
INTRODUCTION
Glioblastoma multiforme (GBM), a high-grade glioma (WHO grade IV) is the most common and lethal primary malignant brain tumor in adults, with a median survival of only 16 months. Despite current intensive therapy regimes including surgery, radiotherapy (RT) and temozolomide (TMZ)-based adjuvant chemotherapy (CT), disease progression occurs in almost all patients [1, 2]. Therefore, the improvement of therapy for GBM patients is in the focus of recent research, which also includes targeted therapeutics to inhibit cellular signaling pathways [3, 4].
This includes the promising approach of using the multi-kinase inhibitor sorafenib. Sorafenib has been shown not only to block the members of the MAPK pathways Raf-1 and p38 but also receptor tyrosine kinases like VEGFR, cKit or PDGFR [5] and it is already approved for the treatment of various tumor entities [6–8]. For GBM cells sorafenib has been shown to induce apoptosis, to deplete tumor initiating cells and to reduce proliferation in cell culture and in xenograft models [9–11]. Despite these promising results sorafenib showed only very limited effects as a mono-therapeutic drug, in combination with TMZ or other targeted therapeutics such as erlotinib in clinical studies with patients having progressive or recurrent diseases [12–16]. However, for other entities we and others have already reported that sorafenib induces cellular radiosensitization, arguing for a combination of radiotherapy and sorafenib to improve the treatment of radioresistant tumors [17–21]. For GBM cells so far only two studies exist which tested the combination of sorafenib and X-irradiation by determining the number of viable cells or by using the MTT assay respectively [22, 23]. Despite some promising results, these data certainly do not answer the question of cellular radiosensitization by sorafenib.
Because of the importance of sorafenib for current targeted therapy approaches and the lack of solid data on the effects of sorafenib on X-irradiation and TMZ in GBM we investigated in this study the potential of sorafenib to radiosensitize and chemosensitize GBM cells. This study was performed using six individual GBM cell lines with differences in the p53 status, because the p53 status is known to be important for cell survival. Furthermore, we only used O6-methylguanine-methyltransferase (MGMT) negative cells since the TMZ sensitivity is known to depend strongly on MGMT status [24].
RESULTS
To test if sorafenib is a potential therapeutic drug to improve radio-chemotherapy of GBM we wanted to analyze the influence of sorafenib on cellular radio- and chemosensitivity in various GBM cells lines. To this end, we chose the colony-forming assay, because this assay is able to directly measure the ability of tumor cells for self-renewal (clonogenicity). This is of special importance since effects on proliferation or metabolism might not truly reflect cell inactivation but could be also be caused by prolonged growth arrest. Withdrawal of the inhibitor, re-stimulating events or extended culture times might lead to a restart in growth of solely arrested but not truly inactivated cells.
Impact of sorafenib on proliferation, clonogenicity and MAPK signaling
Because the colony forming assay can be influenced by the proliferation rate we first investigated the effect of sorafenib on cell proliferation. In these experiments we observed a strong decrease in the proliferation rate for all cell lines in a concentration-dependent manner (Figure 1A).
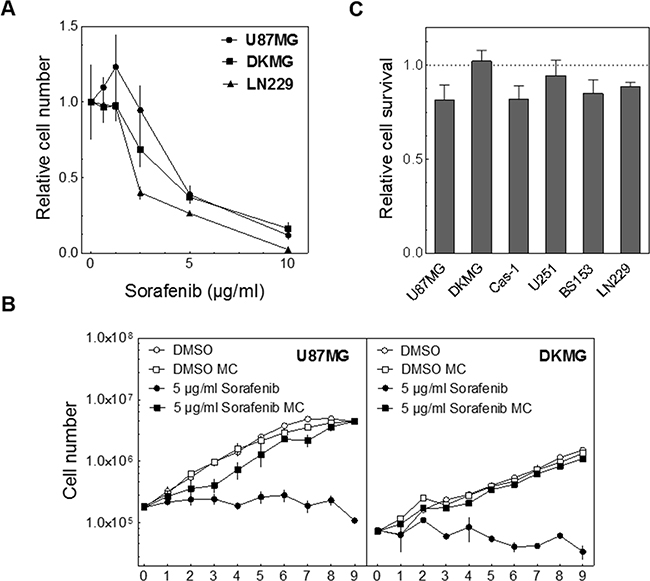
Figure 1: Effect of sorafenib on proliferation & clonogenicity. A. To determine the effect of sorafenib on proliferation U87MG, DKMG and LN229 cells were incubated with different concentrations of sorafenib for 3 days as indicated. The number of cells measured in treated cultures was divided by the number of cells determined for untreated cultures, both counted after 3 days of incubation. The relative cell number is depicted. B. Proliferation of U87MG and DKMG cells in the presence of sorafenib (n=2). Twenty-four hours after seeding the cells were treated with 5 μg/ml sorafenib either for 24 h (media change, MC) or for up to 9 d. C. Relative cytotoxicity as determined by colony forming assay. Cells were treated with 5 μg/ml sorafenib for 24 h and cultivated for 10-25 days to allow for colony formation.
Because this block in proliferation might reduce clonogenicity and thereby mimic cell inactivation we tested if the inhibitory effect of sorafenib persists. For these and further experiments we chose a concentration of 5 μg/ml sorafenib tosylate which matches 7.8 μM and therefore approximately reflects the serum level in patients [12, 13, 25]. When the cells were exposed continuously sorafenib caused a complete block of proliferation for up to 9 days which was statistically significant from the 2nd day on (Figure 1B). In contrast, when sorafenib was removed after 24 h the cells were able to proliferate again. Under these conditions only moderate cytotoxic effects were observable using the colony forming assays (Figure 1C, pre-plating). Therefore the medium was changed 24 h after the treatment in the following assays.
Next, the MAPK signaling pathway was analyzed, since Raf-1 is a main target of sorafenib [5]. We observed reduced MEK1/2 and ERK1/2-phosphorylation in at least three cell lines using Western blot analysis (Figure 2A), with significant inhibition of ERK being detectable for DKMG (p=0.003) and Cas-1 (p=0.043) cells (Figure 2B). For DKMG cells, which displayed the most impressive inhibition, we also analyzed the phosphorylation of Akt, STAT3 and VEGFR. We detected no reduction in protein phosphorylation except for Akt, albeit only when using higher concentrations of sorafenib (Supplementary Figure S1). Interestingly neither the cytotoxic effect of sorafenib nor the inhibition of proliferation correlated with the inhibition of MAPK-signaling.
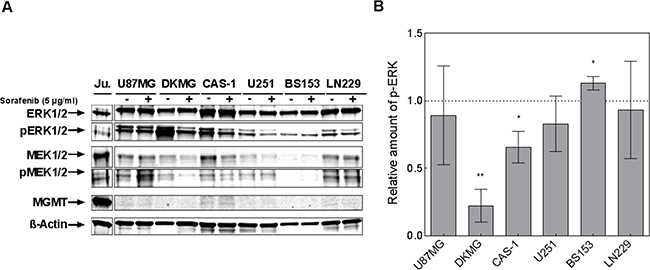
Figure 2: Effect of sorafenib on MAPK signaling. A. Phosphorylation of ERK1/2 (T202/Y204) and MEK1/2 (S217/S221) was determined by Western blot analysis using phosphospecific antibodies. Cells were treated with 5 μg/ml sorafenib for 2 h. The detection of total ERK, MEK and β-actin served as controls. MGMT was detected using MGMT-specific antibodies while lysates from Jurkat cells were used as a positive control. B. Quantification of ERK1/2 phosphorylation after sorafenib treatment. Corrected pERK1/2 levels (pERK/ERK) of sorafenib-treated samples were normalized to the corrected pERK1/2 levels of untreated samples. Depicted are the results of three independent Western blots.
Impact of sorafenib on cellular radiosensitivity
The central aim of this study was to determine whether sorafenib enhances radiosensitivity of GBM cells. Using the colony-forming assay we observed no increase in cellular radiosensitivity following sorafenib treatment in U87MG cells (Figure 3A) or in any of the other GBM cell lines as represented by the survival fraction at 6 Gy in Figure 3B. Remarkably, sorafenib might induce some radioresistance which, however, was significant only in DKMG cells (p=0.047). As reported previously [26] a huge variation in cellular radiosensitivity was seen for the six tested GBM cell lines, with LN229 cells being the most resistant and BS153 the most sensitive.
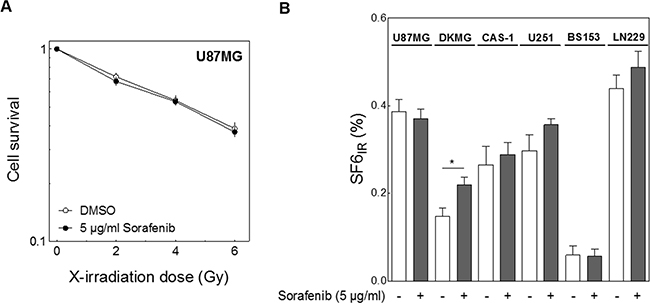
Figure 3: Effect of sorafenib on cellular radiosensitivity. Cells were treated with 5 μg/ml sorefenib for 2 h before irradiation with 0, 2, 4 and 6 Gy. Cell survival was assessed by colony forming assay. A. Relative cell survival of U87MG cells. B. Relative surviving fraction of all six GBM cell lines after 6 Gy (SF6IR).
Impact of sorafenib on chemosensitivity
Besides irradiation TMZ is used in the standard treatment regime of GBM patients. Therefore we also analyzed the effect of sorafenib on TMZ-sensitivity using a clinically relevant dose range of 1 to 10 μM TMZ. Cells were treated with TMZ for 1-3 days according to their proliferation rate (Figure 4A; Supplementary Table S1). During this time sorafenib was present for the first 24 h. The addition of sorafenib did not influence the cell inactivation by TMZ and therefore had no impact on cellular chemosensitivity (Figure 4A, 4B).
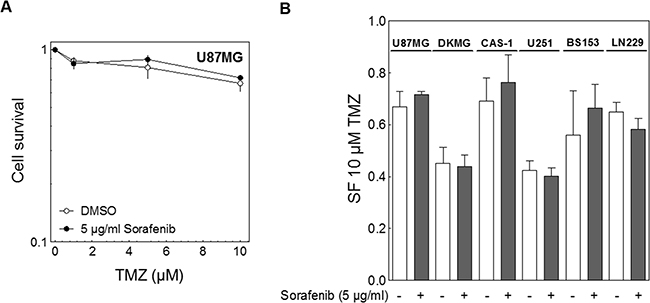
Figure 4: Effect of sorafenib on chemosensitivity. Cells were treated with different concentrations of TMZ as indicated for 1-3 days according to their doubling time. The cells were also treated with 5 μg/ml sorafenib for the first 24 h. The medium was changed and cell survival was assessed by colony forming assay. A. Relative cell survival of U87MG cells. B. Relative survival fraction of all six GBM cell lines after 10 μM TMZ (SF10TMZ).
Interestingly, the TMZ sensitivity varied quite strongly among the different cell lines although none of them expressed MGMT (Figure 1D). Furthermore, the TMZ sensitivity clearly did not correlate with cellular radiosensitivity (Supplementary Figure S2).
Impact of sorafenib on combined treatment with irradiation and TMZ
Because GBM patients are generally treated with radiotherapy and adjuvant TMZ we also evaluated the effect of sorafenib on the combined treatment. In this experimental set up we also tested the effect of TMZ on cellular radiosensitivity alone and detected a radiosensitizing effect of TMZ in three of the six cell lines (Figure 5A, 5C; DKMG p6Gy=0.0252, U251 p6Gy=0.0041, LN229 p6Gy=0.0193) (Figure 5B, 5C). However, when sorafenib was added there was no further decrease in cell survival, but instead a significant increase was seen in Cas-1 cells (p6Gy=0.0303) (Figure 5C).
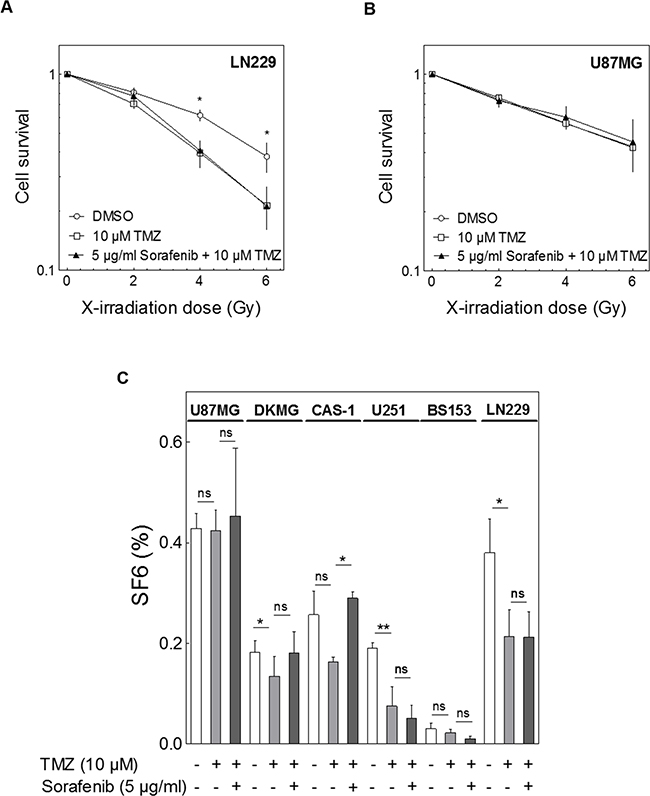
Figure 5: Impact of sorafenib on combined treatment with irradiation and TMZ. Cells were treated with 10 μM TMZ with or without 5 μg/ml sorafenib 2 h before irradiation. Cellular survival was analyzed by colony formation. A, B. Relative cell survival of (A) LN229 and (B) U87MG cells. C. Relative surviving fraction at 6 Gy (SF6) with combined sorafenib and/or TMZ treatment as detected for all six cell lines.
Taken together these results demonstrate that, although sorafenib inhibits proliferation of GBM cells and causes a modest cell inactivation as a single agent, it does not enhance cell inactivation by irradiation, TMZ or combined treatment.
DISCUSSION
In this study, we analyzed the impact of sorafenib on cellular radiosensitivity, chemosensitivity (TMZ) and on the combined treatment of GBM cell lines. To consider potential variations which might affect the outcome of this study, we analyzed six individual GBM cell lines with differences in p53 and PTEN status, but all negative for MGMT expression (Supplementary Table S1).
Surprisingly, we observed no impact of sorafenib on cellular radiosensitivity in any of the cell lines. Instead, there was a trend towards radioresistance (Figure 3). This result is in contrast to the sorafenib-mediated cellular radiosensitization observed for other entities such as head and neck cancer, colorectal carcinoma, breast cancer and hepatocellular carcinoma [17–21]. Consequently, radiosensitization by sorafenib seems to be a frequent phenomenon in specific entities but not a general one.
Our results do not agree with data from former studies reporting an effect of sorafenib on the radiation response of GBM cell lines [22, 23]. However, these studies did not address radiosensitization in terms of clonogenicity. That radiosensitizing effects can in fact be observed using the clonogenic assay is demonstrated by the successful radiosensitization of three of the six cell lines by TMZ (Figure 5). This observation is in line with previous data demonstrating the radiosensitizing effect of TMZ [27].
Similar to the cellular radiosensitivity results we also detected no chemosensitization by sorafenib in combination with TMZ, but again a trend towards resistance (Figure 4). The resistance towards IR or TMZ&IR was unrelated to the ability of sorafenib to block MAPK-signaling (Supplementary Figure S3).
So far there are no other preclinical data available addressing this question. However, in combination with other drugs sorafenib was reported to enhance cytotoxicity in GBM cells but in these studies cellular survival was not analyzed in terms of clonogenicity [28–30]. Moreover, sorafenib might also have negative effects in combination with additional drugs, since it has been shown to reduce drug uptake [12, 31].
Although we demonstrate here, that sorafenib does not increase radiation- or TMZ-induced cell inactivation we observed a strong inhibition of proliferation which might translate into reduced tumor growth. But when sorafenib was removed after 24 h, proliferation restarted, resulting only in a minor delay of cell growth and a small reduction in clonogenicity for most of the cell lines (Figure 1C), which will likely have no strong influence on tumor control. However, sorafenib might improve tumor control in a clinical setting via additional mechanisms like targeting specifically tumor stem cells or influencing tumor angiogenesis [4, 32].
Sorafenib also had no positive effect on the double treatment with irradiation and TMZ. In fact, it instead made two of the six cell lines more resistant, which was significant for Cas-1 cells. These data are in line with recent clinical studies testing sorafenib in the first-line treatment in combination with TMZ after RCT (maintenance therapy) or in combination with TMZ and radiotherapy. These studies also do not support these treatment combinations because they did not seem to improve the efficacy of the treatment but caused increased side effects [33, 34].
Taken together, we have systematically demonstrated here for the first time that for GBM sorafenib induces only minor cell inactivation as a single treatment and has no benefit in respect to cell inactivation when combined with TMZ and/or irradiation. Moreover, sorafenib seems to cause resistance towards TMZ-based RCT as detected by increased cell survival in some cell lines. Therefore, our data do not support the use of sorafenib for GBM with the intention to increase cellular inactivation by RT or TMZ-based RCT.
MATERIALS & METHODS
Inhibitors and reagents
Small molecule inhibitor sorafenib (sorafenib tosylate, Nexavar®, Bayer HealthCare), alkylating agent temozolomide (10 μM, Sigma-Aldrich), solvent DMSO (Sigma-Aldrich).
Cell culture
All the cell lines had already been used in the lab as reported earlier [26]. The GBM cell lines U87MG, CAS-1, U251, BS153 and LN229 were grown in DMEM (Sigma-Aldrich) supplemented with 10% FCS (Biochrom), 2 mM L-glutamine and 1 mM sodium pyruvate (Sigma-Aldrich). The GBM cell line DKMG was cultured in RPMI with 10% FCS, 2 mM L-glutamine, 1 mM sodium pyruvate. All cells were cultured at 37°C, 5% CO2 and 100% humidification and were identified by a short tandem repeat multiplex assay (Applied Biosystems). Gene sequencing and literature search revealed p53 wildtype expression in LN229, U87MG and DKMG cells and expression of mutant p53 in CAS-1, U251 and BS153 cells. Wildtype PTEN was detected only in LN229 cells (Supplementary Table S1). As for BS153 and DKMG cells the EGFRvIII-negative sublines were used [26].
Cell proliferation
To analyse proliferation, 1×105 cells were seeded. One day later, the cells were treated with sorafenib or DMSO (day 0) as indicated and the cell number was determined in parallel cultures by counting trypsinised cells using a Coulter counter (Beckman Coulter GmbH) every day henceforward.
Irradiation
Cells were irradiated at room temperature with 200 kV X-rays (Gulmay RS225, Gulmay Medical Ltd., 15 mA, 0.8mm Be + 0.5 mm Cu filtering; dose rate of 1.2 Gy/min).
Western blot
Proteins from whole cell extracts were detected by Western blot according to standard protocols. Primary antibodies: anti-ERK, anti-pERK (T202/Y204), anti-MEK1/2, anti-pMEK1/2 (S217/S221), anti-MGMT (Cell Signaling Technology) and anti-β-actin (Sigma-Aldrich). Secondary antibodies: anti-mouse and anti-rabbit (Li-COR Biosciences). The Odyssey® CLx Infrared Imaging System (LI-COR) was utilized for signal detection and quantification.
Colony forming assay (clonogenicity)
To analyse the capacity of the GBM cells for self-renewal (clonogenicity), the colony-forming assay was used. To this end, 200-350 cells (depending on the cell line) were seeded in triplicates 24 h prior to treatment with sorafenib (5 μg/ml), TMZ (10 μM) and/or irradiation. Twenty-four hours after treatment the medium was changed and TMZ was added to the corresponding samples for additional 1-2 days according to the doubling time of each cell line (Supplementary Table S1). The cells were grown until the colonies of all treatment arms had reached equal colony size (approximately 10-25 days; pre-plating conditions). Colony formation of U87MG, DKMG, CAS-1 and BS153 cells had to be promoted by replacing the medium in all colony assays by AmnioMax C-100 Basal Medium (Life Technologies) containing 10% FCS and C-100 supplement (Life Technologies) 24 h after irradiation. The cells were fixed with 70% ethanol, stained with crystal violet and colonies of more than 50 cells were counted manually. The surviving fraction of treated cells was normalized to the plating-efficiency of non-treated cells. For combination treatments involving radiation, the cells were irradiated 2 h after adding sorafenib and TMZ to minimise changes in the cell cycle distribution (verified by flow cytometry, data not shown). Sorafenib was removed 24 h later and TMZ was added for additional 1-2 days as mentioned above.
Data evaluation
The experiments were repeated at least three times. The data were presented as mean values (±SEM). Prism software (GraphPad Prism 5) was used for analyzing and graphing the data. Student’s t-tests were performed for the statistical analysis. P-values were calculated using unpaired two-sided tests (* p < 0.05, **p < 0.01).
ACKNOWLEDGMENTS
The authors gratefully acknowledge the technical assistance of K. Hoffer and B. Riepen.
CONFLICTS OF INTEREST
All authors declare no conflict of interest.
FUNDING
N. Struve, M. Kriegs & E. Dikomey: City of Hamburg, Landesexzellenzinitiative Hamburg (LEXI 2012; Tumor targeting via cell surface molecules essential in cancer progression and dissemination); M. Kriegs and E. Dikomey:BMBF (02NUK032); M. Kriegs & N. Struve: Forschungsförderung der medizinischen Fakultät am UKE, Hamburg; M. Kriegs: Erich und Gertrud Roggenbuck-Stiftung; Spierling Stiftung;
REFERENCES
1. Ohgaki H, Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009; 100: 2235-41. doi: 10.1111/j.1349-7006.2009.01308.x.
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352: 987-96. doi: 10.1056/NEJMoa043330.
3. Wick W, Weller M, Weiler M, Batchelor T, Yung AW, Platten M. Pathway inhibition: emerging molecular targets for treating glioblastoma. Neuro Oncol. 2011; 13: 566-79. doi: 10.1093/neuonc/nor039.
4. Wurth R, Barbieri F, Florio T. New Molecules and Old Drugs as Emerging Approaches to Selectively Target Human Glioblastoma Cancer Stem Cells. Biomed Research International. 2014. doi: Artn 12658610.1155/2014/126586.
5. Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006; 5: 835-44.
6. Tuttle RM, Haddad RI, Ball DW, Byrd D, Dickson P, Duh QY, Ehya H, Haymart M, Hoh C, Hunt JP, Iagaru A, Kandeel F, Kopp P, et al. Thyroid carcinoma, version 2.2014. J Natl Compr Canc Netw. 2014; 12: 1671-80; quiz 80.
7. Benson AB, 3rd, Abrams TA, Ben-Josef E, Bloomston PM, Botha JF, Clary BM, Covey A, Curley SA, D’Angelica MI, Davila R, Ensminger WD, Gibbs JF, Laheru D, et al. NCCN clinical practice guidelines in oncology: hepatobiliary cancers. J Natl Compr Canc Netw. 2009; 7: 350-91.
8. Motzer RJ, Agarwal N, Beard C, Bolger GB, Boston B, Carducci MA, Choueiri TK, Figlin RA, Fishman M, Hancock SL, Hudes GR, Jonasch E, Kessinger A, et al. NCCN clinical practice guidelines in oncology: kidney cancer. J Natl Compr Canc Netw. 2009; 7: 618-30.
9. Carra E, Barbieri F, Marubbi D, Pattarozzi A, Favoni RE, Florio T, Daga A. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle. 2013; 12: 491-500. doi: 10.4161/cc.23372.
10. Siegelin MD, Raskett CM, Gilbert CA, Ross AH, Altieri DC. Sorafenib exerts anti-glioma activity in vitro and in vivo. Neurosci Lett. 2010; 478: 165-70. doi: 10.1016/j.neulet.2010.05.009.
11. Yang F, Brown C, Buettner R, Hedvat M, Starr R, Scuto A, Schroeder A, Jensen M, Jove R. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol Cancer Ther. 2010; 9: 953-62. doi: 10.1158/1535-7163.MCT-09-0947.
12. Peereboom DM, Ahluwalia MS, Ye X, Supko JG, Hilderbrand SL, Phuphanich S, Nabors LB, Rosenfeld MR, Mikkelsen T, Grossman SA, New Approaches to Brain Tumor Therapy C. NABTT 0502: a phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol. 2013; 15: 490-6. doi: 10.1093/neuonc/nos322.
13. Lee EQ, Kuhn J, Lamborn KR, Abrey L, DeAngelis LM, Lieberman F, Robins HI, Chang SM, Yung WK, Drappatz J, Mehta MP, Levin VA, Aldape K, et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro Oncol. 2012; 14: 1511-8. doi: 10.1093/neuonc/nos264.
14. Hassler MR, Ackerl M, Flechl B, Sax C, Wohrer A, Widhalm G, Dieckmann K, Hainfellner J, Preusser M, Marosi C. Sorafenib for patients with pretreated recurrent or progressive high-grade glioma: a retrospective, single-institution study. Anticancer Drugs. 2014; 25: 723-8. doi: 10.1097/CAD.0000000000000077.
15. Zustovich F, Landi L, Lombardi G, Porta C, Galli L, Fontana A, Amoroso D, Galli C, Andreuccetti M, Falcone A, Zagonel V. Sorafenib plus daily low-dose temozolomide for relapsed glioblastoma: a phase II study. Anticancer Res. 2013; 33: 3487-94.
16. Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, Marcello J, Herndon JE, 2nd, McLendon RE, Janney D, Friedman AH, Bigner DD, Friedman HS. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neurooncol. 2011; 101: 57-66. doi: 10.1007/s11060-010-0217-6.
17. Dai XF, Ding J, Zhang RG, Ren JH, Ma CM, Wu G. Radiosensitivity enhancement of human hepatocellular carcinoma cell line SMMC-7721 by sorafenib through the MEK/ERK signal pathway. Int J Radiat Biol. 2013; 89: 724-31. doi: 10.3109/09553002.2013.791405.
18. Heravi M, Tomic N, Liang L, Devic S, Holmes J, Deblois F, Radzioch D, Muanza T. Sorafenib in combination with ionizing radiation has a greater anti-tumour activity in a breast cancer model. Anticancer Drugs. 2012; 23: 525-33. doi: 10.1097/CAD.0b013e32834ea5b3.
19. Laban S, Steinmeister L, Gleissner L, Grob TJ, Grenman R, Petersen C, Gal A, Knecht R, Dikomey E, Kriegs M. Sorafenib sensitizes head and neck squamous cell carcinoma cells to ionizing radiation. Radiother Oncol. 2013; 109: 286-92. doi: 10.1016/j.radonc.2013.07.003.
20. Kim YB, Jeung HC, Jeong I, Lee K, Rha SY, Chung HC, Kim GE. Mechanism of enhancement of radiation-induced cytotoxicity by sorafenib in colorectal cancer. J Radiat Res. 2013; 54: 52-60. doi: 10.1093/jrr/rrs074.
21. Kuo YC, Lin WC, Chiang IT, Chang YF, Chen CW, Su SH, Chen CL, Hwang JJ. Sorafenib sensitizes human colorectal carcinoma to radiation via suppression of NF-kappaB expression in vitro and in vivo. Biomed Pharmacother. 2012; 66: 12-20. doi: 10.1016/j.biopha.2011.09.011.
22. Den RB, Kamrava M, Sheng Z, Werner-Wasik M, Dougherty E, Marinucchi M, Lawrence YR, Hegarty S, Hyslop T, Andrews DW, Glass J, Friedman DP, Green MR, et al. A phase I study of the combination of sorafenib with temozolomide and radiation therapy for the treatment of primary and recurrent high-grade gliomas. Int J Radiat Oncol Biol Phys. 2013; 85: 321-8. doi: 10.1016/j.ijrobp.2012.04.017.
23. Sherman JH, Kirzner J, Siu A, Amos S, Hussaini IM. Sorafenib tosylate as a radiosensitizer in malignant astrocytoma. J Clin Neurosci. 2014; 21: 131-6. doi: 10.1016/j.jocn.2013.04.010.
24. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005; 352: 997-1003. doi: 10.1056/NEJMoa043331.
25. Nabors LB, Supko JG, Rosenfeld M, Chamberlain M, Phuphanich S, Batchelor T, Desideri S, Ye X, Wright J, Gujar S, Grossman SA, New Approaches to Brain Tumor Therapy CNSC. Phase I trial of sorafenib in patients with recurrent or progressive malignant glioma. Neuro Oncol. 2011; 13: 1324-30. doi: 10.1093/neuonc/nor145.
26. Struve N, Riedel M, Schulte A, Rieckmann T, Grob TJ, Gal A, Rothkamm K, Lamszus K, Petersen C, Dikomey E, Kriegs M. EGFRvIII does not affect radiosensitivity with or without gefitinib treatment in glioblastoma cells. Oncotarget. 2015; 6: 33867-77. doi: 10.18632/oncotarget.5293.
27. Bobola MS, Kolstoe DD, Blank A, Silber JR. Minimally cytotoxic doses of temozolomide produce radiosensitization in human glioblastoma cells regardless of MGMT expression. Mol Cancer Ther. 2010; 9: 1208-18. doi: 10.1158/1535-7163.MCT-10-0010.
28. Hamed HA, Tavallai S, Grant S, Poklepovic A, Dent P. Sorafenib/regorafenib and lapatinib interact to kill CNS tumor cells. J Cell Physiol. 2015; 230: 131-9. doi: 10.1002/jcp.24689.
29. Aldea MD, Petrushev B, Soritau O, Tomuleasa CI, Berindan-Neagoe I, Filip AG, Chereches G, Cenariu M, Craciun L, Tatomir C, Florian IS, Crivii CB, Kacso G. Metformin plus sorafenib highly impacts temozolomide resistant glioblastoma stem-like cells. J BUON. 2014; 19: 502-11.
30. Jakubowicz-Gil J, Langner E, Badziul D, Wertel I, Rzeski W. Quercetin and sorafenib as a novel and effective couple in programmed cell death induction in human gliomas. Neurotox Res. 2014; 26: 64-77. doi: 10.1007/s12640-013-9452-x.
31. Heim M, Scharifi M, Zisowsky J, Jaehde U, Voliotis D, Seeber S, Strumberg D. The Raf kinase inhibitor BAY 43-9006 reduces cellular uptake of platinum compounds and cytotoxicity in human colorectal carcinoma cell lines. Anticancer Drugs. 2005; 16: 129-36.
32. Bambury RM, Morris PG. Novel investigational approaches for inhibiting angiogenesis in recurrent glioblastoma. Anticancer Drugs. 2014; 25: 611-3. doi: 10.1097/CAD.0000000000000104.
33. Hainsworth JD, Ervin T, Friedman E, Priego V, Murphy PB, Clark BL, Lamar RE. Concurrent radiotherapy and temozolomide followed by temozolomide and sorafenib in the first-line treatment of patients with glioblastoma multiforme. Cancer. 2010; 116: 3663-9. doi: 10.1002/cncr.25275.
34. Hottinger AF, Aissa AB, Espeli V, Squiban D, Dunkel N, Vargas MI, Hundsberger T, Mach N, Schaller K, Weber DC, Bodmer A, Dietrich PY. Phase I study of sorafenib combined with radiation therapy and temozolomide as first-line treatment of high-grade glioma. Br J Cancer. 2014; 110: 2655-61. doi: 10.1038/bjc.2014.209.