
INTRODUCTION
One of the hallmarks of cancer cells is their increased reliance on glycolysis for their bioenergetic and biosynthetic requirements, even in the presence of oxygen [1, 2]. In addition to maintaining aerobic glycolysis, cancer cells display a characteristic reprogramming of metabolic pathways and glutamine-dependent anaplerosis to maintain the high flux of intermediates required for their upregulated nucleotide, protein and fatty-acid biosynthesis [3]. Glutaminase is a mitochondrial enzyme that catalyzes the first step in glutamine-dependent anaplerosis by converting glutamine to glutamate, more simply referred to as glutaminolysis. GLS1 and GLS2 encode two primary isoforms of human glutaminase. GLS1 has two full-length splice variants, kidney-type glutaminase (KGA) and glutaminase-C (GAC), whereas GLS2 encodes liver-type glutaminase (LGA) [4]. Both GLS1 isoforms share an identical catalytic domain.
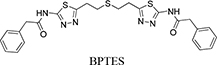
Glutaminase is also reportedly upregulated in response to oncogenes such as c-Myc, Raf, Ras and the Rho GTPase [5–8] and, because of this, glutaminase is an emerging target for cancer therapeutics [7, 9–11]. Indeed, treating tumor cells with an antisense glutaminase mRNA induces apoptosis under oxidative stress [12]. To date, several approaches using small molecules have been used to inhibit glutaminase [8, 13]. Competitive inhibitors, such as 6-diazo-5-oxo-L-norleucine (DON) and azaserine, have had limited success against tumors owing to their severe toxicity and non-specificity [13–15]. Two classes of allosteric inhibitors have been reported, and these vary in their inhibition mechanism: (1) compound-968 (5-(3-bromo-4-(dimethylamino)phenyl)-2,2-dimethyl-2,3,5,6 tetrahydrobenzo[a]phenanthridin-4(1H)-one) and (2) BPTES (bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide). Compound-968 is specific to GLS but shows limited potency in the presence of the inorganic phosphates that promote GLS activation by tetramerization [7]. BPTES, on the other hand, is a GLS1-specific inhibitor that is active even in the presence of inorganic phosphates [16], with some anti-tumoral success against breast, glioblastoma and B lymphoma cancer cell lines [10, 17–19]. A recent study suggests that the concomitant use of glutaminolysis inhibitors and antagonists against certain tumor-derived chemokines, such as IL-8 [20], could improve the antitumor effect.
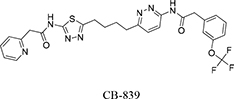
In the past, we have demonstrated the inhibition mechanisms for an active site inhibitor, DON, and an allosteric inhibitor, BPTES [8, 13]. Others and we have reported that BPTES inhibits KGA by binding to an allosteric site between a pair of homodimers, and this results in a dramatic conformational change in key loop residues (Glu312-Pro329) that render the tetramer inactive [8, 21]. Although, BPTES has shown promise in the treatment of a range of cancer types, its low solubility and moderate potency has limited its pharmacological applications [22]. In an attempt to circumvent the potency and solubility limitations of BPTES, various BPTES analogs have been developed. One promising candidate, CB-839, first characterized by Gross et al., is currently undergoing clinical trials. Compared with BPTES, CB-839 is reported to have higher solubility and superior antiproliferative activity in triple-negative breast cancer cells [19]. Furthermore, CB-839 has a 13-fold lower IC50 value, slower reversibility kinetics, and, most importantly, a stronger affinity for GLS1 over BPTES [23]. The compound also demonstrates antitumor efficacy in acute myeloid leukemia (AML), multiple myeloma, solid tumors and hematological malignancies [24–26]. Despite its current successes, the structural basis for this potent inhibitory activity of CB-839 has not been determined.
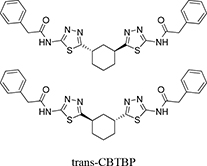
As a part of our continued efforts to understand the allosteric inhibitory mechanism of KGA and develop superior inhibitors, here we characterized the inhibitory efficacy and toxicity of a new BPTES analog developed by Agios (Cambridge, MA), N,N′-(5,5′-(trans-cyclohexane-1,3-diyl)bis(1,3,4-tiadiazole-5,2-diyl))bis(2-phenylacetamide), hereafter referred to as trans-CBTBP [27], and compared it against CB-839 and BPTES. Additionally, we determined the crystal structures of the cKGA (catalytic domain Ile221-Leu533 of KGA) in complex with the trans-CBTBP enantiomer (1S, 3S-CBTBP), and CB-839. These studies have enhanced our understanding of how specific modifications can affect the inhibitory potency of the compound and will aid in the design of new lead molecules for therapeutics.
RESULTS
Exploring the allosteric interactions involved in the inhibition of KGA
Our previous structural studies on the allosteric inhibition of cKGA with BPTES reported two primary interactions between cKGA and BPTES: (1) Hydrogen bonding contacts between cKGA and the thiadiazole moiety and the amide nitrogen of BPTES; (2) Hydrophobic interactions between the cKGA allosteric pocket and aliphatic BPTES linker [8]. In an attempt to explore the role of these interactions in terms of their effect on inhibitor affinity, we chose two representative BPTES analogs, trans-CBTBP and CB-839 to determine the mode of cKGA inhibition. Trans-CBTBP is structurally similar to BPTES, with the exception of a 1,3-di-substituted cyclohexyl ring in place of the acyclic aliphatic linker on BPTES. CB-839, unlike BPTES and trans-CBTBP, is asymmetric across its center and has multiple unique moieties, including the replacement of the terminal phenylacetyl groups with a pyridylacetyl group and a trifluoromethoxylphenyl acetyl group, as well as the substitution of a pyridazine ring for one of the two thiadiazole rings (Figure 1A).
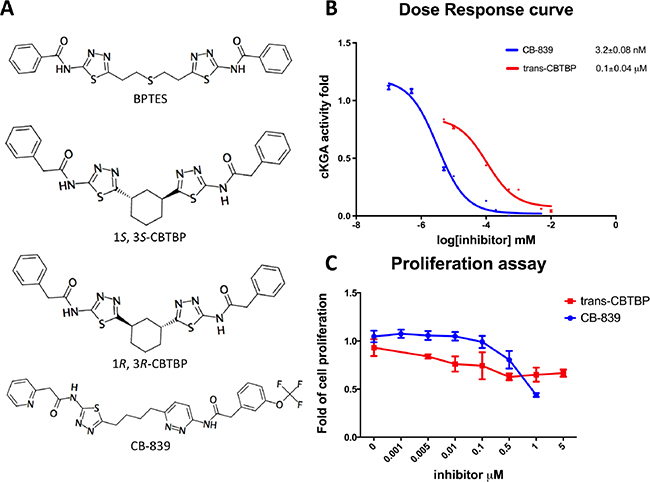
Figure 1: Inhibition assay for cKGA with trans-CBTBP and CB-839. A. Chemical structures for BPTES, trans-CBTBP and CB-839. Trans-CBTBP used for the studies is a racemic mixture of the 1S, 3S and 1R, 3R enantiomers. B. Dose response curves for trans-CBTBP and CB-839. C. Cell viability assay for trans-CBTBP and CB-839.
Inhibition of cKGA by trans-CBTBP and CB-839
In order to compare the inhibitory efficacies of the three inhibitors: BPTES, trans-CBTBP and CB-839, we performed both in-vitro and in-vivo inhibition assay with recombinant cKGA (Figure 1B; Supplementary Figure S3). Among the three inhibitors, CB-839 showed the lowest IC50 values. Although the in-vitro assay shows that both BPTES and trans-CBTBP demonstrate similar IC50 values; in comparison with BPTES, trans-CBTBP displays a smaller number of rotatable bonds (NRB). The reduction in NRBs (8 in trans-CBTBP vs. 12 in BPTES) in trans-CBTBP would improve the probability of good absorption [28].
Figure 1B shows the dose-response curves for these two inhibitors in 293T epithelial cells. The IC50 values for glutaminase inhibition for trans-CBTBP and CB-839 were determined to be 0.1 μM and 3.2 × 10−3 μM, respectively, with a 30-fold difference in activity between the two compounds. Further, we note that the IC50 for trans-CBTBP is only a moderate improvement over that for BPTES (IC50= 0.16 μM) [8] and could be due to greater cell permeability of trans-CPTBP over BPTES. To confirm that glutaminase activities measured were attributed from ectopic cKGA, glutaminase activity were assessed in the absence of ectopic KGA. The endogenous glutaminase activity was 3% of the ectopic expressed cKGA (Supplementary Figure S4). In addition, the endogenous glutaminase was completely inhibited at very low concentrations (1nM) of inhibitors.
Next, to verify the toxicity of the inhibitors to non-tumorigenic cells, we conducted a cell viability assay (Figure 1C). We found that neither CB-839 nor trans-CBTBP caused substantial cytotoxicity at concentrations up to 0.1 μM, suggesting that both inhibitors are safe at their effective concentration range.
Overall, we find CB-839 to be a more potent inhibitor than trans-CBTBP and BPTES, with trans-CBTBP showing only a small improvement in its in vivo inhibitory activity over BPTES. To further understand these differences in potency, we solved the complex crystal structures of both inhibitors with cKGA.
cKGA: 1S,3S-CBTBP Complex
A racemic mixture of trans-CBTBP enantiomers: 1S, 3S and 1R, 3R were used for co-crystallization with cKGA. The cKGA: 1S, 3S-CBTBP was found to crystallize and the structure was determined at 2.74 Å resolution (Table 1; Figure 2A, 2B). However multiple crystallization conditions did not yield the crystals of cKGA: 1R, 3R-CBTBP complex. This indicated a higher preference of the 1S, 3S stereoisomer for cKGA over the 1R, 3R form. The 1S, 3S-CBTBP molecule was positioned using a Fo-Fc Simulated Annealing omit map (Supplementary Figure S1A).
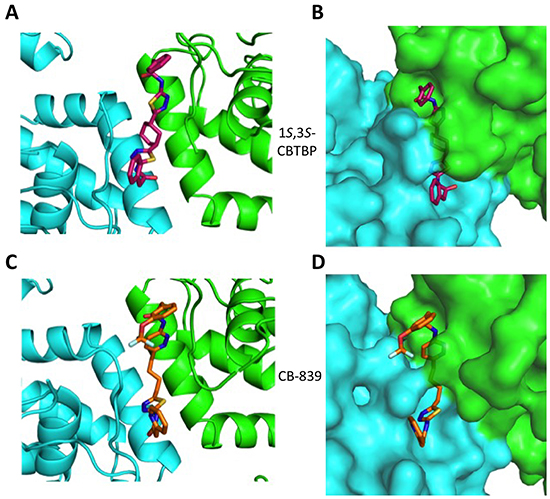
Figure 2: Structure of cKGA in complex with 1S, 3S-CBTBP and CB-839. Close-up view of the inhibitor binding pocket at the dimer interface of cKGA for the A. cKGA:1S, 3S-CBTBP complex and the C. cKGA:CB839 complexes. B. Molecular surface representation of the inhibitor binding pocket for cKGA and stick representation for 1S, 3S-CBTBP. D. Molecular surface representation of the inhibitor binding pocket for cKGA and CB-839. Structure related figures of this manuscript were generated using PyMOL [50].
Table 1: Data collection and refinement statistics for cKGA:1S, 3S-CBTBP and cKGA:CB-839 complexes
cKGA: 1S, 3S-CBTBP complex |
cKGA: CB-839 complex |
|
|---|---|---|
Space group |
I4122 |
P1 |
Cell parameters (Å, °) |
a= b= 139.57,c= 156.61, α= β=γ=90 |
a= 126.40,b= 126.63,c= 126.27, α=112.88, β=102.81,γ=112.74 |
Resolution range (Å) |
30-2.74 (2.79-2.74) |
30-2.1 (2.14-2.10) |
Wavelength (Å) |
1.000 |
1.000 |
Observed hkl |
144179 |
1265236 |
Unique hkl |
20506 |
337350 |
Completeness (%) |
99.5(94.9) |
96.60(93.9) |
Overall I/σI |
20.27(1.75) |
15.65(2.03) |
aRsym |
0.093 (0.572) |
0.108 (0.448) |
Refinement and quality of the model |
||
*Resolution range |
29.29-2.74 |
21.4-2.1 |
bRwork (%) no. reflections |
20.86 (20494) |
18.43 (337261) |
cRfree (%) no. reflections |
24.86 (1905) |
20.50 (1981) |
Root mean square deviation |
||
Bond length (Å) |
0.009 |
0.009 |
Bond angle (°) |
1.17 |
1.08 |
Ramachandran plot (%) |
||
Favored region |
92.0 |
98.0 |
Allowed regions |
7.7 |
2.0 |
Disallowed regions |
0.3 |
0 |
dAverage B-factors (Å2) |
||
Overall average |
65.90 |
48.40 |
Macromolecules |
65.70 |
48.00 |
Ligand |
78.80 |
61.10 |
a Rsym = |Ii − <I>|/|Ii| where Ii is the intensity of the ith measurement, and <I> is the mean intensity for that reflection.
b Rwork = |Fobs − Fcalc|/|Fobs| where Fcalc and Fobs are the calculated and observed structure factor amplitudes respectively.
c Rfree is as for Rwork, but only for approx. 9% and 0.6% of the total reflections chosen at random and omitted from refinement for cKGA: 1S, 3S-CBPTBP and cKGA:CB-839 respectively.
dIndividual B-factor refinement was carried out.
Trans-CBTBP shares an internal symmetry across its 1,3-di-substituted cyclohexyl linker (Figure 1A). BPTES and 1S, 3S-CBTBP share similar symmetric halves, and have identical hydrogen bonding interactions with cKGA (Figure 3A, Supplementary Table S1). However the differences in the hydrophobic interactions of these two inhibitors with cKGA is prominent. Unlike the aliphatic linker of BPTES, the cyclohexane linker from 1S, 3S-CBTBP forms multiple hydrophobic interactions with the side chains of Tyr394, Phe322 and Leu321 from both the neighboring cKGA monomers (see Supplementary Table S2).
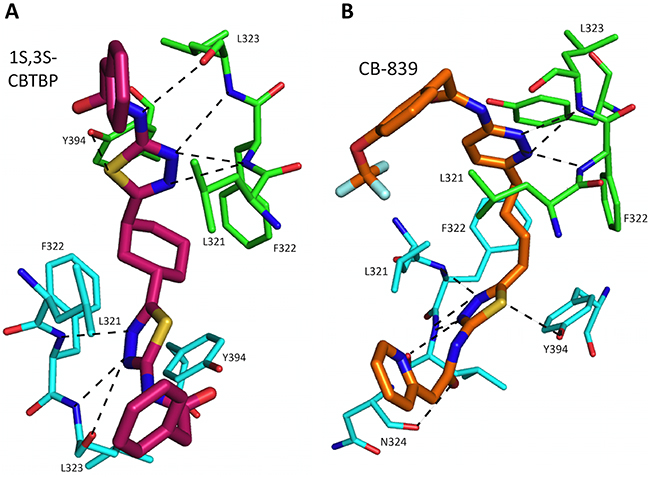
Figure 3: Interactions of 1S, 3S-CBTBP and CB-839 with cKGA. A. Hydrogen bonding interactions involved in cKGA: 1S, 3S-CBTBP binding. B. Interactions between CB-839 and allosteric site of cKGA. For clarity not all the protein side chains are shown in these figures.
cKGA: CB-839 Complex
The complex structure for cKGA:CB-839 was determined at 2.1 Å (Table 1; Figure 2C, 2D). Two molecules of CB-839 are found to be interacting with the cKGA tetramer. Similar to BPTES and 1S, 3S-CBTBP, several hydrogen-bonding interactions mediate the binding of CB-839 with cKGA (Supplementary Table S1). Similar to that observed for the cKGA:1S, 3S-CBTBP complex, the cKGA backbone amide groups of Phe322 and Leu323 are involved in hydrogen bonding contact with CB-839. Notable unique interactions include the hydrogen bonds between the pyridazinyl and acetyl groups of the inhibitor with the side-chains of Try394, Lys320 and Asn324 from cKGA. Furthermore, the thiadiazol group of CB-839 is also involved in a water-mediated interaction with Asp327 of cKGA. The terminal moieties of the inhibitor–fluorine and oxygen atoms of the trifluromethoxy phenyl group, as well as the nitrogen atoms from the pyridine ring of the inhibitor are not engaged in any hydrogen bonding contact with cKGA. It is plausible, however, that the trifluoromethoxy phenyl group could help improve the solubility of CB-839 [29, 30].
Hydrophobic interactions between the aliphatic linker of CB-839 and the side-chains of Phe322 and Tyr394 from cKGA also contribute to the binding (Supplementary Table S2). The pyridinyl moiety and terminal phenyl ring also interact with the hydrophobic side-chain carbon atoms of Glu325 and Leu321, respectively.
Comparison of the binding of BPTES, 1S, 3S-CBTBP and CB-839 with cKGA
A comparison of the cKGA complex structures of BPTES, 1S, 3S-CBTBP, and CB-839 indicates that binding differences are primarily due to the strengths of interactions made with the allosteric pocket of cKGA. The cKGA allosteric pocket resembles a lock, with negligible changes observed in its conformation in complex with the different inhibitors; the inhibitors are analogous to different keys with their moieties acting as levers. The binding affinity is a function of the flexibility and orientation of the interacting moieties from the inhibitors (Figure 4).
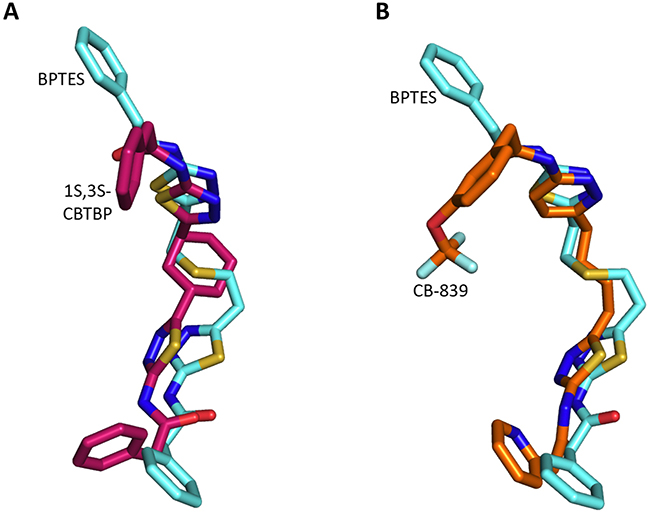
Figure 4: Superposition of inhibitors. A. Comparison of 1S, 3S-CBTBP with BPTES. B. Comparison of CB-839 with BPTES.
The interaction of 1S, 3S-CBTBP with the cKGA allosteric pocket is primarily mediated by hydrogen bonding interactions from the thiadiazol group and hydrophobic interactions from the cyclohexane linker. In comparison, the driving force for CB-839 binding is the higher number of hydrogen bonds and this is assisted by the hydrophobic interactions from the pyridinyl ring. The lower effective concentrations for CB-839 are consistent with an improved cumulative strength of the interactions. The calculated buried surface areas of 520 Å2 for CB-839 and 460 Å2 for 1S, 3S-CBTBP further validate the greater relative strength of the CB-839 interaction [31].
DISCUSSION
Metabolic pathway enzymes are increasingly being investigated as potential drug targets for cancer treatment [32–34]. The mitochondrial enzyme glutaminase in particular is under intense investigation in many tumor models [35], with an immense amount of time invested in the development of selective and non-toxic small-molecule glutaminase inhibitors with superior metabolic stability and pharmacodynamics. Various recent efforts have shown that inhibition of glutaminolysis offers synergistic effects when administered alongside anti-NOTCH1, β-lapachone, or BCL-2 inhibition for the treatment of T cell acute lymphoblastic leukaemia (T-ALL), pancreatic cancer, or acute myeloid leukemia (AML), respectively [36–38]. A structural understanding of the differences in the protein: inhibitor interactions as a result of the small modifications to these inhibitors will help in the development and rational design of more potent inhibitors.
In this study, we report the inhibitory efficacies along with complex structural studies of two analogs of BPTES, trans-CBTBP and CB-839. As shown in Figure 1B, we observe a 50-fold lower IC50 value for CB-839 as compared with BPTES (30-fold as compared with trans-CBTBP), when using a cell-culture based assay to determine the dose response. This is consistent with the inhibition assay performed by Gross et al. for GAC with CB-839 [23]. Further, we have explored the utility of the inhibitors as drugs by correlating their effective concentration range to their toxicities. Both trans-CBTBP and CB-839 were found to be safe at their IC50 concentrations. Intriguingly, increasing CB-839 inhibitor treatment starting from 0.5mM, leads to rapid decrease of proliferation. In contrast, trans-CBTBP treatment does not have a major impact on the proliferation of cells, suggesting lower toxicity of trans-CBTBP over CB-839 (Figure 1C). The mechanism of cell death at higher concentrations of CB-839 is yet to be determined and warrants further investigation.
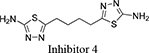
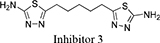
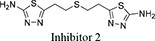
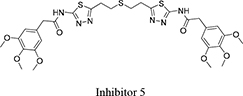
Both 1S, 3S-CBTBP and CB-839 were found to interact with the same allosteric pocket of cKGA, as reported for BPTES. The effective concentrations of trans-CBTBP and CB-839 were compared with BPTES and its other analogs (Table 2). The low affinity of inhibitors 4 and 3 for cKGA could be attributed to the absence of the terminal phenyl ring and acetyl group, which are involved in interactions with the cKGA allosteric pocket as observed in the crystal structure of the cKGA:BPTES complex. Inhibitor 2 likely has better binding because of the central sulfur atom, which restrains the flexibility of the diethyl sulfide linker. The terminal phenyl rings on inhibitor 5 that are substituted with electron-donating methoxy groups, leads to moderate improvements in its affinity over inhibitors 2, 3 and 4. Unlike BPTES, the cyclohexane linker of 1S, 3S-CBTBP improves the hydrophobic interactions and imposes rigidity across the center of the inhibitor. The terminal electron-withdrawing trifluoromethoxy group of CB-839, substituted on the phenyl ring on BPTES, deactivates the aromatic ring system and increases the electronegativity of the pyridazinyl nitrogen atoms, thus strengthening the hydrogen bonding interactions. In addition the trifluoromethoxy group increases the lipophilicity of the inhibitor.
Table 2: A comparison of KGA allosteric inhibitors
Inhibitor Structure |
IUPAC name |
IC50 μM |
Reference |
|---|---|---|---|
|
5-(4-(5-amino-1,3, 4-thiadiazol-2-yl)butyl)-1,3,4-thiadiazol-2-amine |
18.60 |
[7] |
|
5-(5-(5-amino-1,3,4-thiadiazol-2-yl)pentyl)-1,3,4-thiadiazol-2-amine |
11.10 |
[7] |
|
5-(2 -((2-(5-amino-1,3,4-thiadiazol-2-yl)ethyl)sulfanyl)ethyl)-1,3,4-thiadiazol-amine |
8.02 |
[7] |
|
2,3,4-trimethoxy-N-(5-(2-((2-(5-(3,4,5-trimethoxybenzamido)-1,3,4-thiadiazol-2-yl)ethyl)sulfanyl)ethyl)-1,3,4-thiadiazol-2-yl)benzamide |
5.44 |
[7] |
|
bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide |
0.16 |
|
|
N,N′-(5,5′-((1S,3S)-cyclohexane-1,3-diyl)bis(1,3,4-tiadiazole-5,2-diyl))bis(2-phenylacetamide) N,N′-(5,5′-((1R,3R)-cyclohexane-1,3-diyl)bis(1,3,4-tiadiazole-5,2-diyl))bis(2-phenylacetamide) |
0.10 |
This study |
|
2-(pyridin-2-yl)-N-(5-(4-(6-(2-(3-(trifluoromethoxy)phenyl)acetamido)pyridazin-3-yl)butyl)-1,3,4-thiadiazol-2-yl)acetamide |
0.003 |
This study |
The findings from these studies provide further insights into rational design of the next generation of KGA inhibitors by utilizing knowledge garnered from ligand-based interactions. From the activity assay, we have shown that CB-839 has enhanced inhibitory efficacy over trans-CBTBP and BPTES. Since the terminal atoms of the inhibitors do not engage in key interactions with the cKGA, chemical modifications should be targeted around this region to improve the solubility of new inhibitors [39]. Since both GAC and KGA share exactly the same catalytic domain, the structural and inhibition studies performed with cKGA would be similar and applicable to the glutaminase domain of GAC as well. Moreover, building on what we now know, a chimeric inhibitor with a cyclohexane linker and a pyridazinyl ring might improve the interactions of the compound with KGA and GAC [27].
MATERIALS AND METHODS
Inhibitor synthesis
For this study, we used a racemic mixture of two trans-CBTBP enantiomers: 1S, 3S and 1R, 3R. Between the two enantiomers, only 1S, 3S-CBTBP was found to crystallize with cKGA. The racemic mixture of trans-CBTBP was prepared as reported previously [27]. For both the inhibition and proliferation assay, the racemic mixture of trans-CBTBP was used. Likewise, CB-839 was prepared by the previously reported method [40].
Cloning, expression and purification
The cKGA was cloned into pET-28(b) fused to an N-terminal His tag, and expressed in Escherichia coli BL21 (DE3)-RIL-Codon plus cells. The cells were induced with 200 μM IPTG and then sonicated in the lysis buffer consisting of 50 mM HEPES pH 7.5, 500 mM NaCl, 10% glycerol, 1 mM DTT, 0.1% (vol/vol) Triton X-100, and one tablet of complete protease inhibitor mixture (Roche Diagnostics, Risch-Rotkreuz, Switzerland). The cell lysate was bound to Ni-NTA affinity beads (Roche Diagnostics) and eluted with 400 mM imidazole. The protein was further purified using a Superdex-200 column (GE Healthcare, Buckinghamshire, UK) in a buffer containing 20 mM HEPES pH 7.5, 200 mM NaCl, 5% glycerol and 3 mM DTT. The protein was then concentrated to 20 mg/ml concentration using protein concentrators (Vivaspin, Sartorius AG, Goettingen, Germany).
Cell culture, transfection and inhibitors treatment
Human embryonic kidney epithelial 293T cells were maintained in RPMI-1640 (Hyclone, GE Life Sciences) with 10% (vol/vol) FBS (Gibco-BRL, Gaithersburg, MD), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (HyClone). Cells were grown at 37°C in 5% CO2. Confluent cells grown in 12-well plates were transfected using TransIT (Mirus Bio, Madison, WI) with 1 μg of plasmid encoding wild-type KGA and then incubated with the indicated inhibitors for 24 h.
In-vitro glutaminase assay
The glutaminase assay was performed in two steps using a dual enzyme assay [41]. Inhibitors were dissolved in DMSO. 10 μl of DMSO/inhibitor was incubated with 80 μl assay mixture A (1 μM recombinant cKGA, 50 mM Tris-acetate (pH 8.6), 100 mM Potassium phosphate, and 0.2 mM EDTA) for 2 h at room temperature (24°C). 10 μl of 200 mM glutamine was then added prior to a 20 min incubation at 37°C. The reaction was quenched with the addition of 10 μl of 0.6 M HCl. The reaction mixture was then incubated for 30 min at room temperature (24°C) with 100 μl of the assay mix B (3.7 U glutamate dehydrogenase, 160 mM Tris-acetate (pH 9.4), 400 mM hydrazine, 5 mM ADP, and 2 mM nicotinamide adenine dinucleotide) and the absorbance read at 340 nm using an Infinite 200 microplate reader (Tecan, Durham, NC).
Glutaminase assay with 293T cell line
A glutaminase assay was performed using the two-step procedure as described previously [42]. Confluent 293T cells grown in 12-well plate were transfected with cKGA plasmid and incubated with the indicated inhibitors for 24 h. Cells were lysed with 100 μl of Hepes buffer [20 mM Hepes (pH 7.4)], 150 mM NaCl, 1 % NP40, 20 mM glycerol-2-phosphate, 1 mM sodium orthovanadate, and 20mM sodium fluoride and protease inhibitors (Roche Applied Science, Mannheim, Germany). Lysates were quantified for protein amount using BCA protein assay kit (Pierce Biotechnology, Rockford, IL). Cells were incubated with 10 μl cKGA-inhibitor at 37°C for 10 min with 10 μl of assay mix consisting of 20 mM glutamine, 50 mM Tris-acetate (pH 8.6), 100 mM phosphate, and 0.2 mM EDTA. The reaction was quenched with the addition of 2 μl of 3 M HCl. The reaction mixture was then incubated for 30 min at room temperature (24°C) with 200 μl of the second assay mix (2.2 U glutamate dehydrogenase, 80 mM Tris-acetate (pH 9.4), 200 mM hydrazine, 0.25 mM ADP, and 2 mM nicotinamide adenine dinucleotide) and the absorbance read at 340 nm using an Infinite 200 microplate reader (Tecan, Durham, NC).
Cell proliferation assays
293T cells were trypsinized and replated into 96-well plates and then treated with inhibitors for 48 h. 293T cells treated with DMSO served as the control. The amount of cell proliferation was determined using an MTT proliferation assay kit (Promega, Fitchburg, WI) as described by the manufacturer. At least three independent experiments were carried out, each with multiple replicates. Statistical significance was analyzed using ANOVA and the Student-Newman-Kuels multiple range test (StatsDirect, Cheshire, UK). Data are mean ± SD (p < 0.05) and are expressed as fold increase over the control cells.
Crystallization and structure determination
The protein:inhibitor complex was prepared by incubating cKGA (2 mg/ml) with inhibitors (1:10 molar ratio; final concentration of 5% DMSO) at 4°C for 1 h. The complex was then concentrated to 20 mg/ml before crystallization screening using the vapor diffusion method. Crystals for cKGA–CB839 was obtained with 0.1 M Bis-Tris propane (pH 7), 3% DMSO and 1.8 M LiSO4. cKGA–trans-CBTBP crystals were obtained with 0.1 M Bis-Tris propane (pH 7), 4% DMSO and 2 M LiSO4. The crystals were cryopreserved in reservoir solution supplemented with 15% glycerol. For both complexes, the diffraction data sets were collected at the synchrotron beamline 13B1 (wavelength 1.000 Å) at the National Synchrotron Radiation Research Centre (NSRRC, Taiwan). Data sets were processed and scaled using HKL2000 [43].
Structure solution and refinement
Structures of the cKGA:inhibitor complexes were obtained by molecular replacement with the program Phaser-MR [44] using the coordinates of apo cKGA as the search model (Protein data bank, 3VOY). The restraints for the inhibitors were generated using eLBOW program [45, 46]. The model was examined and built in COOT [46] and subsequent refinement was carried out with Phenix-refine [47]. The electron density for the cKGA gating loop (316LRFNKL321) region is disordered in 1S, 3S-CBTBP and CB-839 inhibitor complex structures, and was not modeled [48]. The final structures for cKGA:1S, 3S-CBTBP and cKGA:CB-839 were refined up to 2.74 Å and 2.1 Å resolution, respectively (Table 1). The overall geometry of the final model was analyzed using a Ramachandran plot with the program PROCHECK [49].
ACKNOWLEDGMENT
We would like to thank Dr. Pavel Afonine for the discussion on Phenix refinement for the cKGA: 1S, 3S CBTBP complex. The X-ray diffraction data for this study were collected at the synchrotron beamline 13B1 at the National Synchrotron Radiation Research Centre (NSRRC, Taiwan).
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
GRANT SUPPORT
This work was supported by Ministry of Education (MoE) Singapore Tier-2 grant R154-000-625-112. SCZ is an NIH postdoctoral fellow (F32CA200278). SR is a graduate scholar in receipt of a research scholarship from the National University of Singapore (NUS).
PDB accession codes
The coordinates of cKGA: 1S, 3S-CBTBP and cKGA:CB839 complexes have been deposited in the Protein Data Bank (http://www.pdb.org), accession number 5JYP, and 5JYO respectively.
REFERENCES
1. Vander Heiden MG, Cantley LC and Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029-1033.
2. Warburg O. On the origin of cancer cells. Science. 1956; 123:309-314.
3. DeBerardinis RJ, Lum JJ, Hatzivassiliou G and Thompson CB. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metabolism. 2008; 7:11-20.
4. Aledo JC, Gomez-Fabre PM, Olalla L and Marquez J. Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm Genome. 2000; 11:1107-1110.
5. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB and Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences. 2008; 105:18782-18787.
6. Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009; 458:762-765.
7. Wang J-B, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio ALB, Dias SMG, Dang CV and Cerione RA. Targeting Mitochondrial Glutaminase Activity Inhibits Oncogenic Transformation. Cancer Cell. 2010; 18:207-219.
8. Thangavelu K, Pan CQ, Karlberg T, Balaji G, Uttamchandani M, Suresh V, Schuler H, Low BC and Sivaraman J. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci U S A. 2012; 109:7705-7710.
9. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013; 496:101-105.
10. Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, Dang CV and Riggins GJ. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010; 70:8981-8987.
11. van den Heuvel AP, Jing J, Wooster RF and Bachman KE. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol Ther. 2012; 13:1185-1194.
12. Lora J, Alonso FJ, Segura JA, Lobo C, Marquez J and Mates JM. Antisense glutaminase inhibition decreases glutathione antioxidant capacity and increases apoptosis in Ehrlich ascitic tumour cells. Eur J Biochem. 2004; 271:4298-4306.
13. Thangavelu K, Chong QY, Low BC and Sivaraman J. Structural Basis for the Active Site Inhibition Mechanism of Human Kidney-Type Glutaminase (KGA). Scientific Reports. 2014; 4.
14. Catane R, Von Hoff DD, Glaubiger DL and Muggia FM. Azaserine, DON, and azotomycin: three diazo analogs of L-glutamine with clinical antitumor activity. Cancer Treat Rep. 1979; 63:1033-1038.
15. Kisner DL, Catane R and Muggia FM. The rediscovery of DON (6-diazo-5-oxo-L-norleucine). Recent Results Cancer Res. 1980; 74:258-263.
16. Katt WP, Ramachandran S, Erickson JW and Cerione RA. Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol Cancer Ther. 2012; 11:1269-1278.
17. Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, Hansen JC and Curthoys NP. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J. 2007; 406:407-414.
18. Qie S, Chu C, Li W, Wang C and Sang N. ErbB2 activation upregulates glutaminase 1 expression which promotes breast cancer cell proliferation. J Cell Biochem. 2014; 115:498-509.
19. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, Zimmerman LJ, Liebler DC, Slebos RJ, Lorkiewicz PK, Higashi RM, Fan TW, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012; 15:110-121.
20. Shanware NP, Bray K, Eng CH, Wang F, Follettie M, Myers J, Fantin VR and Abraham RT. Glutamine deprivation stimulates mTOR-JNK-dependent chemokine secretion. Nat Commun. 2014; 5:4900.
21. DeLaBarre B, Gross S, Fang C, Gao Y, Jha A, Jiang F, Song JJ, Wei W and Hurov JB. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry. 2011; 50:10764-10770.
22. Shukla K, Ferraris DV, Thomas AG, Stathis M, Duvall B, Delahanty G, Alt J, Rais R, Rojas C, Gao P, Xiang Y, Dang CV, Slusher BS and Tsukamoto T. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. 2012; 55:10551-10563.
23. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, MacKinnon AL, Parlati F, Rodriguez MLM, Shwonek PJ, Sjogren EB, Stanton TF, et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Molecular Cancer Therapeutics. 2014; 13:890-901.
24. Matre P, Shariati M, Velez J, Qi Y, Konoplev S, Su XP, DiNardo CD, Daver N, Majeti R, Andreeff M, Chan SM and Konopleva M. Efficacy of Novel Glutaminase Inhibitor CB-839 in Acute Myeloid Leukemia. Blood. 2014; 124.
25. Sharma D, Ravillah D, Ray A, Song Y, Munshi NC, Richardson PG, Chauhan D and Anderson KC. Anti-Myeloma Activity of a Novel Glutaminase Inhibitor CB-839. Blood. 2014; 124.
26. Parlati F, Bromley-Dulfano S, Demo S, Janes J, Gross M, Lewis E, MacKinnon A, Rodriguez M, Yang JF, Zhao F and Bennett M. Antitumor Activity Of The Glutaminase Inhibitor CB-839 In Hematological Malignances. Blood. 2013; 122.
27. Lemieux RM, Popovici-Muller J, Salituro FG, Saunders JO, Travins J and Chen Y. (2014). Compounds and their methods of use. United States Patent US20140142081 A1.
28. Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW and Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002; 45:2615-2623.
29. Leroux FR, Manteau B, Vors J-P and Pazenok S. Trifluoromethyl ethers–synthesis and properties of an unusual substituent. Beilstein journal of organic chemistry. 2008; 4:13.
30. Nenajdenko V. (2014). Fluorine in Heterocyclic Chemistry: Springer.
31. Krissinel E and Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007; 372:774-797.
32. Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, Slocum KL, Pu M, Lin F, Vickers C, Joud-Caldwell C, Chung F, Yin H, Handly ED, Straub C, Growney JD, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014; 74:3317-3331.
33. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008; 452:230-233.
34. Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, Lee MA, Hu B, Hu J, Sahin E, Ong D, Fletcher-Sananikone E, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012; 488:337-342.
35. Hensley CT, Wasti AT and DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013; 123:3678-3684.
36. Herranz D, Ambesi-Impiombato A, Sudderth J, Sanchez-Martin M, Belver L, Tosello V, Xu L, Wendorff AA, Castillo M, Haydu JE, Marquez J, Mates JM, Kung AL, Rayport S, Cordon-Cardo C, DeBerardinis RJ, et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med. 2015; 21:1182-1189.
37. Chakrabarti G, Moore ZR, Luo X, Ilcheva M, Ali A, Padanad M, Zhou Y, Xie Y, Burma S, Scaglioni PP, Cantley LC, DeBerardinis RJ, Kimmelman AC, Lyssiotis CA and Boothman DA. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ss-lapachone. Cancer Metab. 2015; 3:12.
38. Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, Saland E, Decroocq J, Trovati Maciel T, Lambert M, Poulain L, Hospital MA, Sujobert P, Joseph L, Chapuis N, Lacombe C, et al. Targeting glutaminolysis has anti-leukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. 2015.
39. Zimmermann SC, Wolf EF, Luu A, Thomas AG, Stathis M, Poore B, Nguyen C, Le A, Rojas C, Slusher BS and Tsukamoto T. Allosteric Glutaminase Inhibitors Based on a 1,4-Di(5-amino-1,3,4-thiadiazol-2-yl)butane Scaffold. ACS Med Chem Lett. 2016; 7:520-524.
40. Li J, Chen L, Goyal B, Laidig G, Stanton TF and Sjogren EB. 2013. Heterocyclic inhibitors of glutaminase. United States Patent US8604016.
41. McDermott LA, Iyer P, Vernetti L, Rimer S, Sun J, Boby M, Yang T, Fioravanti M, O’Neill J, Wang L, Drakes D, Katt W, Huang Q and Cerione R. Design and evaluation of novel glutaminase inhibitors. Bioorg Med Chem. 2016; 24:1819-1839.
42. Kenny J, Bao Y, Hamm B, Taylor L, Toth A, Wagers B and Curthoys NP. Bacterial expression, purification, and characterization of rat kidney-type mitochondrial glutaminase. Protein Expr Purif. 2003; 31:140-148.
43. Minor W and Otwinowski Z. HKL2000 (Denzo-SMN) Software Package. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology, Macromolecular Crystallography, Academic Press, New York. 1997.
44. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC and Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007; 40:658-674.
45. Moriarty NW, Grosse-Kunstleve RW and Adams PD. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr. 2009; 65:1074-1080.
46. Emsley P and Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004; 60:2126-2132.
47. Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH and Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012; 68:352-367.
48. Ferreira AP, Cassago A, Goncalves Kde A, Dias MM, Adamoski D, Ascencao CF, Honorato RV, de Oliveira JF, Ferreira IM, Fornezari C, Bettini J, Oliveira PS, Paes Leme AF, Portugal RV, Ambrosio AL and Dias SM. Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J Biol Chem. 2013; 288:28009-28020.
49. Laskowski RA, Macarthur MW, Moss DS and Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. Journal of Applied Crystallography. 1993; 26:283-291.
50. DeLano WL. The PyMOL molecular graphics system. 2002.