmC levels are reduced in hematological malignancies [17, 18]. A broad spectrum of solid tumors, including colon, breast, liver and prostate tumors, reportedly has low levels of 5hmC compared with normal tissues [19-22] (Table 1). For example, Kraus et al. reported that (i) 5hmC concentrations in brain tumors were distinctly decreased compared with those in normal brain tissues; (ii) levels of 5hmC correlated with WHO grade of brain tumors; (iii) 5hmC levels were associated with isocitrate dehydrogenase 1 (IDH1) mutations in diffuse and anaplastic astrocytomas but not in glioblastoma multiforme (GBM) [23]. However, one study’s findings showed that 5hmC distributions were significantly higher in diffuse intrinsic pontine glioma (DIPG) compared to adult GBM and pediatric GBM [24]. Furthermore, Takai et al. demonstrated that proneural glioblastomas harbor elevated levels of 5hmC and TET1 [25].
5hmC has been shown to be abundant in brain tissue: in DNA from human brain cortex, 5hmC levels are about 1% of all cytosines, indicating that 5hmC plays an important functional role in the mammalian brain [26]. Hahn and colleagues have shown that 5hmC levels increase during neuronal differentiation [27]. In other words, the level of 5hmC is enriched in terminally differentiated neurons, whereas less differentiated neurons, neural precursor cells (NPCs) and neural stem cells (NSCs) have lower 5hmC levels.
Taken together, 5hmC levels differ according to different tumor types, tumor grades and neural developmental cell stages. Even in different molecular subtypes of the same tumor, there are contradictory reports on 5hmC levels. All of these findings suggest that TET family proteins play different roles based on distinct human tumors and stages throughout neural development. In this review, we highlight the dual roles of TET family proteins in human tumors and neural development: enzymatic function and non-enzymatic function.
Table 1: The enzymatic activity of TET family proteins in neural development
TET proteins |
Role in neural development |
Intervention method |
Ref |
TET1 |
OL development |
siRNA |
[67] |
neurogenesis |
shRNA |
[27] |
|
adult hippocampal neurogenesis and cognition |
KO |
[65] |
|
TET2 |
OL development |
siRNA |
[67] |
neurogenesis |
shRNA |
[27] |
|
TET3 |
mOSN development |
overexpression |
[64] |
OL development |
siRNA |
[67] |
|
neurogenesis |
shRNA |
[27] |
|
Xenopus eye and neural development |
cKO |
[66] |
Note: OL: Oligodendrocyte; mOSN: mature olfactory sensory neuron; shRNA: short hairpin RNA; siRNA: small interfering RNA; KO: Knockout; cKO: conditional knockout
Structure of TET family proteins
Three members of the mammalian TET gene family have been defined: TET1, TET2 and TET3 [4, 28]. The TET gene underwent triplication and then generated TET1, TET2 and TET3 in jawed vertebrates [29]. All TET proteins contain a similar catalytic C-terminal CD domain, which is composed of a double-stranded β-helix (DSBH) fold and a cysteine-rich (Cys) region [30, 31]. Hu et al. reported the crystal structure of human TET2-DNA complex [32]. This structure suggests that two zinc fingers bring the DSBH and Cys-rich domains together to facilitate a compact catalytic domain formation. These CD domains of TET proteins confer α-ketoglutarate (α-KG)-and iron (II)-dependent dioxygenase activity [4]. TET family proteins convert 5mC to 5hmC through these CD domains, andα-KG is regarded as a co-substrate for enzymatic activity. Zhang and colleagues reported that TET proteins also oxidize 5mC and 5hmC into 5fC and/or 5caC by these CD domains, but the levels of 5fC and/or 5caC are very low [28]. One recent study showed that TET2 interact with Hdac1/2 via its DSBH domain [12]. In addition, the catalytic DSBH domain of TET2 is also able to interact with O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) [13]. These studies suggest that TET proteins may interact with other epigenetic modifiers through the DSBH domain.
Moreover, Xu and colleagues demonstrated that these CD domains of TET family proteins substantially convert 5mC and 5hmC to 5caC in the presence of ATP in vitro [7]. In addition, TET proteins possess the CXXC zinc finger domain at the amino-terminal region; the CXXC domain can be found in TET1 and TET3, but not TET2 [33, 34]. Several studies reported that the CXXC domain of TET1 recognize unmodified cytosine, 5mC and 5hmC, and it prefers to bind to regions in the genome of high CpG content [35, 36]. During evolution, the TET2 gene splitted into two segments encoding catalytic domain and CXXC domain, respectively. The catalytic domain evolved into TET2, whereas the CXXC domain evolved into a distinct gene, IDAX (also known as CXXC4). IDAX could lead to downregulation of TET2 protein via activation of caspase 3 [37]. TET2 binds different sets of genomic regions, which is depending on whether IDAX is present [29]. IDAX has previously been indicated as an inhibitor of the Wnt signaling pathway that play an important role in modulating cell proliferation, invasion and survival [38, 39]. Unlike TET2 and TET3, TET1 contains three candidate bipartite nuclear localization signal at its amino-terminus [2], indicating a mainly nuclear location of the protein.
Enzymatic activity of TET family proteins: achieving the dynamic conversion cycle of C, 5mC and 5hmC
All three TET proteins possess an intrinsic enzymatic capability to catalyze the successive oxidation of 5mC to yield 5hmC, 5fC and 5caC (Figure 1). The TET proteins enzymatic function has opened up new ways for achieving the dynamic conversion cycle of C, 5mC and 5hmC and regulating the balance of DNA methylation and DNA demethylation (Figure 1). Over the past several decades, DNA methylation at the 5’carbon of the cytosine base (5-methylcytosine, 5mC) has been identified as an important epigenetic modification that plays a vital role in some cellular and molecular mechanisms such as establishment of cellular identity, control of gene expression, suppression of transposon elements, genomic imprinting, X-chromosome inactivation and carcinogenesis [8, 40]. DNA methylation is catalyzed by DNA methyltransterases (DNMTs) through the addition of a methylgroup at the 5-position of cytosine, resulting in 5-methylcycosine (5mC). DNMT3A, DNMT3D and DNMT3L create DNA methylation, while DNMT1 maintain the patterns of methylation [41-44]. During DNA replication, the downregulation or loss of the maintenance activity of DNMT1 dilutes the level of 5mC after each cell cycle, which results in passive demethylation. For instance, 5hmC represses binding of the DNMT1/UHRF1 complex to DNA for methylation maintenance [45, 46]. Chen and co-workers reported that the vertebrate denovo DNA methyltransferases DNMT3A and DNMT3B could also exhibit a reduction-oxidation state-dependent DNA dehydroxymethylase activity, opening a simple pathway for the direct conversion of 5hmC to C [47] (Figure 1).
Many studies showed that there might be multiple pathways or mechanisms by which TET family proteins regulate the cycle of DNA demethylation. The TET oxidation products 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) are able to be recognized and excised by thymine DNA glycosylase (TDG) and replaced with unmethylated cytosines through the base excision repair (BER) machinery, leading to the active demehylation [5-7] (Figure 1). In addition, an alternative active demethylation pathway may be achieved via deamination of 5hmC to 5-hydroxymethyluridine (5hmU) by activation-induced deaminase/apolioprotein B mRNA-editing enzyme catalytic polypeptide (AID/APOBEC) enzymes, followed by BER [9, 10] (Figure 1). However, this deamination mechanism is controversial because of no direct biochemical evidence indicates that these deaminases have their robust activity against 5hmC [10]. Furthermore, Schiesser et al. found that the stem-cell nuclear extracts could be capable of achieving the decarboxylation of 5caC, indicating the stem-cell nuclear extracts may be a decarboxylase [48] (Figure 1).
The enzymatic activity of TET family proteins in tumors
To date, several different mechanisms are reported to be involved in the regulation of the enzymatic activity of TET proteins, which are summarized in Figure 2. The point mutants of isocitrate dehydrogenase type 1 (IDH1) and type 2 (IDH2) were frequently found in human tumors, which exhibits lack of normal enzymatic activity, responsible for 2-oxoglutarate (α-ketoglutarate; 2OG) production [49]. The tumor-associated IDH1/2 mutations gain the ability to generate (R)-2-hydroxyglutarate ((R)-2-HG). 2HG represses the TET-enzymatic reaction via reduction of the key cofactor 2OG and direct impairment of TET catalytic activity by 2HG [50, 51]. For instance, Xu et al. showed that reduced 5hmC levels in a mixture of Grade III astrocytomas and GBMs containing mutant IDH1 [52]. Since it possesses three nuclear localization signals [2], TET1 may be a mainly nuclear localization enzyme and its enzymatic activity of the conversion of 5mC to 5hmC may be a nuclear event. Indeed, in human embryonic kidney cells and human colon cancer cells, overexpressed TET1 and TET2 are predominantly localized in the nucleus and nuclear TET proteins are gradually translocated to the cytoplasm when they are co-expressed with Aid [53]. In colorectal cancer cells, nuclear expression of TET2 is absent and nuclear export inhibitor can increase the 5hmC level [54]. Müller et al. reported that nuclear exclusion of TET1 is related to reduction of 5hmC in IDH1 wild-type gliomas [55]. These findings showed nuclear exclusion of TET proteins may provide a novel mechanism for the reduction in 5hmC from the tumor cells, which is independent from IDH1 status. Moreover, Eleftheriou and co-workers demonstrated that 5caC levels are elevated in a proportion of gliomas and breast cancers. A possible mechanism that leads to decreased levels of 5hmC proposed by Eleftheriou is that TET proteins are preactivated in certain cancer-related environment resulting in the preferential catalytic conversion of 5mC to 5caC, instead of 5hmC [56]. Recently, Xiao et al. showed that tumor-derived SDH and FH mutations also inhibit TET proteins enzymatic activity via accumulation of their respective substrates, succinate and fumarate [57].
Vitamin C is a vital antioxidant that turns Fe(III) to Fe(II) and thereby facilitates the enzymatic integrity of Fe(II)-dependent TET enzymes. Vitamin C has been reported to increase the enzymatic activity of TET family proteins in vitro and vivo [58-60], possibly through binding to the catalytic domain of TET enzymes and directly enhancing their enzymatic activity [58]. Treatment with vitamin C was showed to contribute to higher level of 5hmC in melanoma cell lines and to decrease the degree of malignancy by potentiating TET enzymatic activity [61]. Interestingly, Rampal et al. have found that WT1 mutations reduce TET2 enzymatic activity and then result in decreased 5hmC levels in AML. This study has shown that WT1 interact with TET2 and TET3, and may stimulate TET proteins enzymatic activity at specific sites in the genome [62].
These lines of evidences support the notion that multiple pathways inhibit the TET proteins enzymatic activity in various malignancies and thus lead to the cancer-related 5hmC decrease suggesting that reduced 5hmC may be a valuable tumor hallmark for the early diagnosis and prognosis of cancers. Therefore, the treatment that boost the TET proteins enzymatic activity, such as using vitamin C and promoting WT1 overexpression, is likely to be beneficial to tumor treatment.
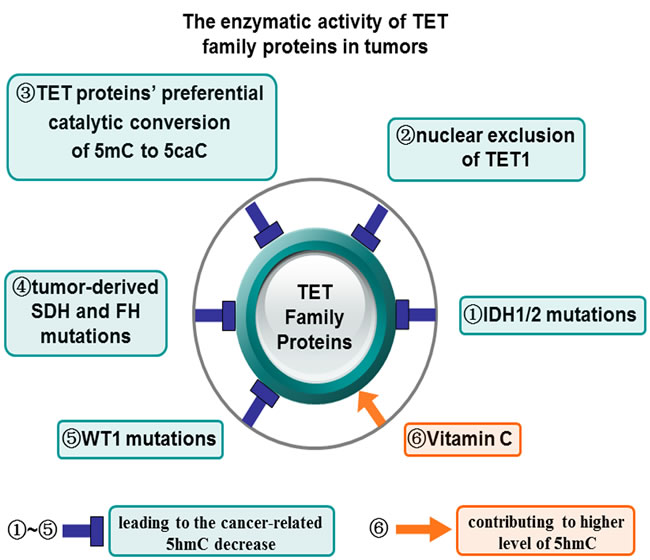
Figure 2: The enzymatic activity of TET family proteins in tumors. There are several different mechanisms were involved in the regulation of the catalytic enzymatic activity of TET proteins in tumors. For example, both the tumor-associated IDH1/2 mutations (1) and nuclear exclusion of TET1 inhibit TET1 catalytic activity and decrease 5hmC levels (2). TET proteins exert the preferential catalytic conversion of 5mC to 5caC, instead of 5hmC (3). Tumor-derived SDH and FH mutations also repress TET proteins enzymatic activity and reduce 5hmC levels (4). WT1 mutations indirectly repress TET2 enzymatic activity and reduce 5hmC levels (5), while pretreatment of Vitamin C increase the enzymatic activity of TET family proteins and 5hmC levels (6). TET proteins enzymatic activity and their oxidation product 5hmC can be reduced (1, 2, 3, 4, 5, 6) and increased (6) in the presence of distinct regulatory elements.
The enzymatic activity of TET family proteins in neural development
In 2009, a seminal paper reported the high levels of 5hmC occur in neurons [63], pointing to a potential role for TET enzymes in the control of neural development. Hahn and colleagues described the roles of TET proteins and 5hmC during neurogenesis [27]. They have reported that the levels of 5hmC elevate during neuronal differentiation and enrichment of 5hmC is not at enhancers but preferentially at gene bodies of many critical neuronal differentiation-related genes. Transcriptional analysis revealed that accumulation of intragenic 5hmC accompanied with loss of H3K27me3 is involved with gene activation during neuronal differentiation. Polycomb complex modulates the switch of NPCs from expansion to differentiation, while TET enzymes are correlated with maintaining the proper progression of the neuronal differentiation process through TET- mediated formation of 5hmC along gene bodies after this process initiation. In addition, as a result of no evidence for substantial DNA demethylation, 5hmC is considered as a stable epigenetic mark. Colquitt et al. found that TET3 oxidation product 5hmC elevates over gene bodies during mature olfactory sensory neurons (mOSNs) development and gene-body 5hmC patterning occurs between the progenitor and mOSNs stages. Additionally, this finding suggested that gene-body 5hmC patterning plays a physiologically significant role in the maintenance of stable cellular identities and transcriptional facilitation independent of its acting as an intermediate in DNA demethylation [64]. Zhang et al have revealed that TET1 possesses a specific function in the control of neural progenitor cells (NPCs) proliferation by promoting DNA demethylation and gene expression in adult brain. They observed that TET1-deficient mice displayed impaired hippocampal neurogenesis and cognitive deficiency such as memory and learning [65]. Xu et al. have uncovered that Xenopus Tet3 plays a vital role in early eye and neural development by two mechanisms. TET3’ enzymatic activity that modulates the 5mC/5hmC status at target gene promoter is an essential mechanism. The TET3 CXXC domain-mediated binding to unmethylated cytosines at target gene promoters provides another mechanism of regulating the transcription of TET3 target genes [66]. During oligodendrocyte (OL) development, three TET family proteins display unique subcellular and temporal expression patterns. 5hmC modification may act on the expression of critical genes required for OL maturation. Furthermore, siRNA-mediated silencing of TET1-3 proteins in OLs demonstrated that three TET proteins are necessary for OL differentiation [67]. As to the other oxidative derivatives of 5hmC, one study showed that 5fC and 5caC are transiently accumulated during differentiation of neural stem cells (NSCs) in vitro and in vivo. Moreover, enrichment of 5caC at the cell-type-specific promoters is observed during lineage specification of NSCs [68].
Taken together, these studies suggest that TET enzymes catalytic products, especially 5hmC, are likely multifunctional modification that is utilized either in the demethylation pathway or as a stable epigenetic mark itself, depending on the cell type and the genomic location of the modification during neural development (Table 1).
The non-enzymatic activity of TET family proteins in tumors and neural development
Epidemiological researches have demonstrated that chronic inflammation predisposes individuals to diverse types of tumor. Inflammation in the tumor microenvironment possesses versatile tumor-promoting effects. It is able to aid in the proliferation and survival of tumor cells, subvert adaptive immune responses, contribute to metastasis and angiogenesis, and alter responses to chemotherapeutic agents [69]. The hallmarks of cancer-related inflammation comprise many key inflammatory factors, such as IL-6. One study has shown that IL-6 level is highest in GBM cerebrospinal fluid (CSF) when compared with normal control low-grade glioma (LGG), and anaplastic astrocytomas (AA) [70]. Moreover, Kraus et al. have reported that 5hmC levels in gliomas are significantly lower compared to normal brain tissue and 5hmC values in GBM are lowest [23].Intriguingly, Cao and his team have demonstrated that TET2 resolves inflammation via recruiting Hdac2 to selectively inhibit IL-6 in innate myeloid cells, which is independent of its enzymatic activity [12]. Since the inflammatory microenvironment contribute to the tumorigenesis and the development of tumors, we speculate that the inhibition of TET2 activities, including non-enzymatic activity and enzymatic activity of TET2 protein, may occur in tumors such as gliomas, and then the repression of TET2’s non-enzymatic activity results in the elevation of IL-6 level in cancers.
In addition, the non-enzymatic role of TET proteins in transcription regulation has also been shown in other studies. Tsai et al. have found that TET1 interacts with hypoxiainducible factors (HIFs) to increase hypoxia-responsive gene expression and epithelial-mesenchymal transition (EMT) independent of its enzymatic activity [14]. TET2 boosts histone O-GlcNAcylation by directly interacting with OGT during gene transcription, which is independent of TET2 protein enzymatic activity [13]. The TET2 or TET3-OGT interaction promote GlcNAcylation and increase H3K4me3 through the SET1/COMPASS complex; OGT does not appear to influence TET proteins enzymatic activity [15]. TET1 interacts with the SIN3A co-repressor complex, leading to transcriptional repression of many TET1- targeted genes [16]. Moreover, Kaas et al. have showed that AAV-mediated overexpression of TET1 in the dorsal hippocampus partially impairs long-term memory formation independent of its enzymatic activity [71], which may be via the interaction between TET1 and some unknown proteins. Ambigapathy et al. showed that TET1 and ERK1/2 are critical partners during learning [72]. Rudenko et al. found that TET1 is critical for memory extinction [73]. However, in the two studies, there is little evidence indicating that we are able to make a distinction between catalytic activity and non- catalytic activity of TET1 in learning, but rather, that it is critical. Additionally, catalytically inactive TET3 mutant is able to partially rescue the effects on Xenopus eye and neural development after TET3 depletion, suggesting a non-enzymatic mechanism in gene regulation through recruiting or interacting with some unknown proteins [66]. Jin and his team have confirmed that Neuro2a cells have lower level of 5hmC; they have found that TET1 inhibits neuronal differentiation of Neuro2a cells independent of its catalytic activity via srGAP3, which is likely to be due to the interaction between TET1 and some unknown proteins [74].
These studies suggest that TET proteins might serve as transcriptional co-activators/co-repressors forming complexes with other transcriptional regulators and scaffolding proteins to perform important non-enzymatic functions in human tumors and neural development (Figure 3).
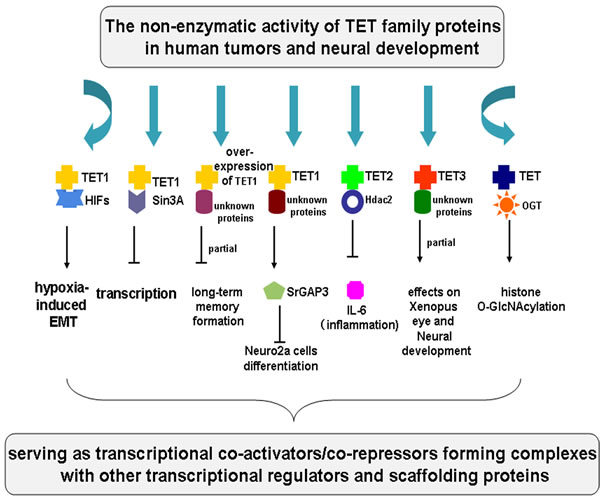
Figure 3: The non-enzymatic activity of TET family proteins in tumors and neural development. The non-catalytic role of TET proteins in tumors and neural development is summarized. TET1 interacts with HIFs to increase hypoxia-responsive gene expression and EMT. TET1 interacts with the SIN3A co-repressor complex, leading to transcriptional repression of many TET1- targeted genes. Overexpression of TET1 impairs long-term memory formation and TET1 inhibits neuronal differentiation of Neuro2a via srGAP3, interacting with some unknown proteins. TET2 resolves inflammation via recruiting Hdac2 to selectively inhibit IL-6 in innate myeloid cells. TET3 is able to partially rescue the effects on Xenopus eye and neural development after TET3 depletion through recruiting or interacting with some unknown proteins. TET2 boosts histone O-GlcNAcylation by directly interacting with OGT during gene transcription. TET proteins might serve as transcriptional co-activators/co-repressors forming complexes with other transcriptional regulators and scaffolding proteins to perform important non-enzymatic functions in human tumors and neural development.
Mechanisms affecting both enzymatic activity and non-enzymatic activity of TET proteins via influencing TET expression in tumors and neural development
A variety of mechanisms that interfere either directly or indirectly with TET expression, including TET mutations, upstream regulators of TET genes, methylation of TET genes, IDAX (also known as CXXC4), and multiple miRNAs, are able to have an effect on both enzymatic activity and non-enzymatic activity of TET family proteins in human tumors and neurodevelopment. In hematopoietic malignancies, including acute myeloid leukemias (AMLs), myeloproliferative neoplasms (MPN), myelodysplastic syndrome (MDS), and chronic myelomonocytic leukemia (CMML), TET2 gene has been shown to be frequently mutated [75-86]. The TET2 mutation frequencies in the different patient groups are MDS (6%-26%), CMML (20%-58%), primary and secondary AML (12%-32%), blastic plasmacytoid dendritic neoplasm (25%-54%), myeloproliferative neoplasms (MPNs) (2%-20%), B-cell (2%-12%) and T-cell (20%-83%) lymphomas, systemic mastocytosis (20-29%), and chronic myeloid leukemia (4%) [87]. However, mutations in TET genes present much less frequently in solid tumors such as clear-cell renal cell cancer, colorectal cancer and prostate cancer [88-91]. Sun et al. have reported that TET1 gene expression is dramatically induced upon depletion of high mobility group AT-hook2 (HMGA2) in invasive human breast cancer cells, suggesting that HMGA2 is an important upstream mediator of TET1 [92]. Furthermore, Huang and co-workers have found that TET1 expression is promoted via MLL-fusion proteins directly binding to the promoter region of TET1 in MLL-rearranged leukemia [93]. Two studies have reported that methylation of TET2 gene is infreguent in MPN and low-grade diffuse gliomas and may result in the inactivation of TET2 gene [94, 95]. Ko et al. have showed that IDAX expression lead to caspase activation and caspase-mediated cleavage could be involved in the degradation of TET2 by ubiquitylation [37]. Cheng et al. have reported that an extensive network of TET2-targeting miRNAs, such as miR-29b and miR-125a, could suppress TET2 expression and induce phenotypes related to hematopoietic malignancy [96]. Additionally, miR-22 mediates the repression of TET by a direct interaction between the TET genes and miR-22 in breast cancer [97]. MiR-494 has been reported to be able to inhibit TET1 gene expression through targeting its 3’UTR region in human hepatocellular carcinoma (HCC) [98]. Interestingly, miR-15b directly reduces TET3 expression to promote neurogenesis and repress neural progenitor proliferation during early neocortical development [99].
In addition to the above findings, TET proteins expression is elevated in proneural glioblastomas, leiomyoma, and diffuse intrinsic pontine glioma (DIPG), but the mechanisms that lead to the elevation of TET proteins are elusive in these studies [24, 25, 100]. In neuroblastoma cells, hypoxia lead to TET1 upregulation, and TET1 is required for full induction of hypoxia-responsive genes and global 5hmC level increases [101].
Clinical implications of TET proteins dual activities in human tumors and neural development
Serials of investigations have discovered that TET proteins dual activities (catalytic or non-catalytic) exert tumor-suppressive or tumor-promoting functions in different types of tumors (Table 2). There are studies where TET proteins dual activities have or may have tumor-suppressing functions. Rasmussen and colleagues have proposed that TET2 catalytic activity prevents leukemic transformation and TET2 mutations promote leukemogenesis [102]. In prostate cancer, compared to patients with high TET1 mRNA levels, patients with low TET1 mRNA levels possess a doubled risk at developing metastases; TET1 catalytic activity plays a suppressive role in prostate cancer [91]. In breast cancer, TET1 suppresses tumor growth, intravasation, and metastasis dependent of its catalytic activity [92]. Moreover, TET1 inhibits tumor development and invasion partly via maintaining the expression of tissue inhibitors of metalloproteinase (TIMP) dependent of its catalytic activity [103]. In addition, TET2 may suppress the tumor-promoting effects of inflammation through recruiting Hda2 to inhibit IL-6 independent of its catalytic activity [12]. Furthermore, TET1 could suppress gastric cancer formation via combining DNA demethylation with inhibition of oncogenic protein EZH2 and DNA-PK activation of p53 [104]. However, there are studies where TET proteins dual activities have or may have tumor-promoting functions. TET1 catalytic activity plays an indispensable oncogenic role through coordination with MLL-fusion proteins in MLL-rearranged leukemia [93]. In proneural glioblastomas, TET1-catalytic production of 5hmC is required for increased tumorigenicity [25]. In DIPG, TET proteins (TET1 and TET3) and 5hmC may underlie the tumor formation and resistance to treatment [24]. Furthermore, TET1 and TET3 protein levels are higher in uterine leiomyoma, and TET1 or TET3 knockdown decrease cell proliferation of leiomyoma cells [100]. Interestingly, TET1 interacts with HIF-1α and HIF-2α to enhance epithelial-mesenchymal transition (EMT) independent of its enzymatic activity and then could contribute to cancer metastasis [14]. Christopher et al. have observed that, in hypoxia, TET1 expression and 5hmC level increase within tumorigenic, N-type neuroblastoma cells [101]. These findings showed that TET proteins dual activities (catalytic or non-catalytic) are capable of playing dual roles (tumor-suppressing or tumor-promoting) via interacting with the TET binding protein partners in different types of tumors. Therefore, TET proteins can be a promising molecular target for tumors therapy. When TET proteins serve as the tumor suppressors, we contribute to the expression of them. When TET proteins serve as the tumors promoters, we suppress the expression of them.
In neural development, TET proteins dual activities (catalytic or non-catalytic) also exert dual functions (promoting or suppressing). TET proteins catalytic activity plays a promoting role in neuronal differentiation, mOSN development, adult neural progenitor cell proliferation, Xenopus eye and neural development, and OL development [27, 64-67]. In addition, TET3 non-catalytic activity plays a promoting role in Xenopus eye and neural development [66]. However, TET1 non-catalytic activity plays a suppressing role in neuronal differentiation of Neuro2a cells [74]. The elucidation of the relationship between the TET proteins dual activities and dual roles in neural development might offer potential advances in treatments for brain disorders.
Table 2: Clinical implications of TET proteins dual roles in tumors
Tumors |
5hmC levels |
TET proteins |
Role in tumorigenesis |
Ref |
many hematopoietic malignancies |
↓ |
(TET2)↓ |
tumor suppression |
|
breast cancer |
↓ |
(TET1)↓ |
tumor suppression |
[92] |
prostate cancer |
↓ |
(TET1)↓ |
tumor suppression |
[91] |
human hepatocellular carcinoma |
↓ |
(TET1)↓ |
tumor suppression |
[98] |
gastric cancer |
↓ |
(TET1)↓ |
tumor suppression |
[104] |
MLL-rearranged leukemia |
↑ |
(TET1)↑ |
tumor promotion |
[93] |
diffuse intrinsic pontine glioma |
↑ |
(TET1 and TET3)↑ |
tumor promotion (not determined) |
[24] |
proneural glioblastomas |
↑ |
(TET1)↑ |
tumor promotion |
[25] |
uterine leiomyoma |
↑ |
(TET1 and TET3)↑ |
tumor promotion |
[100] |
neuroblastoma in hypoxia |
↑ |
(TET1)↑ |
tumor promotion |
[101] |
inflammation-induced tumors |
— |
(TET2)— |
tumor suppression (not determined) |
[12] |
Note: ↑: higher levels compared to normal tissue; ↓: lower levels compared to normal tissue; —: possible no alteration compared to normal tissue
Conclusions and outlooks
As summarized in this review, extensive researches on TET family proteins have shed light on their enzymatic activity and non-enzymatic activity in neural development and human tumors. Further understanding of TET enzymes dual activities in normal neurodevelopment could make us realize that defects or even subtle aberrations of TET proteins in neurological disorders, such as human tumors. TET proteins dual activities are capable of playing dual roles in human tumors, including tumor-suppressive and tumor-enhancing effects. Some of presented findings indicate that the great potential and relevance of TET family proteins as the novel classes of therapeutic target in human tumors.
Abbreviations
TET, ten-eleven translocation; 5mC, 5-methylcytosine; 5hmC, 5-hydroxymethylc-ytosine; 5fC, 5-formylcytosine; 5caC, 5-carboxylcytosine; TDG, thymine DNA g-lycosylase; BER, base-excision repair; MLL, mixed lineage leukemia; AML, acute myeloid leukemia; 5hmU, 5-hydroxymethyluridine; AID, activation-induced deaminase; APOBEC, apolioprotein B mRNA-editing enzyme catalytic polypeptide; Hdac, histone deacetylase; OGT, O-glcNAC- transferase; HIFs, hypoxiainducible factors; IDH, isocitrate dehydrogenase; DIPG, diffuse intrinsic pontine glioma; GBM, glioblastoma multiforme; NPCs, neural precursor cells; OL, oligodendrocyte; NSCs, neural stem cells; DSBH, double-stranded β-helix; α-KG, α-ketoglutarate; DNMTs, DNA methyltransterases; 2-HG, 2-hydroxyglutarate; mOSNs, mature olfactory sensory neurons; CSF, cerebrospinal fluid; LGG, low-grade glioma; AA, anaplastic astrocytomas; MPN, myeloproliferative neoplasms; MDS, myelodysplastic syndrome; CMML, chronic myelomonocytic leukemia; HMGA2, high mobility group AT-hook2; HCC, hepatocellular carcinoma; TIMP, tissue inhibitors of metalloproteinase; EMT, epithelial-mesenchymal transition.
Acknowledgments
This work was in part supported by National Natural Science Foundation of China (No. 81272801).
conflicts of interest
The authors declared no potential financial conflicts of interest.
REFERENCES
1. Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H and Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res. 2002;62:4075-4080.
2. Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST and Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia. 2003;17:637-641.
3. Burmeister T, Meyer C, Schwartz S, Hofmann J, Molkentin M, Kowarz E, Schneider B, Raff T, Reinhardt R, Gokbuget N, Hoelzer D, Thiel E and Marschalek R. The MLL recombinome of adult CD10-negative B-cell precursor acute lymphoblastic leukemia: results from the GMALL study group. Blood. 2009;113:4011-4015.
4. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L and Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930-935.
5. Maiti A and Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334-35338.
6. Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu GL, Luo C, Jiang H and He C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol. 2012;8:328-330.
7. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303-1307.
8. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C and Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300-1303.
9. Guo JU, Su Y, Zhong C, Ming GL and Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423-434.
10. Wu H and Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436-2452.
11. Cartron PF, Nadaradjane A, Lepape F, Lalier L, Gardie B and Vallette FM. Identification of TET1 Partners That Control Its DNA-Demethylating Function. Genes Cancer. 2013;4:235-241. doi: 10.1177/1947601913489020.
12. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, Su X, Liu J, Ge W, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389-393.
13. Chen Q, Chen Y, Bian C, Fujiki R and Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561-564.
14. Tsai YP, Chen HF, Chen SY, Cheng WC, Wang HW, Shen ZJ, Song C, Teng SC, He C and Wu KJ. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014;15:513.
15. Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, Shih AH, Levine RL, Bernard O, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645-655.
16. Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J and Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343-348.
17. Huang Y and Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014;30:464-474.
18. Ko M, An J, Pastor WA, Koralov SB, Rajewsky K and Rao A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol Rev. 2015;263:6-21.
19. Jin SG, Jiang Y, Qiu R, Rauch TA, Wang Y, Schackert G, Krex D, Lu Q and Pfeifer GP. 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 2011;71:7360-7365.
20. Kudo Y, Tateishi K, Yamamoto K, Yamamoto S, Asaoka Y, Ijichi H, Nagae G, Yoshida H, Aburatani H and Koike K. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012;103:670-676.
21. Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, Xu ZD, Zhu HG, Ling ZQ, Ye D, Guan KL and Xiong Y. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32:663-669.
22. Jawert F, Hasseus B, Kjeller G, Magnusson B, Sand L and Larsson L. Loss of 5-hydroxymethylcytosine and TET2 in oral squamous cell carcinoma. Anticancer Res. 2013;33:4325-4328.
23. Kraus TF, Globisch D, Wagner M, Eigenbrod S, Widmann D, Munzel M, Muller M, Pfaffeneder T, Hackner B, Feiden W, Schuller U, Carell T and Kretzschmar HA. Low values of 5-hydroxymethylcytosine (5hmC), the “sixth base,” are associated with anaplasia in human brain tumors. Int J Cancer. 2012;131:1577-1590.
24. Ahsan S, Raabe EH, Haffner MC, Vaghasia A, Warren KE, Quezado M, Ballester LY, Nazarian J, Eberhart CG and Rodriguez FJ. Increased 5-hydroxymethylcytosine and decreased 5-methylcytosine are indicators of global epigenetic dysregulation in diffuse intrinsic pontine glioma. Acta Neuropathol Commun. 2014;2:59.
25. Takai H, Masuda K, Sato T, Sakaguchi Y, Suzuki T, Suzuki T, Koyama-Nasu R, Nasu-Nishimura Y, Katou Y, Ogawa H, Morishita Y, Kozuka-Hata H, Oyama M, et al. 5-Hydroxymethylcytosine plays a critical role in glioblastomagenesis by recruiting the CHTOP-methylosome complex. Cell Rep. 2014;9:48-60.
26. Szwagierczak A, Bultmann S, Schmidt CS, Spada F and Leonhardt H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010;38:e181.
27. Hahn MA, Qiu R, Wu X, Li AX, Zhang H, Wang J, Jui J, Jin SG, Jiang Y, Pfeifer GP and Lu Q. Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep. 2013;3:291-300.
28. Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC and Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129-1133.
29. Pastor WA, Aravind L and Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341-356.
30. Kriukiene E, Liutkeviciute Z and Klimasauskas S. 5-Hydroxymethylcytosine—the elusive epigenetic mark in mammalian DNA. Chem Soc Rev. 2012;41:6916-6930.
31. Tan L and Shi YG. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development. 2012;139:1895-1902.
32. Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, Shi YG, Zhu J, Wang P and Xu Y. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013;155:1545-1555.
33. Frauer C, Rottach A, Meilinger D, Bultmann S, Fellinger K, Hasenoder S, Wang M, Qin W, Soding J, Spada F and Leonhardt H. Different binding properties and function of CXXC zinc finger domains in Dnmt1 and Tet1. PLoS One. 2011;6:e16627.
34. Iyer LM, Tahiliani M, Rao A and Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698-1710.
35. Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J, Barbera AJ, Zheng L, Zhang H, Huang S, Min J, Nicholson T, Chen T, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451-464.
36. Zhang H, Zhang X, Clark E, Mulcahey M, Huang S and Shi YG. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010;20:1390-1393.
37. Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, Segal MF, Li H, Koh KP, Lahdesmaki H, Hogan PG, Aravind L and Rao A. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013;497:122-126.
38. Hino S, Kishida S, Michiue T, Fukui A, Sakamoto I, Takada S, Asashima M and Kikuchi A. Inhibition of the Wnt signaling pathway by Idax, a novel Dvl-binding protein. Mol Cell Biol. 2001;21:330-342.
39. Kojima T, Shimazui T, Hinotsu S, Joraku A, Oikawa T, Kawai K, Horie R, Suzuki H, Nagashima R, Yoshikawa K, Michiue T, Asashima M, Akaza H, et al. Decreased expression of CXXC4 promotes a malignant phenotype in renal cell carcinoma by activating Wnt signaling. Oncogene. 2009;28:297-305.
40. Bergman Y and Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol. 2013;20:274-281.
41. Bestor T, Laudano A, Mattaliano R and Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol. 1988;203:971-983.
42. Hermann A, Goyal R and Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem. 2004;279:48350-48359.
43. Bourc’his D, Xu GL, Lin CS, Bollman B and Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536-2539.
44. Okano M, Xie S and Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219-220.
45. Valinluck V and Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946-950.
46. Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, Zhang X and Cheng X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012;40:4841-4849.
47. Chen CC, Wang KY and Shen CK. The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J Biol Chem. 2012;287:33116-33121.
48. Schiesser S, Hackner B, Pfaffeneder T, Muller M, Hagemeier C, Truss M and Carell T. Mechanism and stem-cell activity of 5-carboxycytosine decarboxylation determined by isotope tracing. Angew Chem Int Ed Engl. 2012;51:6516-6520.
49. Dang L, Jin S and Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med. 2010;16:387-397.
50. Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL and Kaelin WG Jr. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621-1625.
51. Ye D, Ma S, Xiong Y and Guan KL. R-2-hydroxyglutarate as the key effector of IDH mutations promoting oncogenesis. Cancer Cell. 2013;23:274-276.
52. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17-30.
53. Arioka Y, Watanabe A, Saito K and Yamada Y. Activation-induced cytidine deaminase alters the subcellular localization of Tet family proteins. PLoS One. 2012;7:e45031.
54. Huang Y, Wang G, Liang Z, Yang Y, Cui L and Liu CY. Loss of nuclear localization of TET2 in colorectal cancer. Clin Epigenetics. 2016;8:9.
55. Muller T, Gessi M, Waha A, Isselstein LJ, Luxen D, Freihoff D, Freihoff J, Becker A, Simon M, Hammes J, Denkhaus D, zur MA, Pietsch T, et al. Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. Am J Pathol. 2012;181:675-683.
56. Eleftheriou M, Pascual AJ, Wheldon LM, Perry C, Abakir A, Arora A, Johnson AD, Auer DT, Ellis IO, Madhusudan S and Ruzov A. 5-Carboxylcytosine levels are elevated in human breast cancers and gliomas. Clin Epigenetics. 2015;7:88.
57. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326-1338.
58. Yin R, Mao SQ, Zhao B, Chong Z, Yang Y, Zhao C, Zhang D, Huang H, Gao J, Li Z, Jiao Y, Li C, Liu S, et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc. 2013;135:10396-10403.
59. Minor EA, Court BL, Young JI and Wang G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013;288:13669-13674.
60. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martinez JA, Goyal P, Mahapatra S, Tam A, Laird DJ, Hirst M, Rao A, Lorincz MC and Ramalho-Santos M. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222-226.
61. Gustafson CB, Yang C, Dickson KM, Shao H, Van Booven D, Harbour JW, Liu ZJ and Wang G. Epigenetic reprogramming of melanoma cells by vitamin C treatment. Clin Epigenetics. 2015;7:51.
62. Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, Li Y, Ahn J, Abdel-Wahab O, Shih A, Lu C, Ward PS, Tsai JJ, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9:1841-1855.
63. Kriaucionis S and Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929-930.
64. Colquitt BM, Allen WE, Barnea G and Lomvardas S. Alteration of genic 5-hydroxymethylcytosine patterning in olfactory neurons correlates with changes in gene expression and cell identity. Proc Natl Acad Sci U S A. 2013;110:14682-14687.
65. Zhang RR, Cui QY, Murai K, Lim YC, Smith ZD, Jin S, Ye P, Rosa L, Lee YK, Wu HP, Liu W, Xu ZM, Yang L, et al. Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell. 2013;13:237-245.
66. Xu Y, Xu C, Kato A, Tempel W, Abreu JG, Bian C, Hu Y, Hu D, Zhao B, Cerovina T, Diao J, Wu F, He HH, et al. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell. 2012;151:1200-1213.
67. Zhao X, Dai J, Ma Y, Mi Y, Cui D, Ju G, Macklin WB and Jin W. Dynamics of ten-eleven translocation hydroxylase family proteins and 5-hydroxymethylcytosine in oligodendrocyte differentiation. Glia. 2014;62:914-926.
68. Wheldon LM, Abakir A, Ferjentsik Z, Dudnakova T, Strohbuecker S, Christie D, Dai N, Guan S, Foster JM, Correa IR Jr, Loose M, Dixon JE, Sottile V, et al. Transient accumulation of 5-carboxylcytosine indicates involvement of active demethylation in lineage specification of neural stem cells. Cell Rep. 2014;7:1353-1361.
69. Mantovani A, Allavena P, Sica A and Balkwill F. Cancer-related inflammation. Nature. 2008;454:436-444.
70. Shen F, Zhang Y, Yao Y, Hua W, Zhang HS, Wu JS, Zhong P and Zhou LF. Proteomic analysis of cerebrospinal fluid: toward the identification of biomarkers for gliomas. Neurosurg Rev. 2014;37:367-380; discussion 380.
71. Kaas GA, Zhong C, Eason DE, Ross DL, Vachhani RV, Ming GL, King JR, Song H and Sweatt JD. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron. 2013;79:1086-1093.
72. Ambigapathy G, Zheng Z and Keifer J. Regulation of BDNF chromatin status and promoter accessibility in a neural correlate of associative learning. Epigenetics. 2015;10:981-993.
73. Rudenko A, Dawlaty MM, Seo J, Cheng AW, Meng J, Le T, Faull KF, Jaenisch R and Tsai LH. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. 2013;79:1109-1122.
74. Gao J, Ma Y, Fu HL, Luo Q, Wang Z, Xiao YH, Yang H, Cui DX and Jin WL. Non-catalytic roles for TET1 protein negatively regulating neuronal differentiation through srGAP3 in neuroblastoma cells. Protein Cell. 2016;7:351-361.
75. Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R, Huberman K, Thomas S, Dolgalev I, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144-147.
76. Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M, Sainty D, Olschwang S, Xerri L, Chaffanet M, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145:788-800.
77. Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, Quesnel B, Fenaux P, Bastie JN, Beyne-Rauzy O, Stamatoulas A, Dreyfus F, Ifrah N, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94:1676-1681.
78. Langemeijer SM, Aslanyan MG and Jansen JH. TET proteins in malignant hematopoiesis. Cell cycle (Georgetown, Tex.). 2009;8:4044-4048.
79. Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, Kamping EJ, Verhoef GE, Verburgh E, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838-842.
80. Mohamedali AM, Smith AE, Gaken J, Lea NC, Mian SA, Westwood NB, Strupp C, Gattermann N, Germing U and Mufti GJ. Novel TET2 mutations associated with UPD4q24 in myelodysplastic syndrome. J Clin Oncol. 2009;27:4002-4006.
81. Beer PA, Delhommeau F, LeCouédic JP, Dawson MA, Chen E, Bareford D, Kusec R, McMullin MF, Harrison CN, Vannucchi AM, Vainchenker W and Green AR. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood. 2010;115:2891-2900.
82. Colaizzo D, Tiscia GL, Pisanelli D, Bafunno V, Amitrano L, Grandone E, Guardascione MA and Margaglione M. New TET2 gene mutations in patients with myeloproliferative neoplasms and splanchnic vein thrombosis. J Thromb Haemost. 2010;8:1142-1144.
83. Couronné L, Lippert E, Andrieux J, Kosmider O, Radford-Weiss I, Penther D, Dastugue N, Mugneret F, Lafage M, Gachard N, Nadal N, Bernard OA and Nguyen-Khac F. Analyses of TET2 mutations in post-myeloproliferative neoplasm acute myeloid leukemias. Leukemia. 2010;24:201-203.
84. Nibourel O, Kosmider O, Cheok M, Boissel N, Renneville A, Philippe N, Dombret H, Dreyfus F, Quesnel B, Geffroy S, Quentin S, Roche-Lestienne C, Cayuela JM, et al. Incidence and prognostic value of TET2 alterations in de novo acute myeloid leukemia achieving complete remission. Blood. 2010;116:1132-1135.
85. Schaub FX, Looser R, Li S, Hao-Shen H, Lehmann T, Tichelli A and Skoda RC. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood. 2010;115:2003-2007.
86. Szpurka H, Jankowska AM, Makishima H, Bodo J, Bejanyan N, Hsi ED, Sekeres MA and Maciejewski JP. Spectrum of mutations in RARS-T patients includes TET2 and ASXL1 mutations. Leuk Res. 2010;34:969-973.
87. Rasmussen KD and Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733-750.
88. Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki H, Nagata Y, Yoshida K, Kon A, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860-867.
89. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, Guillory J, Ha C, Dijkgraaf GJ, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660-664.
90. Nickerson ML, Im KM, Misner KJ, Tan W, Lou H, Gold B, Wells DW, Bravo HC, Fredrikson KM, Harkins TT, Milos P, Zbar B, Linehan WM, et al. Somatic alterations contributing to metastasis of a castration-resistant prostate cancer. Hum Mutat. 2013;34:1231-1241.
91. Spans L, Van den Broeck T, Smeets E, Prekovic S, Thienpont B, Lambrechts D, Karnes RJ, Erho N, Alshalalfa M, Davicioni E, Helsen C, Gevaert T, Tosco L, et al. Genomic and epigenomic analysis of high-risk prostate cancer reveals changes in hydroxymethylation and TET1. Oncotarget. 2016; 7:24326-38. doi: 10.18632/oncotarget.8220.
92. Sun M, Song CX, Huang H, Frankenberger CA, Sankarasharma D, Gomes S, Chen P, Chen J, Chada KK, He C and Rosner MR. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci U S A. 2013;110:9920-9925.
93. Huang H, Jiang X, Li Z, Li Y, Song CX, He C, Sun M, Chen P, Gurbuxani S, Wang J, Hong GM, Elkahloun AG, Arnovitz S, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci U S A. 2013;110:11994-11999.
94. Chim CS, Wan TS, Fung TK and Wong KF. Methylation of TET2, CBL and CEBPA in Ph-negative myeloproliferative neoplasms. J Clin Pathol. 2010;63:942-946.
95. Kim YH, Pierscianek D, Mittelbronn M, Vital A, Mariani L, Hasselblatt M and Ohgaki H. TET2 promoter methylation in low-grade diffuse gliomas lacking IDH1/2 mutations. J Clin Pathol. 2011;64:850-852.
96. Cheng J, Guo S, Chen S, Mastriano SJ, Liu C, D’Alessio AC, Hysolli E, Guo Y, Yao H, Megyola CM, Li D, Liu J, Pan W, et al. An extensive network of TET2-targeting MicroRNAs regulates malignant hematopoiesis. Cell Rep. 2013;5:471-481.
97. Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C, Beringer G, Brikbak NJ, Yuan X, Cantley LC, Richardson AL and Pandolfi PP. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. 2013;154:311-324.
98. Chuang KH, Whitney-Miller CL, Chu CY, Zhou Z, Dokus MK, Schmit S and Barry CT. MicroRNA-494 is a master epigenetic regulator of multiple invasion-suppressor microRNAs by targeting ten eleven translocation 1 in invasive human hepatocellular carcinoma tumors. Hepatology. 2015;62:466-480.
99. Lv X, Jiang H, Liu Y, Lei X and Jiao J. MicroRNA-15b promotes neurogenesis and inhibits neural progenitor proliferation by directly repressing TET3 during early neocortical development. EMBO Rep. 2014;15:1305-1314.
100. Navarro A, Yin P, Ono M, Monsivais D, Moravek MB, Coon JS, Dyson MT, Wei JJ and Bulun SE. 5-Hydroxymethylcytosine promotes proliferation of human uterine leiomyoma: a biological link to a new epigenetic modification in benign tumors. J Clin Endocrinol Metab. 2014;99:E2437-2445.
101. Mariani CJ, Vasanthakumar A, Madzo J, Yesilkanal A, Bhagat T, Yu Y, Bhattacharyya S, Wenger RH, Cohn SL, Nanduri J, Verma A, Prabhakar NR and Godley LA. TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep. 2014;7:1343-1352.
102. Rasmussen KD, Jia G, Johansen JV, Pedersen MT, Rapin N, Bagger FO, Porse BT, Bernard OA, Christensen J and Helin K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015;29:910-922.
103. Hsu CH, Peng KL, Kang ML, Chen YR, Yang YC, Tsai CH, Chu CS, Jeng YM, Chen YT, Lin FM, Huang HD, Lu YY, Teng YC, et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012;2:568-579.
104. Fu HL, Ma Y, Lu LG, Hou P, Li BJ, Jin WL and Cui DX. TET1 exerts its tumor suppressor function by interacting with p53-EZH2 pathway in gastric cancer. J Biomed Nanotechnol. 2014;10:1217-1230.