
Introduction
The transcription factor Zinc finger protein 148 (Zfp148, ZBP-89, BFCOL, BERF1, htβ) interacts physically with the tumor suppressor p53, but the significance of this interaction is not known [1]. We have recently shown that Zfp148 deficiency leads to ectopic activation of p53 in mice and cultured fibroblasts, suggesting that Zfp148 represses p53 activity [2]. Knockout of Zfp148 leads to respiratory distress and partial neonatal lethality in mice that are caused by proliferative arrest of pulmonary cells, and to premature senescence in cultured mouse embryonic fibroblasts. The phenotypes are rescued by deletion of one or two copies of Trp53 (the gene encoding mouse p53). Moreover, loss of one copy of Zfp148 reduces proliferation of tissue macrophages and atherosclerosis in Apoe-/- mice by increasing p53 activity [3]. Otherwise mice lacking one copy of Zfp148 are fertile and healthy [2, 4].
Several studies show that Zfp148 is required for the integrity of intestinal epithelium suggesting a possible link to colorectal cancer (CRC) [5-8]. Moreover, Zfp148 expression is increased in human CRC compared to normal mucosa [9]. CRC is one of the most common cancer forms and a leading cause of cancer death in the Western world [10]. 80 percent of CRCs are caused by inherited or somatic mutations in the adenomatous polyposis coli (APC) gene [11]. APC mutations give rise to adenomatous polyps that progress to carcinomas, a process driven by additional mutations including p53-mutations [12]. The APCMin/+ mouse harbours a mis-sense mutation in the APC locus which leads to the production of a truncated APC protein [13]. The mutated APC protein lacks the β-catenin binding domain, which leads to an accumulation of β-catenin in the nucleus and activation of β-catenin target genes [14]. APCMin/+ mice develop numerous adenomatous polyps, predominantly in the small intestine, which leads to intestinal bleedings, anemia and death at about 30 weeks of age [13]. The model is therefore appropriate for studies of early-stage CRC.
Our finding that Zfp148 deficiency activates p53 raises the possibility that Zfp148 promotes cancer by repressing p53 activity. In line with this, high expression of Zfp148 in oesophageal squamous cell cancer and clear cell renal cell carcinoma correlates with cancer progression and poor prognosis [15, 16]. However, the role of Zfp148 and its impact on p53 activity in cancer remains largely unknown. Here we address the role of Zfp148 in the APCMin/+ model of CRC, and hypothesize that reduced expression of Zfp148 confers protection against intestinal adenomas by unleashing p53 activity. For this, we crossed ApcMin/+ mice with mice lacking Zfp148 (Zfp148gt/gt and Zfp148gt/+) and studied the development of intestinal adenomas.
Results
We established Zfp148gt/+ and Zfp148gt/gt mice on the APCMin/+ genetic background (from here on designated Zfp148gt/+ and Zfp148gt/gt mice, respectively) to investigate if Zfp148 is involved in intestinal adenoma formation. Intestines from 12 week old mice were sectioned and stained with haematoxylin and eosin for evaluation of tumor development. Deletion of one or two copies of Zfp148 markedly reduced tumor numbers compared to APCMin/+ controls, without significantly altering the anatomical distribution of tumors (Figure 1A, 1B). Moreover, the tumors in Zfp148gt/gt mice were of lower histopathogical grade compared to APCMin/+ controls (Figure 1C). Thus, Zfp148 is required for the formation of intestinal adenomas.
APCMin/+ mice primarily die from secondary phenotypes including severe, chronic anaemia [17]. Since levels of haemoglobin and hematocrit were higher in Zfp148gt/+ and Zfp148gt/gt mice compared to controls, and the frequency of intestinal bleedings was lower (Supplementary Figure 1A-1C), we argued that deletion of Zfp148 may extend the survival of these mice. Indeed, loss of a single copy of Zfp148 extended the median survival time of ApcMin/+ mice with 69%, from 39 to 66 weeks (Figure 1D). Because the majority of Zfp148gt/gt mice die before 6 months of age, most from respiratory distress and some for unknown reasons [2], we did not set up Zfp148gt/gt mice in the survival study or in mechanistic studies since the results could be skewed by confounding phenotypes.
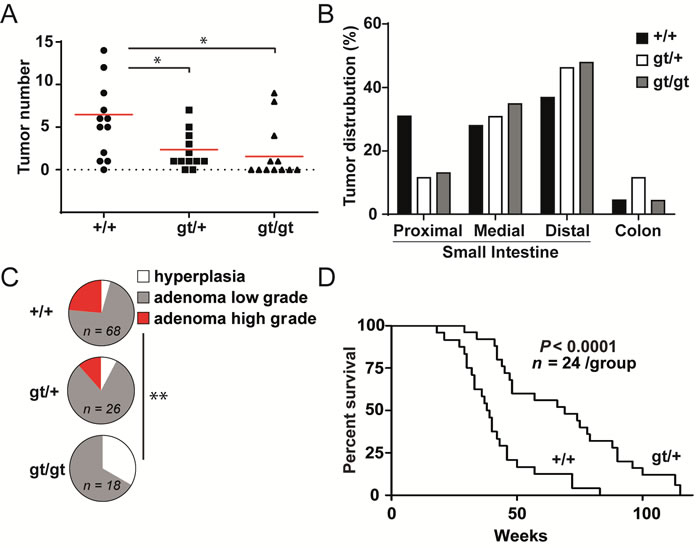
Figure 1: Zfp148 deficiency reduces tumor growth in mice with ApcMin/+-induced intestinal adenomas. A. Tumor count in intestines from ApcMin/+, ApcMin/+Zfp148gt/+, and ApcMin/+Zfp148gt/gt mice at 12 weeks of age (n = 12). B. Percentage of tumors located in proximal, medial and distal third of small intestines and colon of ApcMin/+, ApcMin/+Zfp148gt/+, ApcMin/+Zfp148gt/gt mice at 12 weeks of age. C. Tumor stage (hyperplasia, low grade adenoma, high grade adenoma) in intestines from ApcMin/+, ApcMin/+Zfp148gt/+, ApcMin/+Zfp148gt/gt mice at 12 weeks of age (n = 23-68 tumors in intestines from 12 mice per group). D. Kaplan-Meier plot showing overall survival of ApcMin/+ and ApcMin/+Zfp148gt/+ mice (n = 24 per group). Data are represented as mean ± SEM. *P < 0.05, **P < 0.01.
The extended survival and lower tumor burden of Zfp148-deficient mice raise the possibility that Zfp148 deficiency reduces tumor growth rate. However, as demonstrated by bromodeoxyuridine (BrdU) incorporation, expression of proliferating nuclear cell antigen (PCNA), and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), there was no difference in cell proliferation or apoptosis of tumor cells or normal crypt epithelial cells between 12 weeks old Zfp148gt/+ mice and controls, respectively (Figure 2A-2D and Supplementary Figure 2A, 2B). In accord with this, there was no difference in levels of total or phosphorylated p53 (Figure 2E). Thus, the lower tumor burden of Zfp148-deficient mice is not caused by reduced growth rate. However, the p53-activity was increased in tumors from old Zfp148gt/+ mice (24-26 weeks old) compared to age-matched controls, raising the possibility that Zfp148 deficiency reduces progression of advanced tumors (Figure 2F).
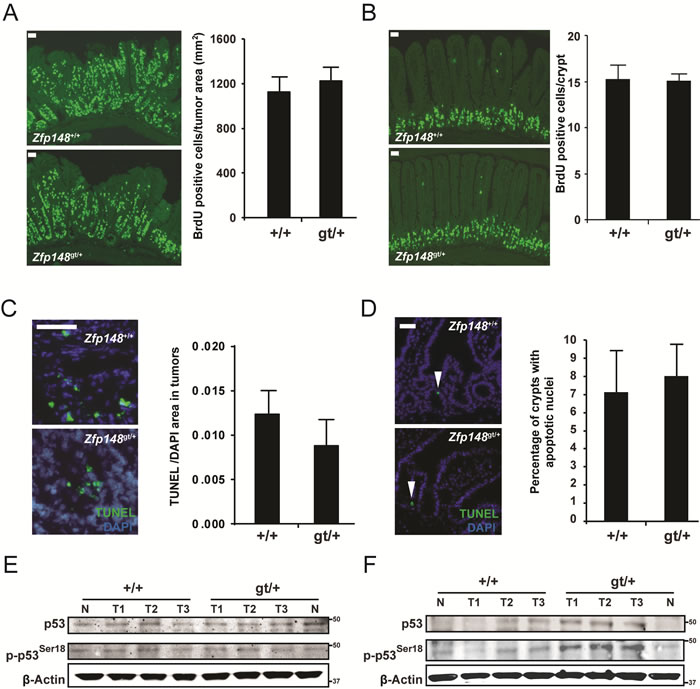
Figure 2: Tumor growth rate is not affected in ApcMin/+Zfp148gt/+ mice. A.-B. Left: Representative immunofluorescence micrographs showing BrdU-positive cells in tumor tissue A. and normal intestinal epithelium B. in ApcMin/+and ApcMin/+Zfp148gt/+ mice. BrdU was injected into the peritoneal cavity 2 hours before the mice were sacrificed. Right: Quantification of BrdU-positive cells in tumor tissue (A; BrdU-positive cells per tumor area; n = 11-18 tumors in intestines from 5-8 mice per group) and normal intestinal epithelium (B; average number of BrdU-positive cells per crypt; n = 60 crypts per mouse in 5-7 mice per group). C.-D. Left: Immunofluorescence micrograph showing TUNEL-positive cells (green) in tumor tissue C. and normal intestinal epithelium D. in ApcMin/+and ApcMin/+Zfp148gt/+ mice. Nuclei are stained blue with DAPI (4’,6-diamidino-2-phenylindole). Right: Quantification of TUNEL-positive cells in tumor tissue (C; TUNEL-positive area per DAPI-positive area; n = 8-9 tumors per group) and normal intestinal epithelium (D; percentage of crypts with TUNEL-positive cells; n = 6-7 mice per group). E.-F. Western blots of phosphorylated and total p53 in tumors dissected from 12 weeks old E. and 24 - 26 weeks old F. ApcMin/+Zfp148gt/+ and ApcMin/+mice (n = 3). β-Actin was used as loading control. Data are represented as mean ± SEM. *P < 0.05, **P < 0.01. Scale bars, 50µm.
An alternative possibility is that Zfp148 deficiency affects expression of key genes in the crypt epithelium where new tumors are initiated. Tumor initiation is driven by β-catenin and suppressed by p53 [14, 18]. To address this, we used laser capture microscopy and gene expression arrays to extract transcriptional profiles from normal crypts (Figure 3A). However, we could not detect any concordant effects of Zfp148 deficiency on β-catenin or p53 signaling in normal crypts, as judged by expression analysis of individual β-catenin or p53 target genes or gene set enrichment analysis (GSEA) of known signaling pathways (Figure 3B, 3C and Supplementary Table 1). Moreover, immunohistological analysis did not reveal any difference in the amount or subcellular localization of β-catenin between Zfp148gt/+intestines and controls (Figure 3D). Thus, Zfp148-decifiency does not affect the activation of β-catenin or p53 in normal crypts.
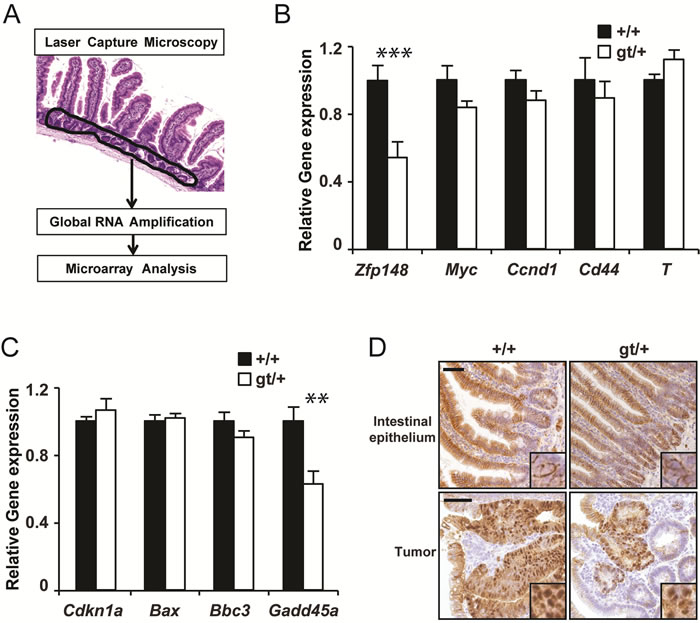
Figure 3: Zfp148 deficiency does not affect basal expression of p53 or β-catenin target genes in crypt epithelium. A. Schematic representation of the gene expression analysis procedure. B.-C. mRNA levels of Zfp148 and four p53-target genes B. and four β-catenin target genes C. in laser microdissected crypt epithelium from ApcMin/+and ApcMin/+Zfp148gt/+ mice at 12 weeks of age (n = 6). D. Representative immunohistochemistry micrographs showing β-catenin (brown) in normal intestinal epithelium (top) and in tumors (bottom) in ApcMin/+and ApcMin/+Zfp148gt/+ mice at 12 weeks of age. Inserts show subcellular localization of β-catenin. Sections are counter stained with Mayers hematoxylin. Data are represented as mean ± SEM. *P < 0.05, **P < 0.01. Scale bars, 50µm.
Tumor initiation is triggered by loss of the second APC allele which leads to constitutive activation of β-catenin. To test whether Zfp148 plays a role downstream of activated β-catenin, we treated intestinal explants from Zfp148gt/+ mice and APCMin/+ controls with a pharmacological inhibitor of glycogen synthase kinase 3β (GSK3β) for 6 and 20 hours. Phosphorylation of β-catenin by GSK3β is required for proteasomal destruction of β-catenin and inhibition of GSK3β activates β-catenin.
As demonstrated by the induction of β-catenin target genes, β-catenin was activated in the explants 6 hours after GSK3β inhibition, but did not affect the expression of p53 target genes at this time point (Figure 4A and Supplementary Figure 3A, 3B). However, p53 targets were robustly induced in explants from Zfp148gt/+ mice at 20 hours after GSK3β inhibition (Figure 4B). Importantly, GSK3β inhibition had no effect on p53 target genes in APCMin/+ controls. Since expression of p53 targets was higher in untreated Zfp148gt/+ explants that were cultured for 20 hours compared to controls (Figure 4A), culture-stress likely contributed to p53-activation in Zfp148gt/+ explants.
In accord with this, protein levels of p53 and p53 target genes and the apoptosis marker cleaved Parp1 were markedly higher in explants from Zfp148gt/+ mice compared to controls (Figure 4C). Collectively, the results suggest that Zfp148 deficiency increases p53 activity in response to constitutively activated β-catenin during tumor initiation, and that newly transformed tumor cells are eliminated by p53-induced apoptosis in Zfp148-deficient mice.
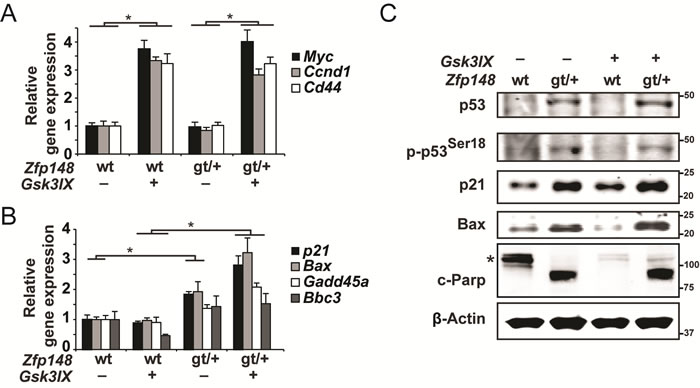
Figure 4: Constitutive activation of β-catenin induces p53-activation and apoptosis in small intestine explants from ApcMin/+Zfp148gt/+ mice. A.-B. Taqman RT-PCR assessment of mRNA levels of three β-catenin target genes A. and four p53-target genes B. in small intestine explants that were dissected from ApcMin/+and ApcMin/+Zfp148gt/+ mice and treated with 2µM of the GSK3β-inhibitor GSK3IX or DMSO. The explants in A. were treated for 6 hours and those in B. for 20 hours (n = 6). C. Western blots of phosphorylated and total p53, the p53 targets p21 and Bax, and the apoptosis marker cleaved Parp1 (89-kD fragment) in small intestine explants that were dissected from ApcMin/+and ApcMin/+Zfp148gt/+ mice and treated with 2µM GSK3IX or DMSO for 20 hours. Asterisk indicates uncleaved Parp1 (116-kD fragment). β-Actin was used as loading control. Data are represented as mean ± SEM. *P < 0.05, **P < 0.01.
To test this idea, Zfp148gt/+ mice were bred onto Trp53+/- and Trp53-/- genetic backgrounds and evaluated for tumor development at 12 weeks of age. There was no difference in tumor count between Zfp148gt/+ and ApcMin/+ controls on Trp53+/- or Trp53-/- genetic backgrounds (Figure 5A). Moreover, levels of haemoglobin and hematocrit were indistinguishable between Zfp148gt/+ and ApcMin/+ controls on Trp53+/- background (Figure 5B, 5C). We conclude that tumor development is restored by deletion of one or both copies of Trp53, and that Zfp148 deficiency suppresses tumor formation by increasing p53 activity. Survival of Zfp148gt/+ mice on p53-deficient background was not investigated since Trp53+/- and Trp53-/- mice develop other types of neoplasia that may confound the result [19, 20].
We finally investigated whether p53 is inactivated by somatic mutations in Zfp148-deficient tumors. We did not find any mutations in exons 5-8 of the Trp53 gene in 5+5 tumors from Zfp148gt/+ mice on Trp53+/+ and Trp53+/- background, respectively. Since exons 5-8 harbour 90 percent of all p53-mutations in human cancers [21, 22], we conclude that functional p53 is retained in the majority of Zfp148-deficient tumors.
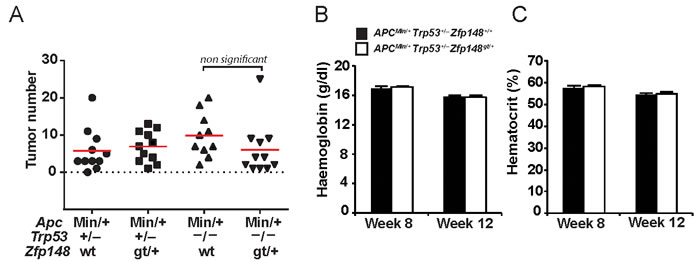
Figure 5: Deletion of one or two copies of Trp53 restores tumor growth in ApcMin/+Zfp148gt/+ mice. A. Tumor count in small intestine of ApcMin/+Trp53+/-, ApcMin/+Zfp148gt/+ Trp53+/-, ApcMin/+Trp53-/- and ApcMin/+Zfp148gt/+ Trp53-/- mice at 12 weeks of age (n = 11). B.-C. Levels of haemoglobin B. and hematocrit C. in blood from ApcMin/+Trp53+/- and ApcMin/+Zfp148gt/+ Trp53+/- mice at 8 and 12 weeks of age (n = 11). Data are represented as mean ± SEM.
Discussion
In this study we show for the first time that Zfp148 deficiency reduces tumor development. Loss of one copy of Zfp148 markedly reduced tumor frequency in the APCMin/+ model of colorectal carcinoma, extended survival of the mice with 69%, and reduced secondary manifestations of the disease including anaemia and intestinal bleedings. The effect was entirely dependent upon functional p53 showing that Zfp148 deficiency reduces tumor development by increasing p53 activity.
Zfp148 deficiency had no effect on cell proliferation or apoptosis in early tumors indicating that Zfp148 does not affect tumor growth rate. Instead, we propose that Zfp148 controls the fate of newly transformed tumor cells by setting a threshold for p53 activation in response to constitutively activated β-catenin. The idea is supported by two lines of evidence. First, levels of phosphorylated and total p53, expression of p53-target genes, and the apoptosis marker cleaved Parp-1 were up-regulated in intestinal explants from Zfp148gt/+ mice in response to pharmacological inhibition of GSK3β. The same treatment had no effect on p53-activity or apoptosis markers in explants from control mice. And second, the effect of Zfp148 deficiency on tumor development was abolished by deletion of one or both copies of Trp53, the gene encoding p53.
Since western blotting revealed increased p53 activity in later-stage adenomas from Zfp148gt/+ mice, Zfp148 deficiency may also reduce progression of later-stage tumors. Thus, the data suggest a model where Zfp148 promotes tumor initiation, has no function early on in progression, and may be reactivated in later stage tumors.
The rescue of tumor development by deletion of one copy of Trp53 is in line with previous data. Breeding onto a Trp53+/- background rescued macrophage proliferation and atherosclerosis in Zfp148gt/+ mice and lung development and neonatal lethality in Zfp148gt/gt mice [2, 3]. And it restored normal cell proliferation and expression of p53-target genes in Zfp148gt/gt MEFs [2]. Thus, reducing p53-expression is sufficient to prevent activation of p53 effector pathways in Zfp148-deficient mice.
We did not detect any somatic p53 mutations in tumors from Zfp148-deficient mice in spite of strong p53-dependent suppression of tumor development. One possible explanation is that p53-activity is transiently increased in these mice during tumor initiation by loss of APC, and that p53 activity is low at other time points, leaving no time for selection of somatic mutations. In support of this, there was no difference in cell proliferation, apoptosis, or levels of total and phosphorylated p53 in tumors from 12 weeks old Zfp148gt/+ mice compared to controls. Moreover, knockout of p53 has no or little impact on tumor development in APCMin/+ mice on Zfp148-wildtype background, suggesting that the selective pressure for accumulating p53 mutations is low in this model [23-25].
The role of Znf148 (human Zfp148 homologue) in human CRC was recently investigated [9]. In familial adenomatous polyposis, expression of Znf148 increased from normal mucosa to adenomas to carcinomas, thus supporting the possibility that Znf148 promotes tumor initiation. Similar results were obtained in sporadic CRC with increased expression in adenomas and carcinomas compared to normal mucosa. Interestingly, the data suggests that Znf148 may play a different role in advanced CRC since Znf148 expression in carcinomas was inversely correlated with TNM stage and patient survival. However, the significance of Zfp148 in advanced tumors remains to be tested in mouse models that develop metastatic CRC.
Cancer chemoprevention is often cited as an effective strategy to reduce cancer mortality. Our finding that global deletion of one copy of Zfp148 reduces tumor initiation is therefore clinically interesting. Since mice lacking one copy of Zfp148 appear to be healthy and have a normal lifespan, our finding suggests that therapeutic targeting of Zfp148 could reduce the incidence of CRC by increasing p53 activity without causing detrimental side effects. However, transcription factors such as Zfp148 are not ideal drug targets although significant progress has been made in this field. A more realistic approach may be to target other proteins in the Zfp148-p53 pathway.
The mechanism by which Zfp148 regulates p53 activity is not known [2, 3]. The physical interaction of Zfp148 with p53 raises the intriguing possibility that Zfp148 might regulate p53 directly. However, Zfp148 deficiency up-regulates Cdkn2a in cultured cells opening up for an alternative mechanism [2, 26]. Since deletion of Wip1 suppresses APCMin-driven polyposis by increasing p53 activity in response to constitutively activated β-catenin [18], it is possible that Wip1 and Zfp148 operate in the same pathway. Defining the mechanism by which Zfp148 suppresses p53 activation in the intestinal epithelium should be a prioritized area for future studies.
In this study we show that Zfp148 deficiency reduces tumor formation in the APCMin/+ model. CRC is one of the most prevalent cancer forms in the Western world and a leading cause of cancer-related death. Mutations in the APC gene is a major causative event of CRC in humans [12] and identification of modifier genes that affect cancer outcome in the APCMin/+ model is of clinical interest. Because deletion of Zfp148 reduces the initiation of tumors, preventive targeting of Zfp148 may be efficient to reduce the incidence of CRC and thereby CRC mortality.
Materials and methods
Mouse breeding
ApcMin/+ and Trp53tm1Tyj (Trp53-/) mice were obtained from The Jackson Laboratory and Zfp148gt/+ mice were produced by us [2]. The mice were kept on a 129/Bl6 mixed genetic background and all experiments were performed with littermate controls. Genotyping was performed by PCR amplification of genomic DNA from mouse tail biopsies. PCR primers used for genotyping are listed in Supplementary Table 2. Mice were fed on a regular diet and had unlimited supply of food and water. All animal procedures used in this study were approved by The Animal Research Ethics Committee in Gothenburg.
Histological analyses
Adenoma frequency was compared in 12 week APCMin/+/Zfp148+/+, APCMin/+/Zfp148gt/+ and APCMin/+/Zfp148gt/gt mice. Mice were dissected, the intestines were removed and separated into four segments; colon and three segments of the small intestines. The segments were rinsed in PBS and prepared using the Swiss roll technique [27]. Tissues were fixed in 4% formaldehyde, imbedded in paraffin and 5 µm thin sections were stained with Haematoxylin and Eosin. Adenoma counts were performed on one single sagittal section from each segment by investigator blinded to the genotype. Histopathological grade was assessed by a pathologist (O.N.).
Survival study
Mice were sacrificed when they became moribund, defined as when they became listless, their haematocrit was below 20%, or their bodyweight was reduced by 15%.
Blood analyses
Blood was drawn from the tail into EDTA-coated tubes (Monovette, Sarstedt). Samples were analysed on a Hemato analyser KX-21N (Sysmex) to determine haemoglobin, hematocrit and leukocyte count.
Gastrointestinal bleeding
Faecal blood was detected by using Hemoccult test for faecal blood (Beckman-Coulter).
Immunohistochemistry
Intestinal tissues were fixed in 4% formaldehyde, imbedded in paraffin and sectioned in 5 um thin sections. Staining was performed using standard protocol. Slides were boiled for 10 min in Citric acid (10 mM, pH = 6) to unmask epitopes. Primary antibody anti-PCNA (Santa Cruz, SC-7907) was diluted 1:1000 and secondary antibody (anti-rabbit-Alexa 546, Molecular Probes) was diluted 1:500 and primary antibody anti-β-catenin (BD, 610154), was diluted 1:200 and visualized with Vectastain Elite ABCkit , Peroxidase, Rabbit IgG, (Vector Laboratories) as before [28].
Cell proliferation and apoptosis
Apoptosis was evaluated with Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) using the ApopTag® Fluorescence In Situ Apoptosis Detection Kit (Millipore). Cell proliferation was evaluated with 5-Bromo-2’-deoxy-uridine (BrdU) Labeling and Detection Kit I (Roche) 2h after intraperitoneal injection of BrdU (75mg/kg) according to manufacturer’s description and as before [2, 28].
Gene expression analysis
Total RNA was extracted from crypt-enriched tissues that were isolated by laser microdissection (PALM) from 10µm thick cryosections of OCT-embedded and snap frozen Swiss roll preparations of small intestines (proximal). RNA concentration was measured with ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) and RNA quality was evaluated using the Agilent 2100 Bioanalyzer system (Agilent Technologies Inc, Palo Alto, CA). 250 nanograms of total RNA from each sample were used to generate amplified and biotinylated sense-strand cDNA from the entire expressed genome according to the GeneChip® WT PLUS Reagent Kit User Manual ( P/N 703174 Rev 1 Affymetrix Inc., Santa Clara, CA). GeneChip® ST Arrays ( GeneChip® XXX Gene 2.0 ST Array) were hybridized for 16 hours in a 45°C incubator, rotated at 60 rpm. According to the GeneChip® Expression Wash, Stain and Scan Manual (PN 702731 Rev 3, Affymetrix Inc., Santa Clara, CA) the arrays were then washed and stained using the Fluidics Station 450 and finally scanned using the GeneChip® Scanner 3000 7G. The gene expression data set has been deposited in the GEO repository with accession number GSE77773.
Microarray data analysis
The raw data was normalized in the free software Expression Console provided by Affymetrix (http://www.affymetrix.com) using the robust (RMA) method first suggested by Li and Wong in 2001 [29, 30]. Subsequent analysis of the gene expression data was carried out in the freely available statistical computing language R (http://www.r-project.org) using packages available from the Bioconductor project (www.bioconductor.org). In order to search for the differentially expressed genes between APCMin/+ and the Zfp148gt/+ groups an empirical Bayes moderated t-test was then applied [31], using the ‘limma’ package [32]. To address the problem with multiple testing, the p-values were adjusted using the method of Benjamini and Hochberg [33].
Gene set enrichment analyses (GSEA)
Differential expression of gene sets was analyzed with the GSEA V2.1.0 software (Broad institute) using gene sets from the KEGG (Kyoto Encyclopedia of Genes and Genomes) repository (c2.cp.kegg.v4.0.symbols.gmt) and default settings.
Intestinal explant experiments
One 2 cm segment of the distal ileum was collected, as previously described [34]. Intestinal contents from the segment were removed through flushing with cold PBS and the segment was divided into four equal longitudinal parts. Each part was placed in cell culture plates containing growth medium (Dulbecco’s modified Eagle’s medium with glutamine and pyruvate, 4.5 g/l glucose, 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin) with or without 2µM GSK3IX inhibitor (Santa Cruz). The plates were then incubated 6-20h at 37°C in 5% CO2. The mixture containing the tissue was transferred to Eppendorf tubes and centrifuged at 14,000 rpm for 5 min. The tissue was collected after removal of the supernatant and used for analysis of mRNA or protein expression. All explant experiments were performed on APCMin/+ background.
Real-time reverse transcription polymerase chain reaction (RT-PCR)
TaqMan assays were performed as described [35] using TaqMan universal polymerase chain reaction mastermix (Life Technologies) and the predesigned TaqMan assays (Life Technologies) listed in Supplementary Table 2.
Western blot analyses
Protein levels was determined as previously described [36] with antibodies against p53 (SC-6243, 1:500, Santa Cruz), p-p53ser15 (9284S 1:500, Cell Signal), p21 (SC-6246, 1:1000, Santa Cruz), BAX (SC-493, 1:500, Santa Cruz), c-PARP (9542 S 1:500, Cell Signal), and β-actin (2228, 1:20000, Sigma Aldrich).
Trp53 sequencing
Exons 5-8 of mouse Trp53 were PCR amplified from genomic DNA isolated from dissected (Zfp148gt/+Trp53+/+) or laser microdissected (Zfp148gt/+Trp53+/) tumors and subjected to Sanger sequencing. Each amplicon was sequenced twice and scanned for deviations from the refseq sequence. Sequencing was performed by Beckman Coulter Genomics using primers listed in Supplementary Table 2.
Statistical analyses
Statistics were performed with non-parametric Mann-Whitney test for tumor count; Chi Square test for tumor distribution and histopathological grade; ANOVA test for haemoglobin; t-test for hematocrit, cell proliferation, apoptosis, and differential gene expression; and Fisher’s exact test for gastrointestinal bleeding. Survival curves were generated using the Kaplan-Meier estimator and statistical significance was calculated using the log rank test (Mantel-Cox). Investigators were blinded to the genotype and values are presented as mean ± SEM. A p-value of < 0.05 was considered statistically significant.
Acknowledgments
The project was supported by the Swedish Research Council, Polysackaridforskning AB, the Swedish Cancer Foundation, the Swedish Heart-Lung Foundation, the Sahlgrenska University Hospital ALF research grants, the University of Gothenburg (to P. Lindahl), Assar Gabriel Foundation and the Swedish Research Council (to V.I. Sayin).
Conflicts of Interest
The authors have no conflict of Interest to declare.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Bai L and Merchant JL. ZBP-89 promotes growth arrest through stabilization of p53. Mol Cell Biol. 2001; 21:4670-4683.
2. Sayin VI, Nilton A, Ibrahim MX, Agren P, Larsson E, Petit MM, Hulten LM, Stahlman M, Johansson BR, Bergo MO and Lindahl P. Zfp148 deficiency causes lung maturation defects and lethality in newborn mice that are rescued by deletion of p53 or antioxidant treatment. PloS one. 2013; 8:e55720.
3. Sayin VI, Khan OM, Pehlivanoglu LE, Staffas A, Ibrahim MX, Asplund A, Agren P, Nilton A, Bergstrom G, Bergo MO, Boren J and Lindahl P. Loss of One Copy of Zfp148 Reduces Lesional Macrophage Proliferation and Atherosclerosis in Mice by Activating p53. Circulation research. 2014.
4. Nilton A, Sayin VI, Staffas A, Larsson E, Rolf J, Petit MM, Palmqvist L, Swolin B, Cardell S and Lindahl P. Zinc finger protein 148 is dispensable for primitive and definitive hematopoiesis in mice. PloS one. 2013; 8:e70022.
5. Bai L, Kao JY, Law DJ and Merchant JL. Recruitment of ataxia-telangiectasia mutated to the p21(waf1) promoter by ZBP-89 plays a role in mucosal protection. Gastroenterology. 2006; 131:841-852.
6. Law DJ, Labut EM, Adams RD and Merchant JL. An isoform of ZBP-89 predisposes the colon to colitis. Nucleic Acids Res. 2006; 34:1342-1350.
7. Law DJ, Labut EM and Merchant JL. Intestinal overexpression of ZNF148 suppresses ApcMin/+ neoplasia. Mammalian genome. 2006; 17:999-1004.
8. Essien BE, Grasberger H, Romain RD, Law DJ, Veniaminova NA, Saqui-Salces M, El-Zaatari M, Tessier A, Hayes MM, Yang AC and Merchant JL. ZBP-89 regulates expression of tryptophan hydroxylase I and mucosal defense against Salmonella typhimurium in mice. Gastroenterology. 2013; 144:1466-1477, 1477 e1461-1469.
9. Gao XH, Liu QZ, Chang W, Xu XD, Du Y, Han Y, Liu Y, Yu ZQ, Zuo ZG, Xing JJ, Cao G and Fu CG. Expression of ZNF148 in different developing stages of colorectal cancer and its prognostic value: a large Chinese study based on tissue microarray. Cancer. 2013; 119:2212-2222.
10. Nelson RS and Thorson AG. Colorectal cancer screening. Current oncology reports. 2009; 11:482-489.
11. Kinzler KW and Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996; 87:159-170.
12. Fearon ER and Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990; 61:759-767.
13. Moser AR, Pitot HC and Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990; 247:322-324.
14. Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B and Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997; 275:1784-1787.
15. Cai MY, Luo RZ, Li YH, Dong P, Zhang ZL, Zhou FJ, Chen JW, Yun JP, Zhang CZ and Cao Y. High-expression of ZBP-89 correlates with distal metastasis and poor prognosis of patients in clear cell renal cell carcinoma. Biochemical and biophysical research communications. 2012; 426:636-642.
16. Yan SM, Wu HN, He F, Hu XP, Zhang ZY, Huang MY, Wu X, Huang CY and Li Y. High expression of zinc-binding protein-89 predicts decreased survival in esophageal squamous cell cancer. The Annals of thoracic surgery. 2014; 97:1966-1973.
17. Shoemaker AR, Gould KA, Luongo C, Moser AR and Dove WF. Studies of neoplasia in the Min mouse. Biochimica et biophysica acta. 1997; 1332:F25-48.
18. Demidov ON, Timofeev O, Lwin HN, Kek C, Appella E and Bulavin DV. Wip1 phosphatase regulates p53-dependent apoptosis of stem cells and tumorigenesis in the mouse intestine. Cell stem cell. 2007; 1:180-190.
19. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT and Weinberg RA. Tumor spectrum analysis in < em>p53 < /em>-mutant mice. Current Biology. 4:1-7.
20. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS and Allan B. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992; 356:215-221.
21. Hollstein M, Sidransky D, Vogelstein B and Harris CC. p53 mutations in human cancers. Science. 1991; 253:49-53.
22. Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, Hovig E, Smith-Sorensen B, Montesano R and Harris CC. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994; 22:3551-3555.
23. Reed KR, Meniel VS, Marsh V, Cole A, Sansom OJ and Clarke AR. A limited role for p53 in modulating the immediate phenotype of Apc loss in the intestine. BMC cancer. 2008; 8:162.
24. Fazeli A, Steen RG, Dickinson SL, Bautista D, Dietrich WF, Bronson RT, Bresalier RS, Lander ES, Costa J and Weinberg RA. Effects of p53 mutations on apoptosis in mouse intestinal and human colonic adenomas. Proceedings of the National Academy of Sciences of the United States of America. 1997; 94:10199-10204.
25. Clarke AR, Cummings MC and Harrison DJ. Interaction between murine germline mutations in p53 and APC predisposes to pancreatic neoplasia but not to increased intestinal malignancy. Oncogene. 1995; 11:1913-1920.
26. Feng Y, Wang X, Xu L, Pan H, Zhu S, Liang Q, Huang B and Lu J. The transcription factor ZBP-89 suppresses p16 expression through a histone modification mechanism to affect cell senescence. FEBS J. 2009; 276:4197-4206.
27. Mahler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO and Sundberg JP. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. The American journal of physiology. 1998; 274:G544-551.
28. Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P and Bergo MO. Antioxidants accelerate lung cancer progression in mice. Science translational medicine. 2014; 6:221ra215.
29. Li C and Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:31-36.
30. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249-264.
31. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004; 3:Article3.
32. Smyth GK. (2005). Limma: linear models for microarray data. In: R. Gentleman VC, S. Dudoit, R. Irizarry, W. Huber ed. Bioinformatics and Computational Biology Solutions using R and Bioconductor. (New York: Springer), pp. 397-420.
33. Benjamini Y HY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society 1995; Series B:289-300.
34. Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyotylainen T, Oresic M and Backhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell metabolism. 2013; 17:225-235.
35. Larsson E, Fredlund Fuchs P, Heldin J, Barkefors I, Bondjers C, Genove G, Arrondel C, Gerwins P, Kurschat C, Schermer B, Benzing T, Harvey SJ, Kreuger J and Lindahl P. Discovery of microvascular miRNAs using public gene expression data: miR-145 is expressed in pericytes and is a regulator of Fli1. Genome medicine. 2009; 1:108.
36. Cisowski J, Sayin VI, Liu M, Karlsson C and Bergo MO. Oncogene-induced senescence underlies the mutual exclusive nature of oncogenic KRAS and BRAF. Oncogene. 2016; 35:1328-1333.