
INTRODUCTION
Myelodysplastic syndromes (MDS), a heterogeneous group of clonal hematopoietic malignancies, are the most frequently encountered acquired bone marrow (BM) failure syndromes in adults [1, 2]. While the disease is classified as normo/hypercellular MDS (NH-MDS) in the majority of patients, about 10-20% of patients have hypoplastic MDS (h-MDS), characterized by a hypocellular BM, traditionally defined as less than 30% cellularity in the BM trephine biopsy specimens [3–5]. Recently, age-adjusted criteria of marrow hypocellularity have also been proposed to define h-MDS, for instance, <30% cellularity in patients younger than 70 years and <20% cellularity in patients older than 70 years, to account for the physiologically deceasing marrow cellularity with increasing age [6–8]. Although h-MDS share a number of similar clinical features with NH-MDS, and both have a propensity to leukemic transformation during clinical follow-up, h-MDS appear to be a distinct clinico-pathological entity [9, 10], suggesting an unique underlying pathogenesis of this disease. Recently, a number of somatic mutations, such as mutations in ASXL1, EZH2, TET2, IDH1/2, DMNT3A, RUNX1, NRAS, KRAS, TP53, and splicing complex genes, which may play important roles in the pathogenesis of MDS, have been described [11]. However, the reports concerning the differences in the genetic alterations between h-MDS and NH-MDS are limited. Further, although the revised international prognostic scoring system (IPSS-R) has been demonstrated to be more powerful than IPSS in prognostication for MDS patients [12, 13], its applicability to h-MDS remains to be explored. The aims of this study were to elucidate the differences in the clinical features, genetic abnormalities, and clinical outcomes between h-MDS and NH-MDS in a large cohort of MDS patients in Taiwan, and to verify the applicability of IPSS-R in this special entity of MDS.
RESULTS
Clinical and laboratory characteristics
A total of 369 MDS patients, diagnosed according to the 2008 WHO classification, were included in our study. Most patients (63.7%) in our cohort received mainly transfusions and/or other supportive care, while 20 (5.4%) patients received hypomethylating agents, 37 (10.0%) received low-dose chemotherapy, 26 (7.1%) received AML-directed intensive chemotherapy, and 51 (13.8%) underwent allogeneic hematopoietic stem cell transplantation. Among the total cohort, 100 (27.1%) patients were diagnosed as having h-MDS. The comparisons of clinical and laboratory features between patients with h-MDS and NH-MDS are listed in Table 1. Patients with h-MDS had lower peripheral blood (PB) white blood cell (WBC) counts (P= 0.030) and blast counts (P=0.006), and BM blast percentages (P=0.001) than those with NH-MDS. There was no difference in the age and gender distribution, hemoglobin, platelet, and serum lactate dehydrogenase levels between these two groups. h-MDS patients had statistically higher proportion of refractory cytopenia with unilineage dysplasia (RCUD) and refractory cytopenia with multilineage dysplasia (RCMD), but lower proportion of refractory anemia with excess blasts-1 (RAEB-1), compared with NH-MDS patients (28.0% vs. 17.5%, P= 0.029; 38.0% vs. 20.8%, P= 0.001; 11.0% vs. 24.9%, P= 0.004, respectively). h-MDS patients were classified more frequently to IPSS low and intermediate-1 risk groups (80.0% vs. 58.6%, P<0.001), or IPSS-R very low, low, and intermediate risk groups (72.2% vs. 52.6%, P= 0.001) than NH-MDS patients.
Table 1: Comparison of clinical and laboratory features between h-MDS and NH-MDS patients
Clinical characters |
Total |
h-MDS |
NH-MDS |
P value |
|---|---|---|---|---|
Sex |
|
|
|
0.324 |
Male |
242 (65.6%) |
70 (70.0%) |
172 (63.9%) |
|
Female |
127 (34.4%) |
30 (30.0%) |
97 (36.1%) |
|
Age (year)* |
65.2 (16.4-94.5) |
61.6 (18.4-94.5) |
65.3 (16.4-90.5) |
0.997 |
Lab data** |
|
|
|
|
WBC (/μL) |
3490 (490-40500) |
3050 (650-9890) |
3660 (490-40500) |
0.030* |
Hb (g/dL) |
8.1 (3.4-14.6) |
8.1 (3.7-14.4) |
8.2 (3.2-14.6) |
0.971 |
Platelet (×1,000 /μL) |
77 (3-931) |
66 (3-618) |
82 (3-931) |
0.179 |
LDH (U/L) |
482 (145-6807) |
463 (145-3122) |
495 (210-6807) |
0.361 |
PB blast count (/μL) |
0 (0-3270) |
0 (0-523) |
0 (0-3270) |
0.006* |
BM blast % |
3.2 (0-19.0) |
2.0 (0-19.0) |
4.5 (0-19.5) |
0.001* |
2008 WHO classification# |
|
|
|
|
RCUD |
75 (20.3%) |
28 (28.0%) |
47 (17.5%) |
0.029* |
RARS |
20 (5.4%) |
3 (3.0%) |
17 (6.3%) |
0.302 |
RCMD |
94 (25.5%) |
38 (38.0%) |
56 (20.8%) |
0.001* |
RCMD-RS |
14 (3.8%) |
3 (3.0%) |
11 (4.1%) |
0.767 |
RAEB1 |
78 (21.1%) |
11 (11.0%) |
67 (24.9%) |
0.004* |
RAEB2 |
85 (23.1%) |
17 (17.0%) |
68 (25.3%) |
0.097 |
MDS-U |
3 (0.8%) |
0 (0%) |
3 (1.1%) |
0.566 |
IPSS‡,#,§ |
|
|
|
<0.001* |
Low+INT-1 |
219 (64.2%) |
72 (80.0%) |
147 (58.6%) |
|
INT-2+High |
122 (35.8%) |
18 (20.0%) |
104 (41.4%) |
|
IPSS-R‡,#,ζ |
|
|
|
0.001* |
Very low+low+INT |
197 (57.8%) |
65 (72.2%) |
132 (52.6%) |
|
High+very high |
144 (42.2%) |
25 (27.8%) |
119 (47.4%) |
|
Treatment modalities |
|
|
|
|
Transfusion/BCS |
235 (63.7%) |
70 (70.0%) |
165 (61.3%) |
0.144 |
HMA |
20 (5.4%) |
4 (4.0%) |
16 (5.9%) |
0.608 |
Intensive chemotherapy |
26 (7.1%) |
2 (2.0%) |
24 (8.9%) |
0.021* |
Lose-dose chemotherapy |
37 (10.0%) |
7 (7.0%) |
30 (11.2%) |
0.329 |
HSCT |
51 (13.8%) |
17 (17.0%) |
34 (12.6%) |
0.309 |
**Median (range).
*Statistically significant if P<0.05.
#Number of patients (% of patients within either hypoplastic or non-hypoplastic MDS subgroups).
‡341patients, including 90 h-MDS and 251 NH-MDS patients, had chromosome data at diagnosis.
§IPSS: Low, 0; intermediate (INT)-1, 0.5-1; INT-2, 1.5-2; and High, ≥ 2.5.
ζIPSS-R: Very low, ≦1.5; Low, >1.5-3; intermediate (INT),>3-4.5; High, >4.5-6; and Vey high, >6.
Abbreviations: h-MDS, hypoplastic MDS; NH-MDS, normo-/hypercellular MDS; FAB, French-American-British classification; RARS, refractory anemia with ring sideroblasts; RAEB, refractory anemia with excess blasts; RCUD, refractory cytopenia with unilineage dysplasia; RCMD, refractory cytopenia with multilineage dysplasia; and MDS-U, MDS (unclassifiable); IPSS, international prognosis scoring system; IPSS-R, revised IPSS.
Comparison of cytogenetic changes and genetic alterations between h-MDS and NH-MDS
Clonal chromosomal abnormalities were detected in 154 (45.2%) of 341 patients who had chromosomal data at diagnosis (Supplementary Table S2). The incidence of cytogenetic abnormalities was similar between the h-MDS and NH-MDS groups (42.2% vs. 46.2%, P= 0.815). We found that h-MDS patients had statistically lower incidence of poor-risk cytogenetic abnormalities than the NH-MDS patients (7.8% vs. 24.3%, P<0.001). However, there was no difference in the distribution of any specific chromosomal abnormalities between patients with h-MDS and NH-MDS.
To investigate the interaction of genetic alterations in the pathogenesis of h-MDS, a mutational screening of 17 genes was performed (Table 2). Overall, 203 (55.0%) patients had at least one gene mutation, 35 (35.0%) in the h-MDS group, compared to 168 (62.5%) in NH-MDS group (P<0.001). The most common mutation in h-MDS was SF3B1 mutation (12.0%), followed by TET2 (11.4%), and ASXL1 mutations (7.1%). Patients with h-MDS had significantly lower incidences of RUNX1, ASXL1, DNMT3A, EZH2, and TP53 mutations than those with NH-MDS (4.0% vs. 14.2%, P= 0.005; 7.1% vs. 21.7%, P=0.001; 3.0% vs. 12.6%, P= 0.006; 0% vs. 5.2%, P= 0.014, 3.0% vs. 10.8%, P= 0.020, respectively). If we consider cytogenetic abnormalities and molecular alterations collectively, 262 (71.0%) of the total cohort had at least one genetic aberration, 57 (57.0%) in the h-MDS subgroup and 205 (76.2%) in the NH-MDS subgroup (P<0.001).
Table 2: Comparison of genetic alterations between patients with h-MDS and NH-MDS
Variables |
Number examined |
Total cohort (%) |
h-MDS (%) |
NH-MDS (%) |
P value |
|---|---|---|---|---|---|
|
|
Mutated |
Mutated |
Mutated |
|
FLT3/ ITD |
366 |
1.1% |
1.0% |
1.1% |
>0.999 |
NRAS |
369 |
2.2% |
1.0% |
2.6% |
0.688 |
KRAS |
367 |
1.1% |
0% |
1.5% |
0.578 |
JAK2 |
368 |
0.8% |
1.0% |
0.7% |
>0.999 |
RUNX1 |
367 |
11.4% |
4.0% |
14.2% |
0.005* |
MLL/ PTD |
352 |
0.6% |
0% |
0.8% |
>0.999 |
IDH1 |
368 |
0.5% |
1.0% |
0.4% |
0.470 |
IDH2 |
366 |
2.2% |
0% |
3.0% |
0.113 |
ASXL1 |
366 |
17.8% |
7.1% |
21.7% |
0.001* |
TET2 |
282 |
12.4% |
11.4% |
12.7% |
>0.999 |
DNMT3A |
369 |
10.0% |
3.0% |
12.6% |
0.006* |
TP53 |
369 |
8.7% |
3.0% |
10.8% |
0.020* |
SETBP1 |
369 |
2.4% |
1.0% |
3.0% |
0.454 |
EZH2 |
369 |
3.8% |
0% |
5.2% |
0.014* |
SF3B1 |
369 |
11.4% |
12.0% |
11.2% |
0.854 |
U2AF1 |
369 |
7.9% |
5.0% |
8.9% |
0.278 |
SRSF2 |
369 |
10.8% |
6.0% |
12.6% |
0.089 |
*Statistically significant if P<0.05.
Survival analysis
With a median follow-up duration of 46.9 months (range, 0.1-250.7 months), the cumulated incidence of acute leukemic transformation at 5 years was 19.3% for h-MDS patients and 40.4% for NH-MDS patients (P= 0.001, Figure 1). In addition, the patients with h-MDS had a longer overall survival (OS) than those with NH-MDS (median, 80.5 months vs. 29.6 months, P=0.001, Figure 2). In subgroup analysis, the survival difference between h-MDS and NH-MDS patients remained statistically significant only among the patients with lower-risk MDS (IPSS-R very low, low and intermediate risks, median 185.5 months vs. 69.9 months, P= 0.024, Figure 3A), but not in those with higher-risk MDS (IPSS-R high and very high risks, median 16.8 months vs. 11.4 months, P=0.057, Figure 3B). Similarly, the lower incidence of acute leukemic transformation in h-MDS, compared to NH-MDS, remained significant only in the lower-risk group (4.8% vs. 31.4%, P= 0.002, Figure 4A), but not in the higher-risk group (55.7% vs. 59.0%, P=0.437, Figure 4B). The IPSS-R could well predict the survival of h-MDS patients: patients with lower-risk MDS had a significantly longer median OS than those with higher-risk MDS (185.5 months vs. 16.8 months, P<0.001, Figure 5A), the same as in NH-MDS patients (69.9 months vs. 11.4 months, P<0.001, Figure 5B). In multivariate analysis, we included gene mutation variables with P values less than 0.1 in the univariate analysis, plus relevant clinical parameters, such as age, sex, BM cellularity, IPSS-R risk stratification, and treatment modality. We found that the BM hypocellularity was an independent favorable prognostic factor for OS (relative risk 0.655, 95% CI 0.431-0.995, P=0.047) irrespective of age, sex, IPSS-R, gene mutations, and treatment modality. (Table 3).
Table 3: Multivariate analysis (Cox regression) for the overall survival in 369 MDS patients
Variable |
RR |
Overall survival |
P value |
|
|---|---|---|---|---|
Lower 95% CI |
Upper 95% CI |
|||
Age† |
1.533 |
1.035 |
2.273 |
0.033* |
Sex |
1.353 |
0.957 |
1.912 |
0.087 |
BM Hypocellularity |
0.655 |
0.431 |
0.995 |
0.047* |
IPSS-Rζ |
3.431 |
2.404 |
4.896 |
<0.001* |
TP53 |
5.904 |
3.442 |
10.130 |
<0.001* |
ASXL1 |
1.394 |
0.892 |
2.179 |
0.144 |
EZH2 |
1.066 |
0.479 |
2.371 |
0.876 |
DNMT3A |
1.367 |
0.847 |
2.204 |
0.200 |
RUNX1 |
0.983 |
0.608 |
1.590 |
0.946 |
SRSF2 |
1.440 |
0.838 |
2.474 |
0.187 |
HSCT or intensive chemotherapy§ |
0.994 |
0.643 |
1.535 |
0.977 |
*Statistically significant if P<0.05.
†Age > 65 relative to Age ≤ 65 (the reference).
ζHigher-risk vs. Lower-risk.
§HSCT or intensive chemotherapy vs. others.
Abbreviations: RR, Relative Risk; CI, confidence interval.
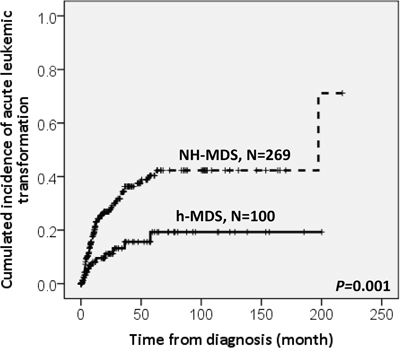
Figure 1: The cumulated incidence of acute leukemic transformation at 5 years was significantly lower in h-MDS patients (19.3%) than in NH-MDS patients (40.4%).
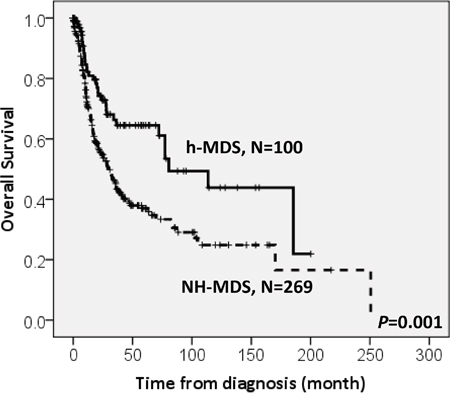
Figure 2: The comparison of overall survival between patients with h-MDS and NH-MDS. Patients with h-MDS had a longer median overall survival than those with NH-MDS (80.5 months vs. 29.6 months, P=0.001).
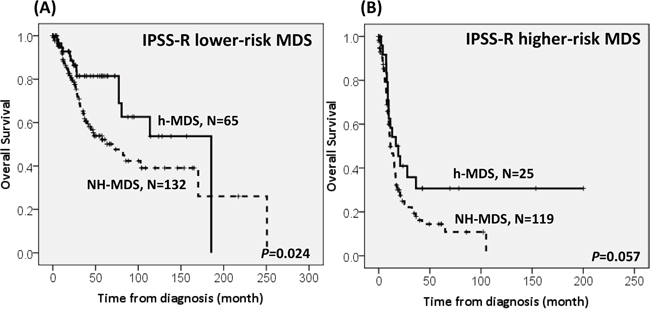
Figure 3: The comparison of overall survival between h-MDS and NH-MDS patients in subgroups of patients with lower-risk and higher-risk MDS. The survival difference between patients with h-MDS and NH-MDS remains statistically significant in lower-risk MDS (IPSS-R very low, low and intermediate risks), with a median OS of 185.5 months and 69.9 months, respectively A., but not among higher-risk MDS (IPSS-R high and very high risks), with a median OS of 16.8 months and 11.4 months, respectively B.
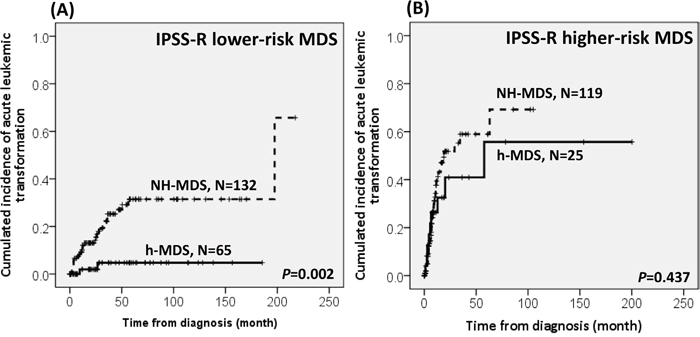
Figure 4: The comparison of the risk of acute leukemic transformation between h-MDS and NH-MDS patients in subgroups of patients with lower-risk and higher-risk MDS. The difference in the incidence of acute leukemic transformation between patients with h-MDS and NH-MDS remains statistically significant in lower-risk MDS, with a 5-year acute leukemic transformation rate of 4.8% and 31.4%, respectively A., but not among higher-risk MDS, with an acute leukemic transformation rate of 55.7% and 59.0%, respectively B.
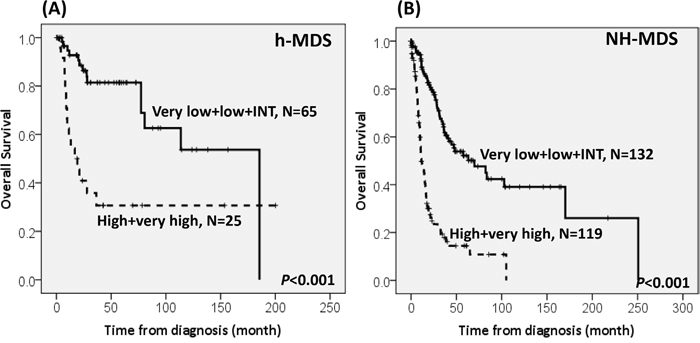
Figure 5: The Kaplan-Meier survival curves of h-MDS and NH-MDS patients, stratified by IPSS-R. The IPSS-R can well predict the survival in h-MDS patients, with the median overall survival of 185.5 months vs. 16.8 months in the IPSS-R lower-risk (very low, low and intermediate risks) and higher-risk (high and very high risks) subgroups, respectively A., the same as in NH-MDS patients, with the median OS of 69.9 months vs. 11.4 months in the IPSS-R lower-risk and higher-risk subgroups, respectively B.
DISCUSSION
We found that h-MDS were associated with distinct clinico-biological features and mutation profile, and that BM hypocellularity represented a favorable prognostic factor in the MDS patients, independent of age, sex, IPSS-R and genetic alterations. Further, IPSS-R could hold its predictive power of discriminating prognosis in the h-MDS patients, as in the NH-MDS patients.
In this study, 27.1% MDS patients had h-MDS, a percentage higher than those (10-20%) reported in literature [3, 4, 6, 14–16]. We speculate that the difference could be explained by the following reasons. First, in some of the previous reports, chronic myelomonocytic leukemia (CMMoL) and refractory anemia with excess blasts in transformation (RAEB-T) were not excluded [4, 7, 8]. Therefore, the higher percentage of the h-MDS patients in this study might represent a renewed epidemiology in the contemporary era of the WHO classification. Second, in our study, we included only the “de novo” MDS patients; while in other studies, therapy-related MDS patients were either enrolled altogether, or not specifically excluded [6, 7, 17]. Third, differences in the genetic and environmental backgrounds and clinico-pathological features of the MDS patients between Asian and Western countries have been reported [13, 18–20], which might contribute to the difference in h-MDS prevalence as well.
To date, there has been no universal consensus on whether h-MDS belong to the lower- or higher-risk subset of MDS. Yue et al reported there was no difference in the distribution of IPSS risk scores between the h-MDS and NH-MDS patients [6], while others demonstrated that the h-MDS patients more frequently had IPSS lower-risk MDS [8], or on the contrary, IPSS higher-risk MDS [7]. In this large homogeneous cohort of consecutive unselected de novo MDS patients classified by the 2008 WHO criteria, we distinctly demonstrated that h-MDS was associated with lower-risk subgroups in both IPSS and IPSS-R, a finding not unequivocally shown previously. The utility of IPSS in the risk stratification of patients with h-MDS has been reported before, both in Asian and Western populations [6, 9]. However, Tong et al. [7], proposed a new prognostic scheme for h-MDS patients because they found IPSS failed to distinguish the outcome of these patients [7]. In 2012, IPSS-R was introduced to better risk-stratify de novo MDS patients [12]. To our knowledge, no study has specifically examined the power of IPSS-R in risk stratification of h-MDS patients. To shed light on the applicability of IPSS-R in this setting, we analyzed the survival of our h-MDS patients according to the IPSS-R, and found that IPSS-R did successfully stratify h-MDS patients into distinct risk groups (Figure 5A).
Although AA and h-MDS share the same hypocellularity in the BM biopsy specimen, the combination of dysplasia of hematopoietic cells, and clonal cytogenetic abnormality can allow a conclusive diagnosis of MDS. In our cohort, the morphologic diagnosis of MDS was made according to the 2008 WHO classification, and all the h-MDS cases had BM morphologies fulfilling the criteria of dysplasia. Besides, the incidence of clonal chromosomal abnormalities was similar between patients with h-MDS and NH-MDS. Taken above, the possibility that we inadvertently included AA patients into our h-MDS cohort should be negligible. On a more detailed inspection, although 5q deletion (as the sole abnormality) appeared more frequently, and loss of chromosome 7 (as the sole abnormality) less frequently in the h-MDS subgroup, the differences did not reach statistical significance (P= 0.073 and P= 0.199, respectively). There were no significant differences in other chromosomal abnormalities between the h-MDS and NH-MDS patients, either, a finding similar to what previous studies had reported [3, 4, 6, 21]. Gene mutations have started to be taken into account for risk stratification of myeloid malignancies recently, and cumulating data are suggesting that certain gene mutations have prognostic significance in MDS [22, 23]. Of particular interest, Nazha et al. recently reported the genomic analysis of 237 MDS patients, in which h-MDS patients comprised 14% [10]. In their study, there was no difference in the distribution of cytogenetic abnormalities between their NH-MDS and h-MDS subgroups. The patients with h-MDS had a lower average number of somatic mutations, and lower incidences of SF3B1 and IDH1/2 mutations. However, they did not find a survival difference between patients with h-MDS and NH-MDS. In our current study, we examined the mutational status of 17 genes relevant to myeloid malignancies, and found that patients with h-MDS had lower number of concurrent genetic alterations, similar to the report by Nazha et al. However, in contrast to their report, we could not find a negative association of SF3B1 and IDH1/2 mutations with h-MDS. Instead, we showed that the h-MDS subgroup had less frequent RUNX1, ASXL1, DNMT3A, EZH2, and TP53 mutations compared with the NH-MDS subgroup. These findings may be of prognostic significance, since mutations in the aforementioned genes have been linked to the pathogenesis, disease progression, and inferior survival of MDS patients [24–37]. In other words, although BM hypocellularity was an independent prognostic factor, the lower frequencies of the poor-risk gene mutations in the h-MDS patients might also contribute to the better survival of this group of patients.
Whether the prognosis of the h-MDS patients differs from that of the NH-MDS patients remains unsettled. Our group previously reported that h-MDS predicted a favorable outcome in a cohort of 189 patients defined by the FAB classification [9], similar to the report by Yue et al [6], while other researchers did not observe such survival benefit [3, 4, 7, 8, 10, 17]. The reason of the variability in prognosis is unknown, but may be due to differences in the ethnic backgrounds, specific patient populations selected (age ranges and FAB subtypes, etc.) and treatment regimens. In this study, we showed that patients with h-MDS had a lower incidence of leukemia transformation and longer OS than those with NH-MDS. And, intriguingly, the prognostic relevance remained significant only in the lower-risk, but not in the higher-risk MDS patients. We postulate that this finding is of particular importance clinically, because although the IPSS and IPSS-R remain two of the most commonly used prognostic scoring systems for MDS [38], the median survival and risk of AML progression may still vary within the same risk group, especially the lower-risk ones [39]. There is clearly an unmet need to identify patients with IPSS/IPSS-R lower-risk MDS that may have an increased risk for disease progression and shortened survival. The fact that we distinctly showed BM hypocellularity is an independent favorable prognostic factor in the MDS patients and that BM cellularity alone could further stratify the lower-risk MDS patient into discrete prognostic subgroups, suggests that BM cellularity could potentially fulfill the unmet need in this group of patients.
In summary, we provide evidence that h-MDS are a distinct clinico-biological subgroup of MDS. h-MDS occur more frequently in patients with lower-risk MDS, have lower number of concurrent gene mutations, and are inversely associated with RUNX1, ASXL1, DNMT3A, EZH2, and TP53 mutations. We demonstrate that BM hypocellularity is an independent favorable prognostic factor which could further risk-stratify MDS patients, especially the lower-risk group. Further prospective studies in larger cohorts of MDS patients are warranted to validate the unique properties and prognostic relevance of h-MDS.
MATERIALS AND METHODS
Patients
A total of 369 adult patients diagnosed consecutively with de novo MDS according to the 2008 WHO classification [40] between 1990 and 2010 at the National Taiwan University Hospital were recruited. The clinical features, laboratory data, and BM findings were reviewed. This study was approved by the Institutional Review Board of the National Taiwan University Hospital; and written informed consents were obtained from all participants in accordance with the Declaration of Helsinki.
Bone marrow aspiration and biopsy
BM aspirate smears were routinely stained with modified Wright–Giemsa stain (Liu's stain) at our institution [41], and biopsy specimens were stained with hematoxylin-eosin after decalcification. BM hypoplasia was assessed based on BM biopsies, and defined as<30% cellularity in patients younger than 70 years and <20% cellularity in patients older than 70 years. The morphological criteria for BM dyspoiesis included megaloblastoid changes, irregularly shaped nuclei or karyorrhexis of erythroblasts, micromegakaryocytes, hypolobated or binucleated megakaryocytes, and pseudo Pelger-Huët anomaly or hypogranulation of granulocytes [40, 42]. Any of the dysplastic features should be present in at least 10% of that particular cell lineage in order to be considered significant.
Cytogenetics
BM cells were harvested directly or after 1-3 days of un-stimulated culture by procedures that had been described previously [43]. Metaphase chromosomes were banded by trypsin-Giemsa technique and karyotyped according to the International System for Human Cytogenetic Nomenclature [44].
Mutation analysis
Analyses of mutations in 17 relevant genes, including FLT3/ ITD [45], NRAS [45], KRAS [45], JAK2 [45], and RUNX1 [46], those genes related to epigenetic regulation, such as MLL/ PTD [47], ASXL1 [48], IDH1 [49], IDH2 [50], EZH2 [51], TET2 [52] and DNMT3A [53], and genes related to the RNA splicing machinery, such as SF3B1 [54], U2AF1 [55], and SRSF2 [56], as well as TP53[57] and SETBP1 [58], were performed as previously described. Please refer to Supplementary Table S1 for a detailed illustration of the sequences and genomic coordinates in these 17 genes. Abnormal sequencing results were confirmed by at least two repeated analyses.
Statistics
Mann-Whitney U test was employed to compare continuous variables such as age and hemogram between h-MDS and NH-MDS patients. Pearson’s chi-square test was utilized to detect the difference of nominal variables such as gender, WHO Classification, cytogenetic changes, IPSS and IPSS-R distribution and genetic alterations between h-MDS and NH-MDS patients. Fisher’s exact test was used as required. OS was measured from the date of initial diagnosis to the date of last follow-up or death from any cause. We used Cox proportional hazards model for multivariate regression analysis. Kaplan-Meier analysis with log-rank test was performed to evaluate the difference in acute leukemic transformation rate and OS between groups. P values less than 0.05 were considered statistically significant. All statistical analyses were performed with the SPSS 17 (SPSS Inc., Chicago, IL, USA) and Statsdirect (Cheshire, England, UK).
ACKNOWLEDGMENTS AND GRANT SUPPORT
This work was partially sponsored by grants MOST 100-2628-B-002-003-MY3, 103-2628-B-002-008-MY3, 104-2923-B-002-001 and 104-2314-B-002-128-MY4 from the Ministry of Science and Technology (Taiwan), MOHW105-TDU-B-211-134005 from the Ministry of Health and Welfare (Taiwan) and NTUH 102P06 from the Department of Medical Research, National Taiwan University Hospital. We would like to acknowledge the service provided by the DNA Sequencing Core of the First Core Laboratory, National Taiwan University College of Medicine.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
Author’s contributions
C.-Y.Y. was responsible for literature collection, data management and interpretation, statistical analysis and manuscript writing; H.-A.H. was responsible for study design and plan, literature collection, data management and interpretation, statistical analysis and manuscript writing; C.-Y.L. was responsible for statistical analysis and interpretation of the statistical findings; H.-A.H., W.-C.C., S.-J.W., X.-W.L., B.-S.K., C.-Y.C., S.-C.H., C.-C.L., S.-Y.H., M.Y., J.-L.T., W.T. and H.-F.T. contributed patient samples and clinical data; T.-Y.L., Y.-Y.K., M.-H.T., C.-F.H., Y.-C.C., C.-Y.L., F.-Y.L. and M.-C.L. performed the gene mutation and chromosomal studies, and H.-F.T. planned, designed, and coordinated the study over the entire period and wrote the manuscript.
REFERENCES
1. Tefferi A, Vardiman JW. Myelodysplastic Syndromes. N Engl J Med. 2009; 361:1872-1885.
2. Adès L, Itzykson R, Fenaux P. Myelodysplastic syndromes. Lancet. 2014; 383:2239-2252.
3. Nand S, Godwin JE. Hypoplastic myelodysplastic syndrome. Cancer. 1988; 62:958-964.
4. Maschek H, Kaloutsi V, Rodriguez-Kaiser M, Werner M, Choritz H, Mainzer K, Dietzfelbinger M, Georgii A. Hypoplastic myelodysplastic syndrome: incidence, morphology, cytogenetics, and prognosis. Ann Hematol. 1993; 66:117-122.
5. Toyama K, Ohyashiki K, Yoshida Y, Abe T, Asano S, Hirai H, Hirashima K, Hotta T, Kuramoto A, Kuriya S. Clinical and cytogenetic findings of myelodysplastic syndromes showing hypocellular bone marrow or minimal dysplasia, in comparison with typical myelodysplastic syndromes. Int J Hematol. 1993; 58:53-61.
6. Yue G, Hao S, Fadare O, Baker S, Pozdnyakova O, Galili N, Woda BA, Raza A, Wang SA. Hypocellularity in myelodysplastic syndrome is an independent factor which predicts a favorable outcome. Leuk Res. 2008; 32:553-558.
7. Tong WG, Quintas-Cardama A, Kadia T, Borthakur G, Jabbour E, Ravandi F, Faderl S, Wierda W, Pierce S, Shan J, Bueso-Ramos C, Kantarjian H, Garcia-Manero G. Predicting survival of patients with hypocellular myelodysplastic syndrome: development of a disease-specific prognostic score system. Cancer. 2012; 118:4462-4470.
8. Marisavljevic D, Cemerikic V, Rolovic Z, Boskovic D, Colovic M. Hypocellular myelodysplastic syndromes: clinical and biological significance. Med Oncol. 2005; 22:169-175.
9. Huang TC, Ko BS, Tang JL, Hsu C, Chen CY, Tsay W, Huang SY, Yao M, Chen YC, Shen MC, Wang CH, Tien HF. Comparison of hypoplastic myelodysplastic syndrome (MDS) with normo-/hypercellular MDS by International Prognostic Scoring System, cytogenetic and genetic studies. Leukemia. 2008; 22:544-550.
10. Nazha A, Seastone D, Radivoyevitch T, Przychodzen B, Carraway HE, Patel BJ, Carew J, Makishima H, Sekeres MA, Maciejewski JP. Genomic Patterns Associated with Hypoplastic Compared to Hyperplastic Myelodysplastic Syndromes. Haematologica. 2015; 100:e434-437.
11. Bejar R, Steensma DP. Recent developments in myelodysplastic syndromes. Blood. 2014; 124:2793-2803.
12. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, Bennett JM, Bowen D, Fenaux P, Dreyfus F, Kantarjian H, Kuendgen A, Levis A, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012; 120:2454-2465.
13. Yang YT, Hou HA, Liu CY, Lin CC, Chou WC, Lee FY, Liu MC, Liu CW, Tang JL, Yao M, Li CC, Kuo YY, Huang SY, et al. IPSS-R in 555 Taiwanese patients with primary MDS: Integration of monosomal karyotype can better risk-stratify the patients. Am J Hematol. 2014; 89:E142-149.
14. Bartl R, Frisch B, Baumgart R. Morphologic classification of the myelodysplastic syndromes (MDS): combined utilization of bone marrow aspirates and trephine biopsies. Leuk Res. 1992; 16:15-33.
15. Wong KF, So CC. Hypoplastic myelodysplastic syndrome-a clinical, morphologic, or genetic diagnosis? Cancer Genet Cytogenet. 2002; 138:85-88.
16. Garcia-Manero G. Prognosis of Myelodysplastic Syndromes. ASH Education Program Book. 2010; 2010:330-337.
17. Saad ST, Vassallo J, Arruda VA, Lorand-Metze I. The role of bone marrow study in diagnosis and prognosis of myelodysplastic syndrome. Pathologica. 1994; 86:47-51.
18. Chen B, Zhao WL, Jin J, Xue YQ, Cheng X, Chen XT, Cui J, Chen ZM, Cao Q, Yang G, Yao Y, Xia HL, Tong JH, et al. Clinical and cytogenetic features of 508 Chinese patients with myelodysplastic syndrome and comparison with those in Western countries. Leukemia. 2005; 19:767-775.
19. Lee JH, Lee JH, Shin YR, Lee JS, Kim WK, Chi HS, Park CJ, Seo EJ, Lee KH. Application of different prognostic scoring systems and comparison of the FAB and WHO classifications in Korean patients with myelodysplastic syndrome. Leukemia. 2003; 17:305-313.
20. Matsuda A, Germing U, Jinnai I, Araseki K, Kuendgen A, Strupp C, Iwanaga M, Miyazaki Y, Hata T, Bessho M, Gattermann N, Tomonaga M. Differences in the distribution of subtypes according to the WHO classification 2008 between Japanese and German patients with refractory anemia according to the FAB classification in myelodysplastic syndromes. Leuk Res. 2010; 34:974-980.
21. Tuzuner N, Cox C, Rowe JM, Watrous D, Bennett JM. Hypocellular myelodysplastic syndromes (MDS): new proposals. Br J Haematol. 1995; 91:612-617.
22. Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011; 364:2496-2506.
23. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, Shlien A, Groves MJ, Forbes SA, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013; 122:3616-3627.
24. Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, Morel P, Fenaux P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994; 84:3148-3157.
25. Kita-Sasai Y, Horiike S, Misawa S, Kaneko H, Kobayashi M, Nakao M, Nakagawa H, Fujii H, Taniwaki M. International prognostic scoring system and TP53 mutations are independent prognostic indicators for patients with myelodysplastic syndrome. Br J Haematol. 2001; 115:309-312.
26. Horiike S, Kita-Sasai Y, Nakao M, Taniwaki M. Configuration of the TP53 gene as an independent prognostic parameter of myelodysplastic syndrome. Leuk Lymphoma. 2003; 44:915-922.
27. Harada H, Harada Y, Kimura A. Implications of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome (MDS): future molecular therapeutic directions for MDS. Curr Cancer Drug Targets. 2006; 6:553-565.
28. Chen CY, Lin LI, Tang JL, Ko BS, Tsay W, Chou WC, Yao M, Wu SJ, Tseng MH, Tien HF. RUNX1 gene mutation in primary myelodysplastic syndrome—the mutation can be detected early at diagnosis or acquired during disease progression and is associated with poor outcome. Br J Haematol. 2007; 139:405-414.
29. Boultwood J, Perry J, Pellagatti A, Fernandez-Mercado M, Fernandez-Santamaria C, Calasanz MJ, Larrayoz MJ, Garcia-Delgado M, Giagounidis A, Malcovati L, Della Porta MG, Jadersten M, Killick S, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010; 24:1062-1065.
30. Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010; 42:665-667.
31. Lin J, Yao DM, Qian J, Chen Q, Qian W, Li Y, Yang J, Wang CZ, Chai HY, Qian Z, Xiao GF, Xu WR. Recurrent DNMT3A R882 mutations in Chinese patients with acute myeloid leukemia and myelodysplastic syndrome. PLoS One. 2011; 6:e26906.
32. Thol F, Friesen I, Damm F, Yun H, Weissinger EM, Krauter J, Wagner K, Chaturvedi A, Sharma A, Wichmann M, Gohring G, Schumann C, Bug G, et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol. 2011; 29:2499-2506.
33. Thol F, Winschel C, Ludeking A, Yun H, Friesen I, Damm F, Wagner K, Krauter J, Heuser M, Ganser A. Rare occurrence of DNMT3A mutations in myelodysplastic syndromes. Haematologica. 2011; 96:1870-1873.
34. Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, Fulton R, Schmidt H, Kalicki-Veizer J, O'Laughlin M, Kandoth C, Baty J, Westervelt P, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011; 25:1153-1158.
35. Xu F, Li X. The role of histone methyltransferase EZH2 in myelodysplastic syndromes. Expert Rev Hematol. 2012; 5:177-185.
36. Saft L, Karimi M, Ghaderi M, Matolcsy A, Mufti GJ, Kulasekararaj A, Gohring G, Giagounidis A, Selleslag D, Muus P, Sanz G, Mittelman M, Bowen D, et al. p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q). Haematologica. 2014; 99:1041-1049.
37. Tsai SC, Shih LY, Liang ST, Huang YJ, Kuo MC, Huang CF, Shih YS, Lin TH, Chiu MC, Liang C. Biological activities of RUNX1 mutants predict secondary acute leukemia transformation from chronic myelomonocytic leukemia and myelodysplastic syndromes. Clin Cancer Res. 2015; 21:3541-3551.
38. Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, Sanz M, Vallespi T, Hamblin T, Oscier D, Ohyashiki K, Toyama K, Aul C, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997; 89:2079-2088.
39. Garcia-Manero G, Shan J, Faderl S, Cortes J, Ravandi F, Borthakur G, Wierda WG, Pierce S, Estey E, Liu J, Huang X, Kantarjian H. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008; 22:538-543.
40. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellstrom-Lindberg E, Tefferi A, Bloomfield CD. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009; 114:937-951.
41. Tien HF, Wang CH, Chen YC, Shen MC, Lin DT, Lin KH. Characterization of acute myeloid leukemia (AML) coexpressing lymphoid markers: different biologic features between T-cell antigen positive and B-cell antigen positive AML. Leukemia. 1993; 7:688-695.
42. Vardiman JW. Hematopathological concepts and controversies in the diagnosis and classification of myelodysplastic syndromes. Hematology Am Soc Hematol Educ Program. 2006; 2006:199-204.
43. Tien HF, Tang JH, Tsay W, Liu MC, Lee FY, Wang CH, Chen YC, Shen MC. Methylation of the p15(INK4B) gene in myelodysplastic syndrome: it can be detected early at diagnosis or during disease progression and is highly associated with leukaemic transformation. Br J Haematol. 2001; 112:148-154.
44. Mitelman F. (1995). ISCN: an international system for human cytogenetic nomenclature: Basel: S. Karger.
45. Hou HA, Huang TC, Lin LI, Liu CY, Chen CY, Chou WC, Tang JL, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Yao M, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood. 2010; 115:5222-5231.
46. Tang JL, Hou HA, Chen CY, Liu CY, Chou WC, Tseng MH, Huang CF, Lee FY, Liu MC, Yao M, Huang SY, Ko BS, Hsu SC, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009; 114:5352-5361.
47. Shiah HS, Kuo YY, Tang JL, Huang SY, Yao M, Tsay W, Chen YC, Wang CH, Shen MC, Lin DT, Lin KH, Tien HF. Clinical and biological implications of partial tandem duplication of the MLL gene in acute myeloid leukemia without chromosomal abnormalities at 11q23. Leukemia. 2002; 16:196-202.
48. Chen TC, Hou HA, Chou WC, Tang JL, Kuo YY, Chen CY, Tseng MH, Huang CF, Lai YJ, Chiang YC, Lee FY, Liu MC, Liu CW, et al. Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood Cancer J. 2014; 4:e177.
49. Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, Chang YC, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010; 115:2749-2754.
50. Lin CC, Hou HA, Chou WC, Kuo YY, Liu CY, Chen CY, Lai YJ, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Liu CW, et al. IDH mutations are closely associated with mutations of DNMT3A, ASXL1 and SRSF2 in patients with myelodysplastic syndromes and are stable during disease evolution. Am J Hematol. 2014; 89:137-144.
51. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010; 42:722-726.
52. Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, Lee MC, Ko BS, Tang JL, Yao M, Tsay W, Wu SJ, Huang SY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011; 118:3803-3810.
53. Hou HA, Kuo YY, Liu CY, Chou WC, Lee MC, Chen CY, Lin LI, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Liu CW, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012; 119:559-568.
54. Lin CC, Hou HA, Chou WC, Kuo YY, Wu SJ, Liu CY, Chen CY, Tseng MH, Huang CF, Lee FY, Liu MC, Liu CW, Tang JL, et al. SF3B1 mutations in patients with myelodysplastic syndromes: the mutation is stable during disease evolution. Am J Hematol. 2014; 89:E109-115.
55. Hou HA, Liu CY, Kuo YY, Chou WC, Tsai CH, Lin CC, Lin LI, Tseng MH, Chiang YC, Liu MC, Liu CW, Tang JL, Yao M, et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget. 2016; 7:9084-9101. doi: 10.18632/oncotarget.7000.
56. Wu SJ, Kuo YY, Hou HA, Li LY, Tseng MH, Huang CF, Lee FY, Liu MC, Liu CW, Lin CT, Chen CY, Chou WC, Yao M, et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood. 2012; 120:3106-3111.
57. Hou HA, Chou WC, Kuo YY, Liu CY, Lin LI, Tseng MH, Chiang YC, Liu MC, Liu CW, Tang JL, Yao M, Li CC, Huang SY, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015; 5:e331.
58. Hou HA, Kuo YY, Tang JL, Chou WC, Yao M, Lai YJ, Lin CC, Chen CY, Liu CY, Tseng MH, Huang CF, Chiang YC, Lee FY, et al. Clinical implications of the SETBP1 mutation in patients with primary myelodysplastic syndrome and its stability during disease progression. Am J Hematol. 2014; 89:181-186.