
INTRODUCTION
Head and neck squamous cell carcinomas (HNSCC) originate from various anatomic sites in the upper aerodigestive epithelium, i.e. the oral and (para) nasal cavities, pharynx and larynx. HNSCC is the sixth most common cancer worldwide with an estimated incidence of 4.8% of all malignancies in the entire body [1]. Etiologically, these tumors roughly fall into two main distinct groups: tumors induced either by tobacco smoking or chewing (e.g. betelnut) and alcohol abuse, or by viral infection with Human Papilloma Virus (HPV) or Epstein Barr Virus (EBV). Alcohol and/or tobacco induced HNSCCs are strongly associated with somatic mutations in tumor suppressor genes (TSG) such as TP53, CDNK2A, PTEN, and oncogenes (OG) such as HRAS and PIK3CA [2–5]. TP53 inactivating HPV oncoproteins E6 and E7 are the main cancer initiators in an increasing number of oropharyngeal squamous cell carcinoma (OPSCC) cases [6]. The overall survival of advanced cases still remains poor. This is especially true for HPV-negative tumors as compared to HPV-positive tumors [7, 8]. Furthermore, the mutational profile of HNSCC appears to significantly affect its disease course and prognosis [9–12]. Although disease outcome of HNSCC depends on multiple levels of disease processes (e.g. pathogenesis, molecular characteristics, and TNM-stage), estimation of its prognosis is still largely based on the tumor stage at clinical presentation and relapse after initial treatment. Furthermore, possibility of successful salvage treatment is largely dependent on early detection and the extent of the locoregional disease [13–16]. This underlines the need to explore new possibilities for improving diagnostics on a molecular level. The use of diagnostic biomarkers could enable detection of tumor specific mutations in order to monitor tumor response after treatment with curative intent. Ultimately, this might improve treatment outcome of HNSCC patients, while avoiding unnecessary (over) treatment and its associated morbidity and accompanying hindrance to the patient.
Technological advances over recent decades have improved the understanding of tumor genetics. Consequently, targeted profiling of tumor genetics is gradually shifting from an experimental setting towards its use in routine clinical practice in fields such as breast and lung oncology [17]. Although no common ground exists yet for the use of biomarkers in clinical decision making for HNSCC patients, evidence for future use is arising [18–20]. As previous studies have shown, TP53 is highly prone to loss of heterozygosity. This leads to the presence of inactivating non-hotspot mutations of TP53 that occur early in HNSCC carcinogenesis [21–24]. Subsequently, subclonal cells from the primary tumor either proliferate towards metastases or locally reside after treatment and develop into recurrences. These clonal expansions are likely to contain the early onset mutations found initially in the primary tumor [25–27]. By using dedicated and clinically accessible gene panels based on NGS, these mutations can reliably be detected and selected as targets. Circulating tumor DNA (ctDNA) released by clonal expansion cells contain these targets and could be quantified using minimally invasive blood samples, as there appears to be a relation between ctDNA plasma concentrations and tumor burden [28–31].
However, the significance of ctDNA in correlation with actual tumor burden and/or tumor growth still needs to be proven for HNSCC patients. This requires research on the identification of early driver gene mutations of HNSCC tumors. Therefore, we retrospectively analyzed a large dataset of sequenced HNSCCs, to map their mutational profile and to explore TP53 and possible other genes as potential diagnostic biomarkers in HNSCC.
RESULTS
Patient and tumor characteristics
A total of 110 patients accounted for 239 tumor samples that remained for analysis. Eighty (73%) patients were male. Of all patients, 76 (69%) had a history of smoking tobacco and 67 (61%) had a history of alcohol consumption. Eleven (10%) patients never used tobacco or alcohol. For 18 patients, either or both tobacco smoking and alcohol use was unknown. Table 1 summarizes patient and tumor characteristics of our study group.
Table 1: Patient and tumor characteristics
Patients |
110 |
Tumor samples |
239 |
Mean age, years (range) |
66 (45-90) |
Sex |
n (%) |
Male |
80 (73) |
Smoking history |
n (%) |
Yes |
76 (69) |
Alcohol use |
n (%) |
Former/active |
67 (61) |
Clinical stage* |
n (%) |
T1-2 |
100 (70) |
Primary tumor sites |
n (%) |
Oral cavity |
53 (36) |
Tumor subtype |
n (%) |
Primary |
148 (62) |
HPV status** |
n (%) |
Positive |
4 (12) |
* Included all primary and secondary primary tumors
** All positive tumors were OPSCCs
Of all 239 tumor samples, 148 (62%) were primary site squamous cell carcinomas. Of the primary tumor samples, 53 (36%) originated from the oral cavity, 37 (25%) from the oropharynx, 16 (11%) the hypopharynx, and 28 (19%) from the larynx. Fourteen (9%) primary tumors originated from miscellaneous sites (i.e. nasopharynx and upper esophagus and trachea). Of the 37 OPSCCs, 33 were tested for HPV-status. Only four (12%) samples proved HPV-positive. The remaining 91 out of 239 tumor samples comprised of 29 (12%) recurrences and 62 (26%) metastases. The latter could be subdivided into 38 (62%) nodal metastases and 23 (38%) distant metastases in the lung, liver, bones, or skin.
Mutational analysis
Sequencing was based on Cancer Hotspot Panel v2 (CHPv2) for 160 tumor samples, OncoAmp Panel v2 (OAPv2) was used for 40 samples, and Cancer Hotspot Panel v2+ (CHPv2+) was used for sequencing of 11 samples (Table 2). Additional Sanger sequencing was performed in 28 cases, in which NGS failed due to insufficient DNA quantity. NGS of the exons that are included in the three different gene panels (as described in our method section) yielded mutations in 26 different genes. No mutations were detected in ABL1, MYD88, NOTCH1, AKT1, ARAF, GNAS, GNA11, NRAS, PDGFRA, CALR, CDH1, IDH1, PTPN11, RET, SMO, SRC, STK11, VHL, MLH1, MPL, JAK3, JAK2, IDH2, CRAF, CSF1R, CTNNB1, and EZH2. TP53 had the highest mutation rates in recurrences (83%), metastases (82%) and primary tumor samples (76%). These rates compared to mutation rates of CDKN2A, PIK3CA, HRAS, CDK4, FBXW7 and RB1 of between 0 and 21% (Figure 1A). Furthermore, TP53 was found mutated most frequently in OPSCC (81%), OSCCs (64%), HPSCCs (81%), LSCC (86%), and in miscellaneous tumors (79%). On average, other frequently mutated genes in HNSCC sites were PIK3CA (11%), CDKN2A (10%), HRAS (8%), FGFR3 (3%) and FBXW7 (3%) (Figure 1B). In the 130 successfully sequenced primary tumor samples, no mutations were detected in 21 (16%) samples. In one of the HPV-positive tumor samples, a single TP53 mutation (c.225-35G>C) was found. In the other HPV-positive samples, no mutations were detected. Full range of mutated genes with prevalence rates for all subgroups is provided in Supplementary Tables S1 and S2.
Table 2: Used gene panels for sequencing of tumor samples
|
CHPv2 |
CHPv2+ |
OAv2 |
Sanger |
Total |
|---|---|---|---|---|---|
Primary |
99 |
9 |
22 |
18 |
148 |
OPSCC |
24 |
3 |
4 |
6 |
37 |
Recurrence |
19 |
2 |
7 |
1 |
29 |
Metastasis |
42 |
- |
11 |
9 |
62 |
Total |
160 |
11 |
40 |
28 |
239 |
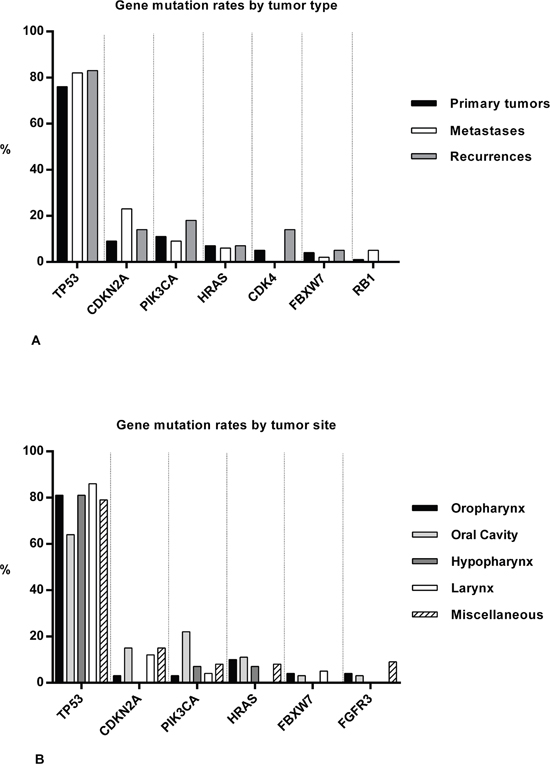
Figure 1: Summary of gene mutations in HNSCC samples. Bar charts showing mutation rates (%) of genes in all tumor samples ordered by tumor type A., and in 148 primary tumor samples ordered by HNSCC sites B.
The 92 patients with a history of alcohol use and/or smoking accounted for 121 primary tumor samples in total. TP53 was sequenced in all 121 samples. CDKN2A, PIK3CA, and HRAS were sequenced in 110 of 121 samples (Table 3). In all tumor samples of patients with or without a history of smoking and alcohol use, highest mutation rates were found in TP53, CDKN2A, PIK3CA, and HRAS. Mutation rates for TP53 were between 56 and 88%, for CDKN2A between 4 and 24%, for PIK3CA between 0 and 43%, and for HRAS between 2 and 21%. Overall, tumors exclusively related to a history of smoking had the highest mutation rates compared to the other subgroups (Figure 2A-2D).
Table 3: Prevalence of gene mutations in alcohol and/or smoking related tumor samples
|
Smoking/alcohol |
Non- smoking/alcohol |
Smoking |
Alcohol |
Total |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
No. |
(%) |
No. |
(%) |
No. |
(%) |
No. |
(%) |
No. |
(%) |
|
TP53 |
55/69 |
(80) |
9/16 |
(56) |
22/25 |
(88) |
9/11 |
(82) |
96/121 |
(79) |
CDKN2A |
2/60 |
(4) |
1/14 |
(7) |
6/25 |
(24) |
1/10 |
(10) |
11/110 |
(10) |
PIK3CA |
2/60 |
(4) |
6/14 |
(43) |
3/25 |
(12) |
0/10 |
- |
11/110 |
(10) |
HRAS |
1/60 |
(2) |
3/14 |
(21) |
2/25 |
(8) |
2/10 |
(20) |
8/110 |
(7) |
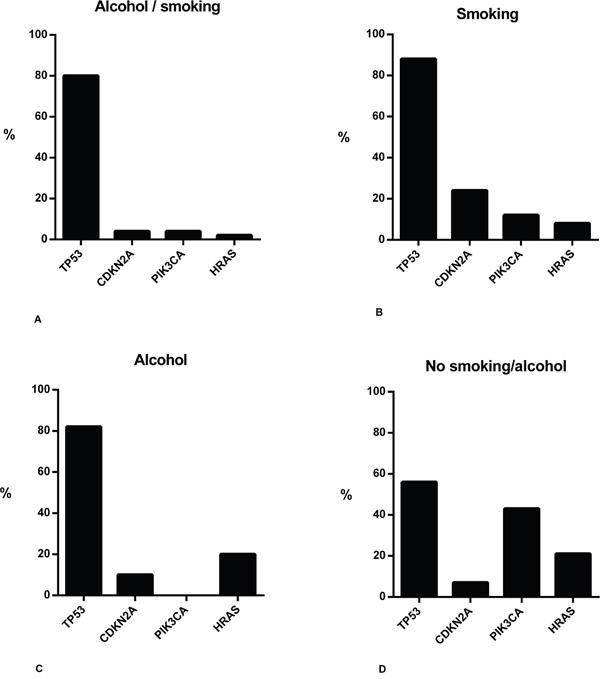
Figure 2: Summary of gene mutations in HNSCC samples of patients with or without a history of smoking and/or alcohol use. Bar charts showing mutation rates of primary tumor samples related to a history of smoking and alcohol use A., exclusively smoking B., exclusively alcohol use C., and samples not related to a history of smoking and alcohol use D.
Mutational co-occurrence
All tumor samples accounted for 171 mutational co-occurrences of two genes within one tumor sample. Co-occurrences were mostly found between TP53 and CDKN2A (16%), TP53 and PIK3CA (9%), TP53 and HRAS (5%), PIK3CA and HRAS (4%), and PIK3CA and CDKN2A (3%), as shown in Figure 3 and Supplementary Table S3.
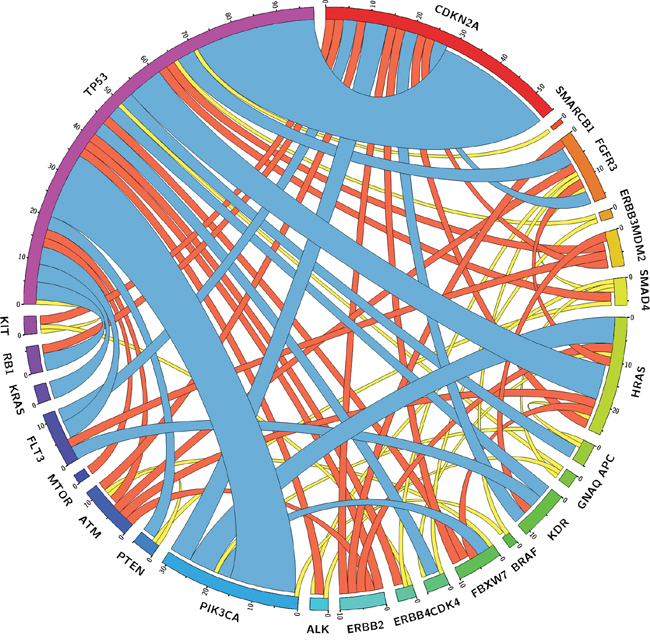
Figure 3: Circle plot illustrating co-mutations between genes found in HNSCC samples. Outer bars showing the absolute total amount of co-mutations found for genes entitling the bars. The curved ribbons inside the circle depict absolute number of co-mutations with the genes they are connected with (ribbon thickness corresponds with number of co-mutations). Ribbons are color coded by quartiles Q1 (yellow), Q2 (red), and Q3 (blue).
Mutational synonymy
Sequencing data of the associated (second) primary tumor were available for 51 of the 62 regional and distant metastases (the remaining 11 metastases were either associated to unsequenced primary tumors or were recurrent tumors only). By comparing the mutational profiles of the matched tumor pairs, we outlined the mutational heterogeneity of HNSCCs (Figure 4A). In 5 clinically related tumor pairs, no mutations were detected at all. The remaining 46 matched tumor pairs allowed for analysis of mutational synonymy, revealing 92% (81/88) of the analyzed gene mutations to be concordantly present in the associated metastasis. One discordant mutation was found in PTEN. A different single somatic nucleotide variant was detected in the metastasis (c.316G>T) compared to its associated primary tumor (c.892C>T). Six mutations were exclusively detected in the primary tumor: HRAS (c.38G>T), TP53 (c.192_217del26), PIK3CA (c.3140A>G), CDKN2A (c.247C>G), MDM2 (c.158G>A). Additionally, 2 new mutations were detected in exclusively the metastasis: SMAD4 (c.725C>G) and ALK (c.1588G>C). TP53 mutations were detected in 43 matched tumor pairs and were concordantly present in the associated metastasis in 95% (41/43). Mutational concordance of CDKN2A, HRAS and PIK3CA was respectively 92% (11/12), 83% (5/6) and 83% (5/6). An overview of mutational profiles of associated primary tumors and metastases is shown in Supplementary Table S4.
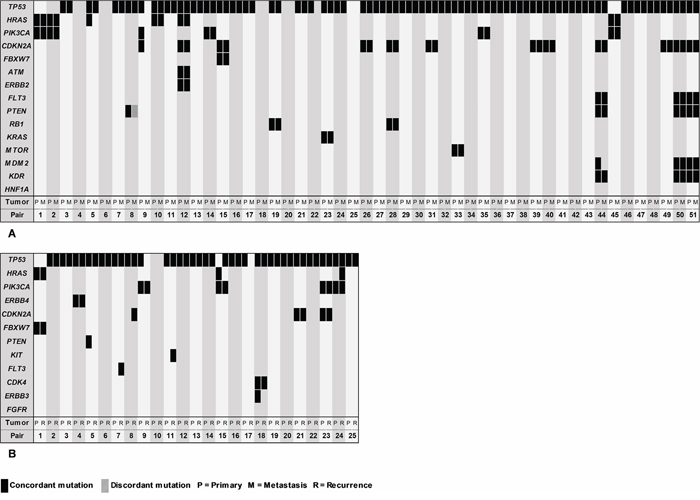
Figure 4: Block chart showing the mutational synonymy within tumor pairs. Mutational profiles of primary tumors (P) on the left side of vertical bars, compared to metastases (M) in A., and recurrences (R) in B. on the right side of the bars. Metastases and recurrences were either matched to a primary tumor or a second primary tumor.
Twenty-five associated pairs of primary tumors and recurrences were available for comparison (Figure 4B). In 2 matched tumor pairs, no mutations were detected. In the other 23 matched tumor pairs, 89% (33/37) of gene mutations found in the primary tumors were also found in the associated recurrences. No discordant mutations were detected. Four mutations were exclusively detected in the primary tumor: PTEN (c.892C>T), TP53 (c.192_217del26), HRAS (c.34G>A), ERBB3 (c.1016G>A). Additionally, 5 new mutations were exclusively detected in the recurrent tumor sample: FLT3 (c.2498C>G), CDKN2A (c.172C>T), KIT (c.1640A>G), TP53 (c.406delC), HRAS (c.38G>T). TP53 mutations were found concordant in 91% (21/23), 100% (4/4) in PIK3CA, 100% (2/2) in CDKN2A, and 50% (1/2) in HRAS. An overview of mutational profiles of associated primary tumors and recurrences is shown in Supplementary Table S5.
DISCUSSION
Despite developments in diagnostic techniques and treatment, prognosis barely improved over the past few decades [8]. This is probably due to likelihood of recurrence (25-50%) after (chemo)radiotherapy and the lack of specific detection methods for recurrent disease [8, 13, 32]. And, although treatment options for recurrences depend on many different factors (e.g. initial disease stage, site of recurrence, previous treatment, expected quality of life), early detection of recurrences using biomarkers might contribute in improving disease outcome. Therefore, we think that the potential role of genetic biomarkers in the management of advanced stage HNSCC is of increasing importance. Although its potential role as a prognostic and predictive biomarker has been debated [33, 34], as well as its use for diagnosing recurrent or minimal residual disease during follow-up after treatment [35–38], TP53 has, to our knowledge, not yet been proposed as a potential diagnostic biomarker for the early detection of locoregional metastases of HNSCC. Our data indicate that, using our 36-54 gene cancer panels, TP53 is the most frequently mutated gene in HNSCC tumor samples. This was the case for primary tumors of all sites, as well as their associated recurrences and metastases. While previous studies of the mutational profile of HPV-negative HNSCC have shown prevalence rates of TP53 mutations of 53%-78% [39, 40], these studies included either smaller sample sizes or were based on less sensitive whole exome sequencing (WES) data. More recent WES data from The Cancer Genome Atlas Network showed TP53 mutations in 86% of 279 HPV-negative HNSCC samples, which is more consistent with our data [5]. Moreover, NGS data, based on clinical assays that were capable of deep coverage of cancer-relevant genes, found TP53 mutations in 87% [4] and 81% [41] of HPV-negative HNSCCs.
In our study, TP53 mutations were detected in only 64% of the primary OSCCs compared to 80% or more in other sites. Because HPV-positive OPSCCs and TP53 mutations are known to be mutually exclusive in the majority of cases, in most studies similarly lowered TP53 mutation rates are being found in OPSCCs [16, 42, 43]. In our study, this difference between OSCCs and other sites may also be due to the low number of HPV-positive OPSCC tumor samples. Another explanation may be the presence of epigenetic aberrations, copy number variations (CNV), or mutations in unknown genes that could have been more abundant in OSCCs than in tumors from other sites [44–46]. Also, differences in exon coverage of our gene panels could have affected the variability in mutation profiles of tumor samples, although the differences we found in TP53 mutations cannot be fully proven because samples were not tested for all possible genetic aberrations and no statistical validation was performed. Additionally, statistical sampling bias could have affected sample sizes of HNSCC sites, since tumor type definition was primarily based on TP53 clonality assessment. Moreover, because distinction between a recurrence and a second primary tumor is not always clear in clinical practice, the parameters of “time span” and “adjacent site”, used to determine whether a tumor is secondary or recurrent, may be interpreted differently in various studies. Re-evaluation of the available clinicopathological data did not yield any changes in TP53 mutation prevalence rates.
Although not identified in hypopharyngeal tumors, we could confirm CDKN2A mutations as second most frequent mutations overall in HPV-negative HNSCCs. This is equivalent to findings in previous studies [4]. Compared to the literature, HRAS mutated less frequently in OSCC [5]. Furthermore, concordant with previous reports on PIK3CA [47], OSCCs contained PIK3CA mutations most frequently compared to other sites. Interestingly, concurrent PI3K pathway mutations such as PIK3R1/PIK3R2 were recently identified as being involved in HNSCC tumor progression. This supports the potential use of mutations in this oncogenic pathway as predictive biomarkers [48]. As expected, fewer TP53 mutations were found in HNSCCs of non-drinking and non-smoking patients. All tumor subgroups exclusively related to alcohol and/or smoking contained the most TP53 mutations. Furthermore, we found a relative increase of PIK3CA mutations in the non-smoking/non-drinking related group. NOTCH1 might also be a potential target as a diagnostic biomarker. NOTCH1 mutations are found in 14-20% of HNSCC and possibly play a role as early drivers in OSCC progression [2-4, 49, 50]. However, we found no aberrations in the NOTCH1 pathway. Aberrations were identified in the study of Agrawal et al., in which tumor specimens were sequenced using WES based on assays that covered NOTCH1 exons 1-34. This difference in results might be because our gene panels only covered NOTCH1 exons 25, 27, and 37.
Clarifying tumor evolution genetically is of great importance, since tumor heterogeneity could seriously challenge the principle of using genetic mutations as (diagnostic) biomarkers [51, 52]. In order to use tumor specific mutations for quantifying purposes, it is essential to target mutations in ctDNA that are contained in both the primary tumor and its clonal expansions. Primary tumor biopsy (e.g. core needle, incision, or excision biopsy) carries the risk of incompletely depicting the mutational profile of primary tumor tissue due to intratumoral heterogeneity, a problem that increases with newly acquired mutations in clonal expansions. As a result, it can lead to tumor specific mutations being selected as biomarkers that are not present in ctDNA from clonal expansions. However, blood testing for these diagnostic biomarkers could identify mutations in TP53 as well as those in other early driver genes that are extensively present in primary tumors and their clonal expansions.
Our data show that in most tumor pairs, mutations are concordant. This is largely consistent with the results of Hedberg et al. [53], who found that the primary tumor transmitted 86% of single somatic nucleotide variants identified in synchronous nodal metastases and 60% of those in recurrences. The relatively higher total amount of concordant mutations in associated recurrences compared to metastases in our study could be explained by the use of targeted gene panels instead of WES, which possibly impeded the detection of (unknown) driver genes that contribute to different pathways in tumor progression towards recurrences. Simultaneously, targeted sequencing could possibly have concealed intertumor heterogeneity, since we found comparable mutational synonymy rates of metastases and recurrences. On the other hand, our gene panels allowed for more sensitive sequencing compared to WES [57]. Thus, the small differences we found in mutational synonymy of TP53 might suggest increased intertumor heterogeneity between primary tumors and recurrences compared to primary tumors and metastases. Another explanation for mutational discordance could be differences in tissue acquisition methods, because sometimes sequencing of primary tumor samples was performed on resection specimens, whereas sequencing of metastases was more often performed on (smaller) biopsies. This could have caused discordance due to intratumor heterogeneity.
Interestingly, the detection of two additional mutations (SMAD4 and ALK) in two metastatic samples, might implicate these mutations to drive metastatic outgrowth, as these mutations, especially SMAD4, contribute to the downregulation of growth inhibitors and increased genomic instability [54]. Though, the number of mutations we found is not definite to draw conclusions. Furthermore, technical difficulties and flaws associated with performing NGS on FFPE material might have biased our results, as fragmented DNA originating from FFPE tissue challenges sequencing. Therefore, used NGS assays are adapted by using small amplicons facilitating shorter fragment sequencing. Also, fixation of tissue is known to potentially deaminate cytosines, possibly leading to more C>T or G>A base transitions [55]. However, recently performed validation of our gene panels revealed minimal FFPE induced DNA damage [56].
Despite their limitations, our findings provide useful information for developing new diagnostic strategies for HNSCC using targeted NGS panels that are easily accessible and capable of deep sequencing. Most investigated gene mutations were found concordantly mutated in the associated metastases and recurrences. Furthermore, TP53 mutations are by far the most frequent. This suggests TP53 mutations have potential value as diagnostic biomarkers in conjunction with subsequent ctDNA detection through liquid biopsy. By depicting these mutations in ctDNA using liquid biopsies, tumor remission after treatment could possibly be monitored non-invasively as compared to repeated biopsy for histological confirmation. This might complement current surveillance methods of clinical evaluation supported by flexible endoscopy and/or imaging such as PET-CT or diffusion weighted MRI, in order to increase accuracy for early detection of recurrent and/or metastatic HNSCC in the future.
MATERIALS AND METHODS
Data collection and analysis
We collected NGS sequencing data of all HNSCC samples, generated through TP53 clonality assessment on clinical request between the period of October 2013 and May 2015. Sequencing results from primary skin tumors of the head and neck region were not included. All samples on which sequencing was performed were formalin-fixed paraffin embedded (FFPE), after being obtained by surgical resection or tissue biopsy for diagnostic purposes between March 1992 to April 2015. Demographic and clinical data, including history of tobacco and alcohol use, were retrieved from hospital charts. Smoking and alcohol consumption habits were classified as previously described [58].
For analysis, samples were grouped and sorted by site of primary tumor (i.e. oral cavity, oropharynx, hypopharynx, larynx, miscellaneous), and tumor subtype (primary, metastasis, recurrence). Definition of tumor type (i.e. primary tumor, metastasis or recurrence) was mainly based on TP53 clonality. If a clonal relationship could not be ruled out, we based subtype determination on clinical suspicion and date of incidence as described previously [58]. In the same manner, distinction was made between second primary tumors, metastases and recurrences. Samples were excluded from analysis if tumor subtype remained unclear. Also, samples of unknown anatomical origin and duplicates of sequencing results were excluded.
Sequencing results were retrieved from the nationwide network and registry of histo- and cytopathology in The Netherlands (PALGA). Descriptive analysis consisted of mutational prevalence, which was determined for each gene in primary tumors and for all tumor types. Because of the use of varying gene panels over time by our molecular diagnostics laboratory, all (average) percentages were weighed for differences in gene coverage of used gene panels. Furthermore, co-occurrence of gene mutations in primary tumors was determined. Mutational synonymy was assessed by comparing the genetic profiles of primary tumors or second primary tumors with matched locoregional and/or distant metastases, if present. Gene mutations within matched tumor pairs were considered concordant when alterations were identical in the primary tumor and its associated metastasis or recurrence. Associated tumor samples within each matched pair were consistently sequenced by the same gene panel.
Molecular analysis
Clonality assessment was based on the presence of TP53 mutations or similar loss of heterozygosity (LOH) profiles using short tandem repeats. Targeted NGS was performed using the Ion Torrent™ PGM platform (Thermo Fisher Scientific, Waltham, MA, USA) as previously described [56]. The following gene panels were used: CHPv2 (Thermo Fisher Scientific, Waltham, MA, USA), CHPv2+ (i.e. CHPv2 supplemented with several extra genes and amplicons) and OAPv2 [59]. Exact genes and exons sequenced are shown in Supplementary Table S6-8. References used for reporting gene mutations were Center for Personalized Cancer Treatment (CPCT, Utrecht, The Netherlands; http://www.cpct.nl), Catalogue Of Somatic Mutations in Cancer (COSMIC; http://cancer.sanger.ac.uk/cosmic), International Cancer Genome Consortium (ICGC; https://icgc.org/) and The Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/).
Sufficient coverage is reached when an amplicon was sequenced at least 500 times. Variants with an allele frequency below 1% were considered as background noise and were not reported. Variants with allele frequency between 1% and 5% were first discussed multidisciplinary before decision to report. Variants with allele frequencies above 5% were reported. The used assay was validated recently according to general rules for diagnostic laboratories through ISO certification [56]. Accordingly, minimum tumor percentage is set at 10%. Gene amplification was indicated when five or more amplicons showed a z-score of 5 or more [59]. Where NGS failed, additional Sanger sequencing of TP53 exon 4-9 in forward and reverse directions was performed to allow for TP53 clonality assessment. The Sanger sequencing products were analyzed on a 3730 DNA Analyzer (Applied Biosystems, Foster city, CA, USA). If sequencing failed altogether, samples were ultimately excluded from analysis.
ACKNOWLEDGMENTS AND FUNDING
S.M.W. is funded by the Dutch Cancer Society (clinical fellowship No. 2011-4964).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. 2015; 136:E359-386.
2. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, Weinstein JN, Trevino L, Drummond JA, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011; 333:1154-1157.
3. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortes ML, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011; 333:1157-1160.
4. Chung CH, Guthrie VB, Masica DL, Tokheim C, Kang H, Richmon J, Agrawal N, Fakhry C, Quon H, Subramaniam RM, Zuo Z, Seiwert T, Chalmers ZR, Frampton GM, Ali SM, Yelensky R, et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Annals of oncology. 2015; 26:1216-1223.
5. Comprehensive genomic characterization of head neck squamous cell carcinomas. Nature. 2015; 517:576-582.
6. Marur S, D'Souza G, Westra WH, Forastiere AA. HPV-associated head and neck cancer: a virus-related cancer epidemic. The Lancet Oncology. 2010; 11:781-789.
7. Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, Westra WH, Chung CH, Jordan RC, Lu C, Kim H, Axelrod R, Silverman CC, Redmond KP, Gillison ML. Human papillomavirus and survival of patients with oropharyngeal cancer. The New England journal of medicine. 2010; 363:24-35.
8. Braakhuis BJ, Leemans CR, Visser O. Incidence and survival trends of head and neck squamous cell carcinoma in the Netherlands between 1989 and 2011. Oral oncology. 2014; 50:670-675.
9. Chung CH, Parker JS, Ely K, Carter J, Yi YJ, Murphy BA, Ang KM, El-Naggar AK, Zanation AM, Cmelak AJ, Levy S, Slebos RJ, Yarbrough WG. Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor-kappa B signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer research. 2006; 66:8210-8218.
10. Chung CH, Parker JS, Karaca G, Wu JY, Funkhouser WK, Moore' D, Butterfoss D, Xiang D, Zonation A, Yin XY, Shockley WW, Weissler MC, Dressler LG, Shores CG, Yarbrough WG, Perou CM. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. 2004; 5:489-500.
11. Graveland AP, Braakhuis BJ, Eerenstein SE, de Bree R, Bloemena E, de Maaker M, van den Brekel MW, Dijk F, Mesker WE, Tanke HJ, Leemans CR, Brakenhoff RH. Molecular diagnosis of minimal residual disease in head and neck cancer patients. Cell Oncol (Dordr). 2012; 35:367-375.
12. Smeets SJ, Brakenhoff RH, Ylstra B, van Wieringen WN, van de Wiel MA, Leemans CR, Braakhuis BJ. Genetic classification of oral and oropharyngeal carcinomas identifies subgroups with a different prognosis. Cellular oncology. 2009; 31:291-300.
13. Ho AS, Kraus DH, Ganly I, Lee NY, Shah JP, Morris LG. Decision making in the management of recurrent head and neck cancer. Head & neck. 2014; 36:144-151.
14. Leemans CR, Tiwari R, Nauta JJP, Vanderwaal I, Snow GB. Recurrence at the Primary Site in Head and Neck-Cancer and the Significance of Neck Lymph-Node Metastases as a Prognostic Factor. Cancer. 1994; 73:187-190.
15. Graveland AP, Golusinski PJ, Buijze M, Douma R, Sons N, Kuik DJ, Bloemena E, Leemans CR, Brakenhoff RH, Braakhuis BJM. Loss of heterozygosity at 9p and p53 immunopositivity in surgical margins predict local relapse in head and neck squamous cell carcinoma. International Journal of Cancer. 2011; 128:1852-1859.
16. Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nature reviews Cancer. 2011; 11:9-22.
17. Mehta S, Shelling A, Muthukaruppan A, Lasham A, Blenkiron C, Laking G, Print C. Predictive and prognostic molecular markers for cancer medicine. Therapeutic advances in medical oncology. 2010; 2:125-148.
18. Noorlag R, van Kempen PM, Stegeman I, Koole R, van Es RJ, Willems SM. The diagnostic value of 11q13 amplification and protein expression in the detection of nodal metastasis from oral squamous cell carcinoma: a systematic review and meta-analysis. Virchows Archiv. 2015; 466:363-373.
19. Kang H, Kiess A, Chung CH. Emerging biomarkers in head and neck cancer in the era of genomics. Nature reviews Clinical oncology. 2015; 12:11-26.
20. Thariat J, Vignot S, Lapierre A, Falk AT, Guigay J, Van Obberghen-Schilling E, Milano G. Integrating genomics in head and neck cancer treatment: Promises and pitfalls. Crit Rev Oncol Hematol. 2015; 95:397-406.
21. Nees M, Homann N, Discher H, Andl T, Enders C, Herold-Mende C, Schuhmann A, Bosch FX. Expression of mutated p53 occurs in tumor-distant epithelia of head and neck cancer patients: a possible molecular basis for the development of multiple tumors. Cancer research. 1993; 53:4189-4196.
22. Califano J, vanderRiet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, Sidransky D. Genetic progression model for head and neck cancer: Implications for field cancerization. Cancer research. 1996; 56:2488-2492.
23. Gollin SM. Chromosomal alterations in squamous cell carcinomas of the head and neck: window to the biology of disease. Head & neck. 2001; 23:238-253.
24. Braakhuis BJ, Leemans CR, Brakenhoff RH. A genetic progression model of oral cancer: current evidence and clinical implications. Journal of oral pathology & medicine. 2004; 33:317-322.
25. Boyle JO, Hakim J, Koch W, van der Riet P, Hruban RH, Roa RA, Correo R, Eby YJ, Ruppert JM, Sidransky D. The incidence of p53 mutations increases with progression of head and neck cancer. Cancer research. 1993; 53:4477-4480.
26. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953; 6:963-968.
27. Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter's concept of field cancerization: evidence and clinical implications. Cancer research. 2003; 63:1727-1730.
28. Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, Del Rio M, Molina F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010; 38:6159-6175.
29. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA. Circulating mutant DNA to assess tumor dynamics. Nature Medicine. 2008; 14:985-990.
30. Gray ES, Rizos H, Reid AL, Boyd SC, Pereira MR, Lo J, Tembe V, Freeman J, Lee JHJ, Scolyer RA, Siew K, Lomma C, Cooper A, Khattak MA, Meniawy TM, Long GV, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget. 2015; 6:42008-42018. doi: 10.18632/oncotarget.5788.
31. Heidary M, Auer M, Ulz P, Heitzer E, Petru E, Gasch C, Riethdorf S, Mauermann O, Lafer I, Pristauz G, Lax S, Pantel K, Geigl JB, Speicher MR. The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Research. 2014; 16.
32. de Bree R, van der Putten L, Brouwer J, Castelijns JA, Hoekstra OS, Leemans CR. Detection of locoregional recurrent head and neck cancer after (chemo)radiotherapy using modern imaging. Oral oncology. 2009; 45:386-393.
33. Nemunaitis J, Clayman G, Agarwala SS, Hrushesky W, Wells JR, Moore C, Hamm J, Yoo G, Baselga J, Murphy BA, Menander KA, Licato LL, Chada S, Gibbons RD, Olivier M, Hainaut P, et al. Biomarkers Predict p53 Gene Therapy Efficacy in Recurrent Squamous Cell Carcinoma of the Head and Neck. Clinical cancer research. 2009; 15:7719-7725.
34. Masica DL, Li S, Douville C, Manola J, Ferris RL, Burtness B, Forastiere AA, Koch WM, Chung CH, Karchin R. Predicting survival in head and neck squamous cell carcinoma from TP53 mutation. Human genetics. 2015; 134:497-507.
35. van Houten VMM, Tabor MP, van den Brekel MWM, Kummer JA, Denkers F, Dijkstra J, Leemans R, van der Waal I, Snow GB, Brakenhoff RH. Mutated p53 as a molecular marker for the diagnosis of head and neck cancer. Journal of Pathology. 2002; 198:476-486.
36. Wang X, Chen S, Chen X, Zhang C, Liang X. Tumor-related markers in histologically normal margins correlate with locally recurrent oral squamous cell carcinoma: a retrospective study. Journal of oral pathology & medicine. 2016; 45:83-88.
37. Poeta ML, Manola J, Goldenberg D, Forastiere A, Califano JA, Ridge JA, Goodwin J, Kenady D, Saunders J, Westra W, Sidransky D, Koch WM. The Ligamp TP53 Assay for Detection of Minimal Residual Disease in Head and Neck Squamous Cell Carcinoma Surgical Margins. Clinical cancer research. 2009; 15:7658-7665.
38. Lopez M, Aguirre JM, Cuevas N, Anzola M, Videgain J, Aguirregaviria J, Castro A, de Pancorbo MM. Use of cytological specimens for p53 gene alteration detection in oral squamous cell carcinoma risk patients. Clin Oncol (R Coll Radiol). 2004; 16:366-370.
39. Mountzios G, Rampias T, Psyrri A. The mutational spectrum of squamous-cell carcinoma of the head and neck: targetable genetic events and clinical impact. Annals of oncology. 2014; 25:1889-1900.
40. Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, Ridge JA, Goodwin J, Kenady D, Saunders J, Westra W, Sidransky D, Koch WM. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. The New England journal of medicine. 2007; 357:2552-2561.
41. Seiwert TY, Zuo ZX, Keck MK, Khattri A, Pedamallu CS, Stricker T, Brown C, Pugh TJ, Stojanov P, Cho J, Lawrence MS, Getz G, Bragelmann J, DeBoer R, Weichselbaum RR, Langerman A, et al. Integrative and Comparative Genomic Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas. Clinical Cancer Research. 2015; 21:632-641.
42. Pickering CR, Zhang JX, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, Zhao M, Alves MVO, Chang K, Drummond J, Cortez E, Xie TX, Zhang D, Chung W, Issa JPJ, Zweidler-McKay PA, et al. Integrative Genomic Characterization of Oral Squamous Cell Carcinoma Identifies Frequent Somatic Drivers. Cancer discovery. 2013; 3:770-781.
43. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nature communications. 2013; 4:2873.
44. Noorlag R, van Kempen PMW, Moelans CB, de Jong R, Blok LER, Koole R, Grolman W, van Diest PJ, van Es RJJ, Willems SM. Promoter hypermethylation using 24-gene array in early head and neck cancer Better outcome in oral than in oropharyngeal cancer. Epigenetics. 2014; 9:1220-1227.
45. van Kempen PM, Noorlag R, Braunius WW, Moelans CB, Rifi W, Savola S, Koole R, Grolman W, van Es RJ, Willems SM. Clinical relevance of copy number profiling in oral and oropharyngeal squamous cell carcinoma. Cancer medicine. 2015; 4:1525-1535.
46. Guerrero-Preston R, Michailidi C, Marchionni L, Pickering CR, Frederick MJ, Myers JN, Yegnasubramanian S, Hadar T, Noordhuis MG, Zizkova V, Fertig E, Agrawal N, Westra W, Koch W, Califano J, Velculescu VE, et al. Key tumor suppressor genes inactivated by “greater promoter” methylation and somatic mutations in head and neck cancer. Epigenetics. 2014; 9:1031-1046.
47. Murugan AK, Hong NT, Fukui Y, Munirajan AK, Tsuchida N. Oncogenic mutations of the PIK3CA gene in head and neck squamous cell carcinomas. International journal of oncology. 2008; 32:101-111.
48. Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, Lu Y, Zhang Q, Du Y, Gilbert BR, Freilino M, Sauerwein S, Peyser ND, Xiao D, Diergaarde B, Wang L, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer discovery. 2013; 3:761-769.
49. William WN, Jr., El-Naggar AK. A novel target for oral cancer chemoprevention? Notch quite, yet. Cancer Prev Res (Phila). 2015; 8:262-265.
50. Izumchenko E, Sun K, Jones S, Brait M, Agrawal N, Koch W, McCord CL, Riley DR, Angiuoli SV, Velculescu VE, Jiang WW, Sidransky D. Notch1 mutations are drivers of oral tumorigenesis. Cancer Prev Res (Phila). 2015; 8:277-286.
51. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012; 366:883-892.
52. Jacoby MA, Duncavage EJ, Walter MJ. Implications of Tumor Clonal Heterogeneity in the Era of Next-Generation Sequencing. Trends in Cancer. 2016; 1:10.
53. Hedberg ML, Goh G, Chiosea SI, Bauman JE, Freilino ML, Zeng Y, Wang L, Diergaarde BB, Gooding WE, Lui VW, Herbst RS, Lifton RP, Grandis JR. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. The Journal of clinical investigation. 2016; 126:169-180.
54. Korc M. Smad4: gatekeeper gene in head and neck squamous cell carcinoma. The Journal of clinical investigation. 2009; 119:3208-3211.
55. Srinivasan M, Sedmak D, Jewell S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. The American journal of pathology. 2002; 161:1961-1971.
56. de Leng WW, Gadellaa-van Hooijdonk CG, Barendregt-Smouter FA, Koudijs MJ, Nijman I, Hinrichs JW, Cuppen E, van Lieshout S, Loberg RD, de Jonge M, Voest EE, de Weger RA, Steeghs N, Langenberg MH, Sleijfer S, Willems SM, et al. Targeted Next Generation Sequencing as a Reliable Diagnostic Assay for the Detection of Somatic Mutations in Tumours Using Minimal DNA Amounts from Formalin Fixed Paraffin Embedded Material. PloS one. 2016; 11:e0149405.
57. Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome Med. 2015; 7:80.
58. van Kempen PM, Verdam FJ, de Poel E, Braunius WW, de Weger RA, van Es RJ, Grolman W, Willems SM. TP53 Y220C Is a Hotspot Mutation in Oropharyngeal Squamous Cell Carcinoma. Pathobiology. 2015; 82:21-27.
59. Hoogstraat M, Hinrichs JW, Besselink NJ, Radersma-van Loon JH, de Voijs CM, Peeters T, Nijman IJ, de Weger RA, Voest EE, Willems SM, Cuppen E, Koudijs MJ. Simultaneous detection of clinically relevant mutations and amplifications for routine cancer pathology. The Journal of molecular diagnostics. 2015; 17:10-18.