
INTRODUCTION
A major challenge in chemotherapy is to change the biodistribution of drugs to reduce free drug toxicity and favor tumor accumulation. Nanomedicines have been designed to overcome this challenge, based on the following advantages: (a) improve the solubility of hydrophobic compounds, (b) protect a molecule from undesirable interactions with biological milieu components and improve its stability, (c) provide controlled release of the contents, and (d) favorably alter the pharmacokinetics and biodistribution [1–3]. One of the nanomedicines that are used clinically in the treatment of cancer is nanoparticle albumin-bound paclitaxel (brand name: Abraxane), which showed less toxic and superior efficacy compared with polyethylated castor oil-based paclitaxel in the treatment of breast cancer [4]. Although high expectations have been placed on that nanomedicines would change the current status quo of cancer treatment, the increase in overall survival of cancer patients is only modest in many cases [5].
Transport of a therapeutic agent from the systemic circulation to cancer cells is a three-step process. First, nanoparticles flow to different regions of tumors via blood vessels. They must then cross the vessel wall, and finally, penetrate through the interstitial space to reach the target cells [5]. The concerted effects of the lack of functional lymphatic vessels and the vascular hyperpermeability result in the elevated interstitial fluid pressure (IFP) [6]. Although nanomedicines could be preferentially delivered to tumors owing to the enhanced permeability and retention effect, the convective transvascular and interstitial transport for them is severely hampered due to the elevated IFP [3, 7], which leads to a poor penetration into and distribution across the interstitium [7]. Therefore, lowering IFP in solid tumors may be a great strategy to improve the uptake and distribution of nanomedicines in tumors.
Many efforts have been made to demonstrate the feasibility of this strategy. For instance, collagenase-induced reductions in IFP can restore convective mass transport, thereby improving the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts [8]. Similarly, hyaluronidase-induced reductions in IFP can enhance transvascular transport of nanoprobes, thereby improving the uptake and distribution of liposomal doxorubicin in the same model [9]. Other agents that have been used to lower tumor IFP include bevacizumab, tumor necrosis factor-alpha, dexamethasone, prostaglandin E1-methyl ester, labradimil, docetaxel, nicotinamide, etc. [3, 10] However, all of these agents have some limitations, including toxicity to normal tissue or lack of tumor selectivity.
Recently, vascular-disrupting strategies have been explored to lower tumor IFP. Vascular disrupting agents (VDAs) are a relatively new class of drugs that target the established tumor vasculature and cause a rapid and selective vascular shutdown and subsequent necrosis in the tumor core [11]. VDAs preferentially target the intratumoral vascular bed due to its fragile phenotype compared to normal tissue blood vessels [12]. One of the most promising VDAs is combretastatin-A4 phosphate (CA4P), which can induce 90–99% of tumor necrosis at well tolerated doses and is currently in phase 3 clinical trials [13]. It has been demonstrated that CA4P could significantly decrease IFP in both C3H mammary carcinomas and KHT sarcomas [14, 15].
Based on the above, we postulated that CA4P might improve the uptake and distribution of nab-paclitaxel in tumor by reducing IFP, thereby enhancing the antitumor efficacy of nab-paclitaxel. Meanwhile, the combination of CA4P and nab-paclitaxel may provide a synergistic and complementary antitumor effect, in which CA4P could induce the destruction of large areas of the interior of tumors while nab-paclitaxel could effectively kill residual tumor cells in the outer layer.
To test this hypothesis, IFP was measured by the wick-in-needle technique after i.v. injection of CA4P in rats bearing W256 breast carcinoma. The whole-body biodistribution and tumor distribution of 131I-nab-paclitaxel injected i.v. at 1 h after CA4P injection was studied by gamma counting and autoradiography, respectively. The intratumoral uptake of paclitaxel (PTX) was determined using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). Finally, the anticancer efficacy of nab-paclitaxel combined with CA4P was evaluated in the same model by magnetic resonance imaging (MRI).
RESULTS
Radiolabeling and in vitro stability
In order to facilitate the biodistribution study, nab-paclitaxel was radiolabeled with iodine 131. The thin layer chromatography (TLC) determination showed that the radiochemical purity (RCP) of 131I-nab-paclitaxel was greater than 95% after purification, and in vitro stabilities of 131I-nab-paclitaxel incubated in rat serum at 37°C were excellent with RCP over 92% up to 24 h.
CA4P reduced tumor IFP
Figure 1 shows the temporal changes in IFP following administration of CA4P with the values normalized to the pretreatment value for each tumor. Tumors treated with 30 mg/kg of CA4P showed IFP levels continued to decline gradually over time, reaching approximately 31% of the initial pretreatment value by 60 min post injection. Tumors treated with PBS did not induce any significant reduction in IFP.
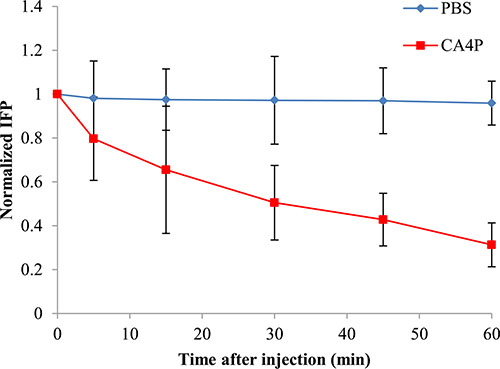
Figure 1: IFP response as a function of time after i.v. injection of PBS or CA4P (30 mg/kg). IFP values were normalized to the pressure values before PBS or CA4P injection. Each value is the mean of n tumors, the error bars represent SD.
Whole-body biodistribution and intratumoral distribution of 131I-nab-paclitaxel
The whole-body biodistribution of 131I-nab-paclitaxel at 24 h post injection is shown in Figure 2. 131I-nab-paclitaxel exhibited significantly higher tumor uptake in CA4P + 131I-nab-paclitaxel group compared with131I-nab-paclitaxel group (0.58 ± 0.07% ID/g vs 0.28 ± 0.06% ID/g, p < 0.01). In all normal organs or tissues, the uptake of 131I-nab-paclitaxel had no significant difference between the 131I-nab-paclitaxel group and CA4P + 131I-nab-paclitaxel group (p > 0.05). This shows that CA4P can significantly improve tumor uptake of 131I-nab-paclitaxel without affecting the biodistribution of 131I-nab-paclitaxel in normal organs or tissues.
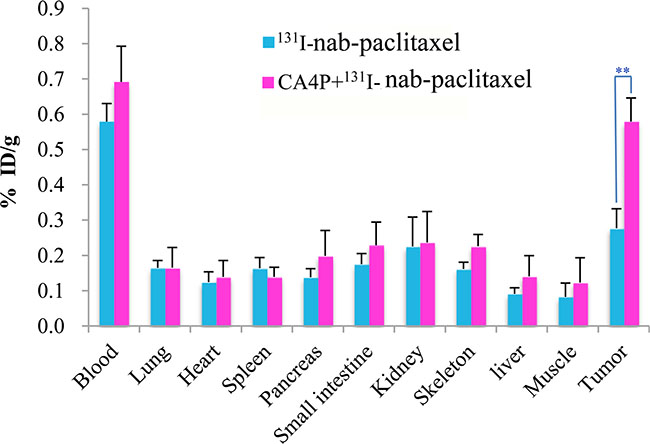
Figure 2: Biodistribution of 131I-nab-paclitaxel inW256 tumor-bearing rats at 24 h post injection in 131I-nab-paclitaxel and CA4P + 131I-nab-paclitaxel group. Data are expressed as percentage injected dose per gram of tissue (%ID/g). **P < 0.01.
Representative autoradiographs of tumor slices from 131I-nab-paclitaxel group and CA4P + 131I-nab-paclitaxel group are displayed in Figure 3A. Tumor uptake was significantly higher in CA4P + 131I-nab-paclitaxel group than that in 131I-nab-paclitaxel group, which is consistent with the results of the gamma counting. The intratumoral distribution of 131I-nab-paclitaxel in 131I-nab-paclitaxel group was primarily localized in the periphery of the tumor, while in CA4P + 131I-nab-paclitaxel group, 131I-nab-paclitaxel penetrated further into the tumor center and its intratumoral distribution is relatively more uniform. This shows that CA4P can not only improve tumor uptake of 131I-nab-paclitaxel, but also increase its penetration into tumor interior.
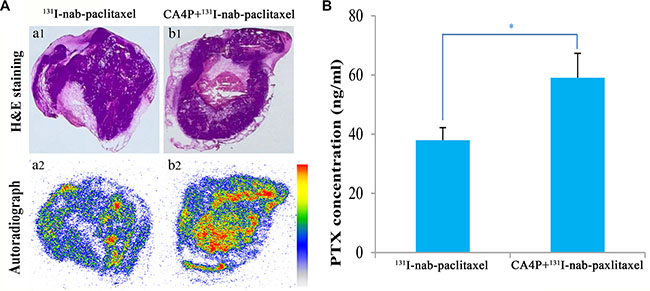
Figure 3: Representative autoradiographs (a2, b2) and corresponding H&E images (a1, b1) of 30 μm tumor slices from W256 tumor-bearing rats (A) and the intratumoral distribution of paclitaxel (B) at 24 h post i.v. of 131I-nab-paclitaxel (14.8 MBq/kg of 131I-nab-paclitaxel, 6 mg/kg of nab-paclitaxel) in 131I-nab-paclitaxel and CA4P + 131I-nab-paclitaxel group. Data represents the mean ± SD. *P < 0.05.
Quantitation of intratumoral PTX
The quantitation of intratumoral PTX at 24 h post injection is presented in Figure 3B. CA4P + 131I-nab-paclitaxel group showed higher PTX concentration in the tumor compared with 131I-nab-paclitaxel group (59.08 ± 8.26 ng/ml vs 37.98 ± 4.23 ng/ml, p < 0.05), which indicated that CA4P improved tumor uptake of PTX.
In vivo MRI
Tumors at baseline appeared slightly hypointense or isointense on T1W images and hyperintense on T2W images. Contrast enhancement was observed on CE-T1Wimages, suggesting hypervascularity of the subcutaneous W256 mass. A nonenhanced central region surrounded by a thin rim enhancement presented on CE-T1W images in CA4P + nab-paclitaxel group after sequential i.v. administrations of CA4P and nab-paclitaxel, suggesting the presence of massive necrosis and minimum tumor residue (Figure 4D2–4D4), which differed from the tumors with spontaneous necrosis in PBS group (Figure 4A2–4A4). This was speculatively attributed to the synergistic effect of CA4P and nab-paclitaxel. The tumor growth in CA4P + nab-paclitaxel group was well inhibited and maintained a stable in size until the endpoint. By contrast, the tumors in the other three groups grew rapidly with the tumor size progressively becoming larger. Irregularly enhanced thick rims suggestive of viable tumor tissue were shown in nab-paclitaxel group and CA4P group at later time points. Necrotic and viable tumor could be determined by histopathological findings (Figure 4A5–4D5, 4A6–4D6). Necrosis occupied a large-area of the tumor mass at the endpoint in all groups except for the PBS group.
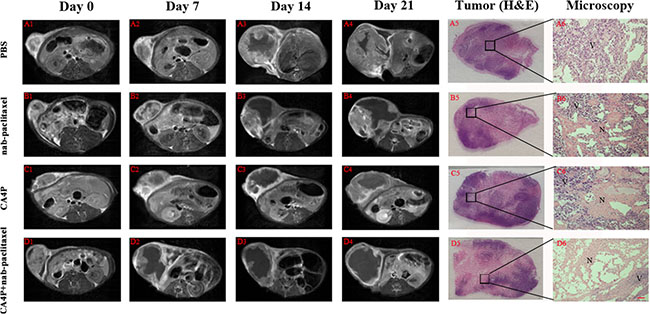
Figure 4: Contrast enhanced T1 (CE-T1) MR images of representative tumor bearing rats from 4 groups at day 0 (A1-D1), 7 (A2-D2), 14 (A3-D3), 21 (A4-D4). Tumors in PBS, nab-paclitaxel and CA4P group grew faster than that of CA4P + nab-paclitaxel group. Macroscopic photographs (A5–D5) showed extensive central necrosis surrounded by viable tumor tissues in the three therapy groups. Microscopic photographs (A6–D6) showed the interface between necrotic (N) and viable (V) tumor tissues in the three therapy groups. Scale bar = 50 μm.
Tumor volume and necrosis ratio
Tumor volumes on day 0, 7, 14, 21 were shown in Figure 5. The tumor volumes at baseline were approximate in the PBS, nab-paclitaxel, CA4P and CA4P + nab-paclitaxel group, respectively (P > 0.05). There was no significant difference (p > 0.05) among the groups in tumor volume on day 7 after different treatments. However, on day 14, mean tumor volumes in PBS, nab-paclitaxel and CA4P group were 4.0, 3.0 and 2.3 times that of CA4P + nab-paclitaxel group, respectively. Sequential administration of CA4P and nab-paclitaxel showed significantly stronger tumor growth inhibition effect than that of administration of nab-paclitaxel alone on day 14 (1.88 ± 0.31 cm3 vs 5.71 ± 0.75 cm3, p < 0.01) and 21 (3.34 ± 0.56 cm3 vs 8.85 ± 0.94 cm3p < 0.01).
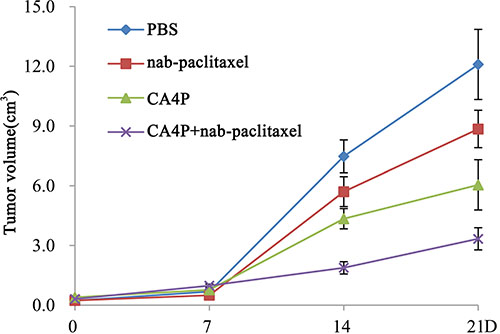
Figure 5: Tumor growth curve on day 0, 7, 14 and 21 post-therapy. Significant difference of tumor volume in CA4P + nab-paclitaxel group was found compared with that of the PBS, CA4P and nab-paclitaxel group (P < 0.01) from day 14 on.
Tumor necrosis ratios on day 0, 7, 14, 21 were shown in Figure 6. Tumor necrosis measured from CE-T1W images at baseline was about 12% in each group because of the existence of spontaneous necrosis. After different treatments, a significantly higher necrosis ratio was observed in CA4P + nab-paclitaxel group compared with the PBS, CA4P and nab-paclitaxel group, respectively (P < 0.05) from day 7 till day 21. On day 21, the necrosis ratio was 16.24 ± 2.10%, 43.36 ± 3.79%, 36.52 ± 3.65% and 72.83 ± 4.64% for the PBS, nab-paclitaxel, CA4P and CA4P + nab-paclitaxel group, respectively, which further demonstrated the advantage of the coadministration of CA4P and nab-paclitaxel.
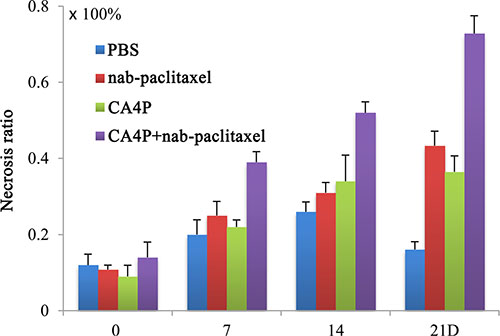
Figure 6: Tumor necrosis ratios on day 0, 7, 14 and 21 post-therapy. A significantly higher necrosis ratio was observed in CA4P + nab-paclitaxel group compared with the PBS, CA4P and nab-paclitaxel group (P < 0.05) from day 7 on.
DISCUSSION
PTX is one of the most effective drugs in the management of breast cancer [16, 17]. Although nanoparticle delivery of PTX improves the tumor uptake compared to free PTX [18, 19], the tumor response to the improved formulation of paclitaxel is typically only 30% to 35% [18–20]. The present work demonstrated that CA4P could improve the uptake and distribution of nab-paclitaxel in W256 breast carcinoma tumor, thereby enhancing the antitumor efficacy of nab-paclitaxel.
Our present study demonstrated that CA4P could significantly reduce IFP in W256 tumor in a nonlinear time-dependent manner within 60 min. This is consistent with previous study by Ley et al. [15], who found that IFP values declined continuously within 90 min following CA4P treatment in C3H mammary carcinoma tumors, however, no further IFP reductions were observed at later time points. Three hours after CA4P treatment, IFP levels of approximately 70% of controls was observed, which indicates that IFP does not recover within that period [14]. Significant reduction in IFP after CA4P treatment may be attributed to the decrease in microvascular pressure (MVP) which is the principal driving force of IFP [21]. CA4P induced intravascular clotting, vessel obstruction, or both could cause a rapid decrease in perfusion [15, 22]. Interruption of blood supply would decrease MVP inside large parts of the tumor vasculature, removing the source of fluid for filtration across the vascular membrane.
Although nab-paclitaxel could be preferentially delivered to tumors, the distribution of nab-paclitaxel in untreated tumor was heterogeneous and mainly located in the periphery of the tumor. This may correlate spatially with the peripheral IFP gradient as measured in subcutaneous OHS xenografts, where the high central IFP decreased rapidly in the periphery of the tumor [8]. In addition to high IFP, heterogeneous vascularization and perfusion together with slow interstitial diffusion may also contribute to the poor distribution [5].
CA4P could increase the tumor uptake and improve intratumor distribution of nab-paclitaxel. The present study indicated that not only was the uptake of nab-paclitaxel in tumor significantly higher, but also the distribution of nab-paclitaxel within tumor was more homogeneous in CA4P treated tumors compared with untreated tumors. This may be mainly attributed to the role of CA4P in reducing IFP, despite that an increase in tumor vascular permeability induced by CA4P [23, 24] would also contribute to the extravasation of macromolecule drugs from blood flow into tumor. As previous studies had shown that reducing IFP could improve the uptake and distribution of monoclonal antibody and liposomal doxorubicin in human osteosarcoma xenografts [8, 9].
There is an extensive clinical development program to evaluate the efficacy and safety of nab-paclitaxel in combination with several agents routinely used in breast cancer to further enhance antitumor efficacy [19]. Our study, for the first time, demonstrated the effectiveness of combination therapy with nab-paclitaxel and CA4P, a vascular disrupting agent in an experimental model of W256 breast cancer. The significant increase in the efficacy of tumor-bearing rats in the nab-paclitaxel/CA4P combined group compared with rats treated with single drugs shows great potential for implementing such strategy in clinics. The superior efficacy of nab-paclitaxel/CA4P combination therapy might be explained by the following mechanisms. First, vascular disrupting agent therapy can reduce IFP leading to increased nab-paclitaxel delivery and improved distribution within tumor, which means that a better antitumor effect can be obtained even excluding the extensive cell kill following the vascular shutdown achieved with CA4P. Second, CA4P combined with nab-paclitaxel may exert a synergistic effect by targeting two distinct cell populations. CA4P can eliminate cells in the central part of the tumor where the delivery of administered nab-paclitaxel is limited, whereas nab-paclitaxel is more likely to kill the actively proliferating cells in the peripheral viable rim that survive CA4P treatment.
There are still some limitations in the present study. Firstly, the effect of CA4P on MVP was not studied because the corresponding experimental instrument is currently not available for us. The transport of nanomedicines across vessel walls and in the tumor interstitium occurs mainly by convection [25, 26], which depends mainly on the pressure gradients between MVP and interstitial pressure [21]. Whether the increased tumor uptake and improved intratumor distribution of nab-paclitaxel is caused by a transcapillary pressure gradient induced by CA4P is still unknown for us. Secondly, we only investigated the effect of a dose of CA4P (30 mg/kg) on tumor IFP. The relationship between the effect of CA4P in reducing tumor IFP and its dosage has not been studied in detail. Related issues will be further explored in the follow-up study.
MATERIALS AND METHODS
Animals and tumor model
Adult male Sprague-Dawley rats (250–300 g) were provided by the Experimental Animal Center, Jiangsu Province Academy of Traditional Chinese Medicine (Nanjing, Jiangsu, China). The rat models of breast cancer were prepared by subcutaneously inoculating W256 tumor cells (5 × 106) at a volume of 0.2 ml into the right flank region of rats. The longest (L) and shortest (W) tumor diameters (mm) were measured by digital caliper and used to calculate tumor volume (V) according to the formula: V = (L × W2)/2. Experiments were performed when tumors had reached a size of approximately 0.25 ~ 0.35 cm3. All the animal experiments were approved by the institutional animal care and use committee.
Drug preparation and administration
CA4P (HuaMei technology Co., Ltd, Wuhan, China) was diluted in phosphate buffered saline (PBS) solution at a concentration of 18.75 mg/ml and injected i.v. at a dose of 30 mg/kg. Nab-paclitaxel (Celgene Corporation, Summit) was diluted in PBS solution at a concentration of 5 mg/ml and injected i.v. at a dose of 6 mg/kg.
The Iodogen coating method was used to radioiodinate nab-paclitaxel to form 131I-nab-paclitaxel. Nab-paclitaxel was dissolved in PBS to form 1 mg/ml solution. Radioiodination was initiated by adding nab-paclitaxel and Na131I solution (volume ratio, 5: 1) into Iodogen (1, 3, 4, 6-tetrachloro-3a, 6a-diphenylglycouril; Pierce Biotechnology, ZI Camp Jouven, France)-coated tube. Afterwards, the mixture was vortexed and incubated at 30°C for 3 ~ 5 min. The radiochemical yield was determined by TLC using 10% trichloroacetate (TCA) as mobile phase. Separation of 131I-nab-paclitaxel from unbound radioactive iodine was performed on Sephadex G-50, eluted with 0.05 M phosphate buffer (pH 7.5) and collected in 0.5 ml fractions. The agent was intravenously injected at a dose of 14.8 MBq/kg.
In vitro stability
131I-nab-paclitaxel mixed with rat plasma (volume ratio, 1: 9) was incubated at 37°C. And then RCP of 131I-nab-paclitaxel was determined by TLC at 0.5, 1, 2, 6, 12 and 24 h, respectively.
IFP measurements
The IFP was measured in subcutaneous tumors using the wick-in-needle technique [27]. In brief, a 23-gauge sensing needle was inserted into the center part of the tumor and connected to a pressure transducer (Changzhou Qianhong Bio-pharma Co., Ltd, Jiangsu, China) via polyethylene tubing filled with sterile heparinized PBS(70 units/ml). Pressures were monitored online using a PowerLab analogue-to-digital recording system (PowerLab/8S, AD Instruments, Hastings, UK). Fluid communication between the needle and the tumor tissue was tested by compressing and decompressing the tubing, and it was accepted when the IFP did not vary by more than 20%. After reaching a stable IFP value, a single dose of CA4P were administered into the tail vein. Control animals received an equal volume of PBS. Subsequent IFP measurements were made at 0, 5, 15, 30, 45, and 60 min post injection in the same tumors.
Whole-body biodistribution and intratumoral distribution of 131I-nab-paclitaxel
Twelve W256 tumor-bearing rats were divided into two groups. CA4P+ 131I-nab-paclitaxel group received i.v. administration of 131I-nab-paclitaxel at 1 h post injection of CA4P, while 131I-nab-paclitaxel group received i.v. administration of 131I-nab-paclitaxel and an equal volume of PBS. At 24 h post injection of 131I-nab-paclitaxel, all rats were euthanized. Blood, lung, heart, spleen, pancreas, small intestine, kidney, skeleton, liver, muscle and tumor were sampled, weighted and then radioactivity was measured with an automatic γ-counter (WIZARD; 2470, Perkin Elmer, New York, USA). The results were expressed as percentage of the injected dose per gram of tissues (% ID/g).
The above tumor tissues were frozen in the cryotome (Shandon FSE, Thermo Fisher Scientific Co., USA) and cut into 30 μm sections along the long axis of the center. Autoradiograms of these slides were obtained by 12 h exposure using a high performance storage phosphor screen (Super resolution screen, Canberra-Packare, Ontario, Canada) and then the screen was read using a Phosphor Imager scanner (Cyclone™, Canberra-Packard) with Optiquant™ software. Subsequently, the same slices were stained with hematoxylin-eosin (H&E) and digitally photographed.
Quantitation of intratumoral PTX by LC-MS/MS [28–30]
An LC-MS/MS assay was employed to determine the concentrations of PTX in the above tumor samples. After the counting, 20 mg of each tumor mass was collected. Ten times volume of methanol was added and homogenized. Then, 60 μl tissue homogenate was mixed with 20 μl of the internal standard (IS) docetaxel solution (50 μg/ml). Next, 3 ml of methyl tert-butyl ether (MTBE) was added to the mixture and vortexed for 5 min. After centrifugation at 3500 g for 5 min, the supernatant was collected and evaporated to dryness under a stream of nitrogen in a water bath at 35°C. The samples were diluted and injected onto the LC-MS/MS system (LC: waters ACQUITY and MS: waters synapt).
Combinational therapy protocols
Twenty-four W256 tumor-bearing rats were randomized into four groups of six animals. The animals in each group received sequential intravenous injections at a 1 h interval at day 0, 6, 13 and 20. CA4P + nab-paclitaxel group received i.v. injection of CA4P and nab-paclitaxel; CA4P group received i.v. injection of CA4P and PBS; nab-paclitaxel group received i.v. injection of PBS and nab-paclitaxel; PBS group received twice i.v. injection of PBS. MRI was performed before administration and every 7 days after administration to monitor and quantify tumor volume and necrosis. At day 21 after MRI scanning, animals in all groups were sacrificed. Then all tumors were excised and cut into 5 μm frozen sections for postmortem histopathology verification.
Magnetic resonance imaging
MRI was performed using a clinical 1.5T MR magnet (Echo speed; GE Co., NY) with a rat coil for rat studies. Under isoflurane gas anesthesia, T1-weighted (T1W) and T2-weighted (T2W) spin-echo multi-slice coronal images were acquired. Then contrast enhanced T1-weighted (CE-T1W) images were obtained immediately after i.v. administration of Gd-DTPA (Bayer Schering Pharma AG, Berlin, Germany) at 0.2 mmol/kg. The related parameters are described below: Field of view (FOV) = 100 mm × 100 mm; T1W: Sequence SE, TR/TE = 550 ms/24 ms; T2W: Sequence FSE, TR/TE = 2920 ms/88 ms; CE-T1W: Sequence SE, TR/TE = 550 ms/60 ms.
Quantifications of tumor area were done by manually delineating the outline of the tumor mass on each T2W MRI slice covering the whole tumor. Tumor volume was calculated using the equation: tumor volume = Σ [tumor area on each slice × (slice thickness)]. The area of central nonenhancing region was delineated from CE-T1W images to estimate necrosis. The ratios of necrosis were defined as the volume of necrosis over that of entire tumor, i.e. necrosis ratio = Σ (area of necrosis × slice thickness)/(area of whole tumor × slice thickness) × 100%.
Statistical analysis
Numerical data were expressed as the mean ± standard deviation. Statistical analysis was carried out with SPSS for Windows software package (version 17.0; SPSS, Chicago, IL, USA). Two-tailed independent samples t-test was used to compare the biodistribution of 131I-nab-paclitaxel and the uptake of intratumoral PTX between 131I-nab-paclitaxel and CA4P + 131I-nab-paclitaxel group. For other comparisons, a one-way ANOVA was used to test differences among groups. P value less than 0.05 was considered to be significant difference.
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interests.
FUNDING
This work was supported by the National Natural Science Foundation of China (No. 81473120), the Ninth Batch of “Six Talent Peaks” Project of Jiangsu Province (No.2012-YY-008), and the Project of Medical and Health Technology Development Program in Shandong Province (No.2013WS0354).
REFERENCES
1. Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release. 2015; 200:138–157.
2. Rizzo LY, Theek B, Storm G, Kiessling F, Lammers T. Recent progress in nanomedicine: therapeutic, diagnostic and theranostic applications. Curr Opin Biotech. 2013; 24:1159–1166.
3. Li Y, Wang J, Wientjes MG, Au JLS. Delivery of nanomedicines to extracellular and intracellular compartments of a solid tumor. Adv Drug Deliver Rev. 2012; 64:29–39.
4. Petrelli F, Borgonovo K, Barni S. Targeted delivery for breast cancer therapy: the history of nanoparticle-albumin-bound paclitaxel. Expert Opin Pharmaco. 2010; 11:1413–1432.
5. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin oncol. 2010; 7:653–664.
6. Chauhan VP, Stylianopoulos T, Boucher Y, Jain RK. Delivery of molecular and nanoscale medicine to tumors: transport barriers and strategies. Annu Rev Chem Biomol. 2011; 2:281–298.
7. Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012; 161:175–187.
8. Eikenes L, Bruland ØS, Brekken C, de Lange Davies C. Collagenase increases the transcapillary pressure gradient and improves the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts. Cancer Res. 2004; 64:4768–4773.
9. Eikenes L, Tari M, Tufto I, Bruland ØS, de Lange Davies C. Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx™) in human osteosarcoma xenografts. Brit J Cancer. 2005; 93:81–88.
10. Marcucci F, Corti A. How to improve exposure of tumor cells to drugs—Promoter drugs increase tumor uptake and penetration of effector drugs. Adv Drug Deliver Rev. 2012; 64:53–68.
11. Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. 2005; 5:423–435.
12. Taylor M, Billiot F, Marty V, Rouffiac V, Cohen P, Tournay E, Opolon P, Louache F, Vassal G, Laplace-Builhé C. Reversing resistance to vascular-disrupting agents by blocking late mobilization of circulating endothelial progenitor cells. Cancer discov. 2012; 2:434–449.
13. Siemann DW, Chaplin DJ, Walicke PA. A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P). Expert Opin Inv Drug. 2009; 18:189–197.
14. Nielsen T, Murata R, Maxwell RJ, Stødkilde-Jørgensen H, Østergaard L, Ley CD, Kristjansen PE, Horsman MR. Non-invasive imaging of combretastatin activity in two tumor models: Association with invasive estimates. Acta Oncol. 2010; 49:906–913.
15. Ley CD, Horsman MR, Kristjansen PE. Early effects of combretastatin-A4 disodium phosphate on tumor perfusion and interstitial fluid pressure. Neoplasia. 2007; 9:108–112.
16. Piccart-Gebhart MJ, Burzykowski T, Buyse M, Sledge G, Carmichael J, Lück H-J, Mackey JR, Nabholtz JM, Paridaens R, Biganzoli L. Taxanes alone or in combination with anthracyclines as first-line therapy of patients with metastatic breast cancer. J ClinOncol. 2008; 26:1980–1986.
17. De Laurentiis M, Cancello G, D’Agostino D, Giuliano M, Giordano A, Montagna E, Lauria R, Forestieri V, Esposito A, Silvestro L. Taxane-based combinations as adjuvant chemotherapy of early breast cancer: a meta-analysis of randomized trials. J ClinOncol. 2008; 26:44–53.
18. Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, Tao C, De T, Beals B, Dykes D. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res. 2006; 12:1317–1324.
19. Guarneri V, Dieci MV, Conte P. Enhancing intracellular taxane delivery: current role and perspectives of nanoparticle albumin-bound paclitaxel in the treatment of advanced breast cancer. Expert Opin Pharmaco. 2012; 13:395–406.
20. Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil–based paclitaxel in women with breast cancer. J Clin Oncol. 2005; 23:7794–7803.
21. Boucher Y, Jain RK. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: implications for vascular collapse. Cancer Res. 1992; 52:5110–5114.
22. Kragh M, Quistorff B, Horsman MR, Kristjansen PE. Acute effects of vascular modifying agents in solid tumors assessed by noninvasive laser Doppler flowmetry and near infrared spectroscopy. Neoplasia. 2002; 4:263–267.
23. Tozer GM, Prise VE, Wilson J, Locke RJ, Vojnovic B, Stratford MR, Dennis MF, Chaplin DJ. Combretastatin A-4 phosphate as a tumor vascular-targeting agent early effects in tumors and normal tissues. Cancer Res. 1999; 59:1626–1634.
24. Tozer GM, Prise VE, Wilson J, Cemazar M, Shan S, Dewhirst MW, Barber PR, Vojnovic B, Chaplin DJ. Mechanisms associated with tumor vascular shut-down induced by combretastatin A-4 phosphate: intravital microscopy and measurement of vascular permeability. Cancer Res. 2001; 61:6413–6422.
25. Kristensen C, Nozue M, Boucher Y, Jain R. Reduction of interstitial fluid pressure after TNF-alpha treatment of three human melanoma xenografts. Brit J cancer. 1996; 74:533.
26. Swabb EA, Wei J, Gullino PM. Diffusion and convection in normal and neoplastic tissues. Cancer Res. 1974; 34:2814–2822.
27. Fadnes H, Reed R, Aukland K. Interstitial fluid pressure in rats measured with a modified wick technique. Microvasc Res. 1977; 14:27–36.
28. Tong X, Zhou J, Tan Y. Liquid chromatography/tandem triple-quadrupole mass spectrometry for determination of paclitaxel in rat tissues. Rapid Commun Mass Sp. 2006; 20:1905–1912.
29. Tong X, Zhou J, Tan Y. Determination of Paclitaxel in Rat Plasma by LC—MS—MS. J Chromatogr Sci. 2006; 44:266–271.
30. Parise RA, Ramanathan RK, Zamboni WC, Egorin MJ. Sensitive liquid chromatography–mass spectrometry assay for quantitation of docetaxel and paclitaxel in human plasma. J Chromatogr B. 2003; 783:231–236.