
Introduction
The glutamatergic system plays an essential role in neural signaling and as such, the ionotropic N-methyl-D-aspartate receptors (NMDARs) influence the operative tone of the synapse by determining the weight assigned to the incoming signals. Unfortunately, a series of neurological disorders concur with dysfunctions of these glutamatergic receptors, such as those produced by the excitotoxicity resulting from their excess activity. As chronic blockade of NMDARs compromises cell viability other approaches must be considered to safely diminish their activity. Accordingly, the regulation of NMDARs by certain G protein-coupled receptors (GPCRs) provides one such therapeutic opportunity. GPCRs and glutamate NMDARs stimulate complex cellular signaling pathways, yet they also exert a mutual regulation on each other’s signaling [1, 2]. In this context, the endocannabinoid system, though the activation of the cannabinoid 1 receptor (CB1R), plays a very relevant role in reducing NMDAR activity [3, 4]. Thus, this endogenous system could be pharmacologically manipulated to re-establish the function of dysregulated NMDARs.
There are several excellent reviews on glutamate [5, 6] and endocannabinoids [7, 8] that will bring the reader up to date on what is currently known about these systems. Recent studies have described how the tandem histidine triad nucleotide-binding protein 1 (HINT1) and the sigma receptor type 1 (σ1R) promote physical coupling and uncoupling between the CB1R and the NR1 subunit of the NMDAR [9, 10]. Thus, this review will analyze the negative control that endocannabinoids exert on NMDAR activity and its potential to reduce the incidence of convulsive syndromes like epilepsy, which are mediated by NMDAR hyperactivity, as well as their possible role in provoking NMDAR hypofunction, such as that accompanying psychosis/schizophrenia.
I. The glutamate NMDA receptor
Glutamate is the major excitatory neurotransmitter in the CNS [11], and it activates both ionotropic and metabotropic receptors. Ionotropic receptors directly gate ion passage and they are divided into three major subclasses: α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA), kainate, and NMDA receptors. Of these, the NMDARs have received much attention because their deregulation is observed in many neurological disorders, such as neurodegenerative diseases [12], neuropathic pain [13, 14], mood disorders and psychosis-schizophrenia [15, 16].
The NMDAR is a ligand-gated cation channel that is highly permeable to monovalent ions and Ca2+. Binding of glutamate opens the channel pore, and the concurrent binding of glycine increases the amplitude and time course of ion flux. NMDARs are composed of NR1, NR2 (A, B, C and D) and NR3 (A and B) subunits, and the functional NMDAR is a tetramer consisting of a pair of NR1 subunits each associated to at least one type of the NR2/3 subunits [17]. NMDAR activation enhances the binding of cytosolic Ca2+ to calmodulin (CaM), propagating this signal through many other proteins, including kinases (e.g., CaMKII), phosphatases (e.g., calcineurin and serine/threonine protein phosphatase 1 -PP1), neural nitric oxide synthase (nNOS) and adenylyl cyclase (types I, III and VI) [18, 19].
II. Cross-regulation between GPCRs and NMDARs
The NMDAR is essential for neuronal plasticity and differentiation, brain development and synaptic plasticity, directly affecting learning and memory consolidation [20]. Temporal and/or spatial coincidence determines the weight that a neural cell assigns to the incoming signals, and this weight is influenced by the degree of excitability that glutamate NMDARs confer to the post-synapse. However, the activity of NMDARs also falls under the influence of GPCRs and for example, the acetylcholine type 1 muscarinic receptor dampens NMDAR function via the activation of tyrosine phosphatases [21]. In addition, the serotonin 5HT1A [22], adrenergic α1 and α2 [23] and group III mGluR7 receptors [24] impair NMDAR turnover, while other receptors like the CB1R can promote the co-internalization of NMDAR NR1 subunits [25]. Other GPCRs exert the opposite effect, enhancing NMDAR calcium flux via Gβγ/PLCβ/PKC signaling and the non-receptor tyrosine kinase Src [26, 1], including the mu-opioid receptor (MOR) [27], the dopamine D1 receptor [28], group I metabotropic glutamate receptors (mGluR1/5), group II mGluR2/3 [29, 30], and the serotonin 5HT2A/C receptor [31]. Accordingly, the activity of neural cells is influenced by the complex array of signals that are tightly integrated, harmonizing GPCR-triggered signaling cascades and NMDAR glutamate responses. For example, MOR activation recruits NMDARs, exerting a negative influence on opioid signaling by restraining their capacity to produce analgesia, thereby contributing to the development of tolerance [32, 33]. Similarly, NMDAR activity provokes endocannabinoid release and cannabinoid receptor stimulation, in turn diminishing NMDAR activity and preventing excitotoxicity [34].
An interaction that has generated significant interest of late is that between GPCRs and NMDARs during the dynamic process that supports their cross-regulation [2]. The C terminus of NMDAR NR1 subunits is composed of C0-C2(C2’) or of C0-C1-C2(C2´) domains, and the NMDAR NR1 subunits that carry the C1 region bind to the C terminus of the dopamine D1 receptor [35], that of group I metabotropic glutamate receptor (mGlu5a) [36], the MOR [37] and the CB1R [25] when studied in vitro and in cell assays. Indeed, ex vivo assays performed on different areas of the mouse brain show that these GPCRs co-precipitate with NMDAR NR1 subunits [37, 38, 25]. Moreover, the physiological relevance of the complexes containing MOR/CB1R-NMDAR NR1 subunits is confirmed by their dynamic arrangement under the control of the HINT1 and σ1R [9, 39].
III. The GPCR-NMDAR connection: the HINT1-σ1R tandem
At the neural plasma membrane, the HINT1 protein forms complexes with cytosolic regions of different GPCRs [40]. In this environment HINT1 serves as a scaffold for signaling proteins that work together to couple GPCR activity with that of glutamate NMDARs. Among the proteins that HINT1 associates with are protein kinases like PKCγ and PKCα [41], and proteins of the Rz subfamily “Regulators of G-protein signaling” (RGS), mostly RGSZ1(20) [42]. These RGS-Rz proteins have a zinc-finger in their N terminal sequence [40] and they bind to the N terminal PDZ domain of nNOS. HINT1 also connects the Raf-1/MEK/ERK1-2 cassette to GPCRs and the NMDAR NR1 subunits that carry the C1 segment [43]. Significantly, the docking of proteins to HINT1 is organized by Redox signaling, zinc metabolism and PKC activity [33].
The σ1R is a linear protein that is widely expressed in nervous tissue [44] and that was initially considered as a type of opioid receptor [45]. However, its amino acid sequence has no significant homology with any other mammalian protein, and it lacks glycosylation sites and a known transducer system [46]. The σ1R interacts with lipid membranes and in the absence of third party proteins this receptor can form oligomers in vitro, probably trimers, with each monomer anchored to the lipid membrane by its N terminal region [47]. In the ER and plasma membrane, the σ1R associates with different signaling proteins and in these interactions it apparently displays two transmembrane domains, adopting different conformations [48, 49, 50, 51]. Thus, the σ1R N and C termini are either cytoplasmic [51, 52], or in the context of its interaction with NMDAR NR1 subunits, both the N and C terminal sequences project into the extracellular space through two transmembrane domains [48, 9] (Figure 1). The σ1R does not fulfill the criteria of a typical membrane receptor but its associations with other signaling proteins may be altered through a series of endogenous and exogenous substances, as well as by calcium [49, 53, 10]. Thus, the molecular structure of the σ1R and its different arrangements suggests it fulfills different functions, most likely that of a ligand-regulated chaperone [46].
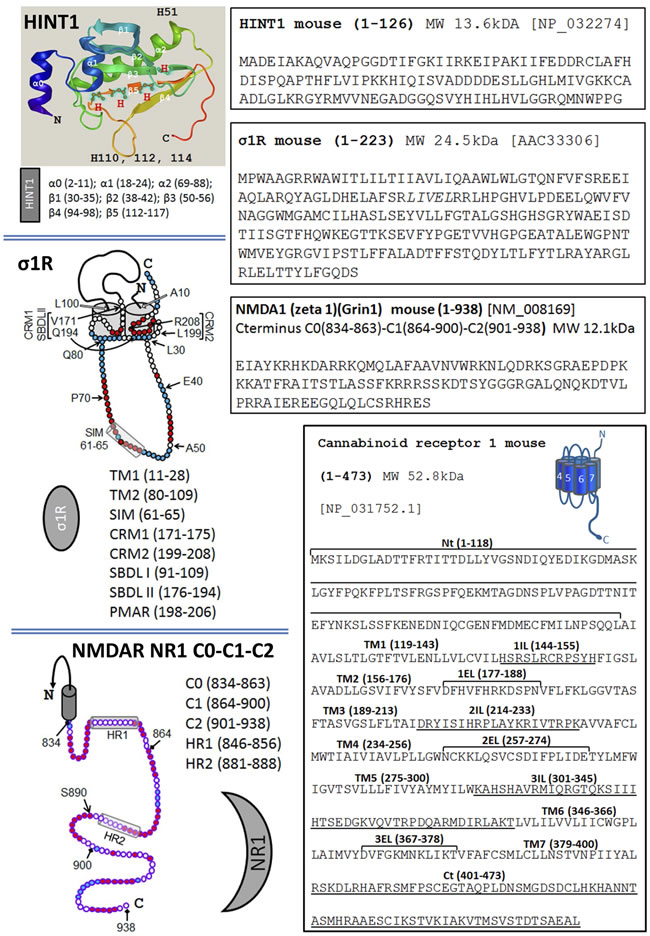
Figure 1: The sequence of HINT1, σ1R, the C terminal of the NMDAR NR1 subunit and the CB1 receptor. Sequence of the murine HINT1 protein. The α and β regions, and the histidines are indicated on the ribbon backbone (Novafold/Protean 3D/DNASTAR v12). The long isoform of the murine σ1R has two hydrophobic transmembrane regions, TM1 and TM2. The σ1R hairpin loop (L30-Q80) contains a SUMO-Interacting motif (SIM: 61-65), while the C-terminal region includes two cholesterol-binding motifs, CRM1 and CRM2, and a potential membrane attachment region (PMAR). The steroid binding site is formed by the SBDL I in TM2 and SBDL II at the C terminus [49, 62, 9]. The C terminal C0-C1-C2 of the NMDAR NR1 subunit contains 104 residues with two hydrophobic regions HR1 and HR2 [9]. The S890 residue is indicated, a PKC regulatory site. In the sequence of the murine CB1R, the extracellular, transmembrane and cytosolic regions are indicated.
The pharmacology of the σ1R is complex, with exogenous ligands showing different profiles depending on the system under study [54]. Thus, σ1R ligands influence NMDAR function in vivo and in vitro [61, 39], and σ1Rs bind to other proteins in the endoplasmic reticulum and plasma membrane in a calcium-dependent manner in cellular expression systems and in vitro assays, NMDARs included [9, 49, 62]. Nevertheless, σ1R ligands are therapeutically interesting to treat neurological diseases [55], substance abuse syndromes [56], and NMDAR-related neural dysfunctions (such as certain neuropsychiatric disorders [53], and the allodynia and hyperalgesia that accompanies neuropathy in different animal models [57, 58], as well as potentially serving as adjuvants of opioid analgesia [59, 60].
The activity of σ1R is coordinated with that of HINT1 to connect GPCRs with NMDARs and promote (e.g., MOR) or reduce (e.g., CB1R) its glutamatergic activity [9, 25, 3]. A series of molecular studies have shed some light on how this molecular switch brings NMDARs under the control of GPCRs. Whilst, HINT1 binds to cytosolic sequences of GPCRs and of NMDAR NR1 subunits in a calcium-independent fashion, the association of the σ1R with these signaling proteins increases greatly in the presence of physiological levels of calcium (i.e.: low mM range). The relationship between both proteins is asymmetric, and whilst the σ1R prevents HINT1 binding to NR1 subunits and it weakens the association of HINT1 with GPCRs, neurosteroids but not HINT1 alter σ1R binding to these proteins [39, 10] (Figures 1 & 2A). In this environment, high calcium and σ1R agonists such as pregnenolone sulfate enhance the association of σ1Rs with the NR1 C1 subunits, whilst they diminish the binding of σ1Rs to GPCRs, consequently strengthening that of HINT1 to GPCRs. Thus, σ1R agonists restrain the control of GPCR-HINT1 complexes to NR1 subunits that are free of σ1Rs, e.g., silent or weakly active NMDARs. In these circumstances, the presence of the σ1R at the GPCR prevents the transfer of HINT1 from the GPCR to the NMDAR [49, 62, 9]. Conversely, regulation by σ1R antagonists like progesterone differs from that of agonists, and whilst antagonists do not alter or only slightly diminish the binding of σ1Rs to GPCRs, weakening the GPCR-HINT1 association, they do drive the removal of σ1Rs from activated NMDARs and they promote the transfer of HINT1 proteins from GPCRs to NR1 C1 subunits [39, 10]. As a result, NMDARs are uncoupled from the influence of GPCRs, be it positive or negative. These observations indicate that the HINT1-σ1R tandem is physiologically driven by calcium and the putative endogenous ligands of σ1Rs are neurosteroids.
IV. Cross-regulation between NMDARs and CB1Rs
IV.1. Molecular aspects
The HINT1-σ1R protein tandem highlighted above works as a flip-flop switch connecting and disconnecting the activity of GPCRs with that of NMDARs carrying the C1 cytosolic segment within the NR1 subunits, and it can enhance (e.g., MOR) or dampen (e.g., CB1R) glutamate signaling. As part of this molecular switch, HINT1 physically connects the GPCR to the NMDAR, the ON situation, and when it moves from the GPCR towards the NMDAR it uncouples both receptors, the OFF state. In the GPCR environment, the σ1R weakens the HINT1-NR1 association and it is crucial to maintain the HINT1 protein bound to the GPCR. Thus, its physiological or pharmacological removal brings about HINT1 transfer to the NMDAR NR1 subunit [10, 39]. Following the formation of the GPCR-HINT1-σ1R-NMDAR complex, the activation of receptors like the MOR increases the activity of the coupled NMDAR via PKC/Src. The action of PKC promotes the separation of the MOR-HINT1 complex from the phosphorylated NR1 C1 region that now carries the σ1R. On the other hand, Src phosphorylates tyrosine residues of NR2 subunits and increases calcium permeation, favoring σ1R binding to the NMDAR. Thus, activated and phosphorylated NMDARs display low affinity for the HINT1 protein and this precludes their unproductive coupling to the MOR. This cycle would commence when a σ1R plus a silent NMDAR (unphosphorylated) reach the MOR-HINT1 complex, and it ends with the release of the phosphorylated and active NMDAR [9]. Notably, antagonists impair σ1R binding to NMDARs, even in the presence of high calcium. In these circumstances, and before PKC reaches all its targets on the NR1 C1 segment, HINT1 rather than σ1R switches from the GPCR to this region of the coupled NMDAR. Thus, σ1R antagonists promote the separation of MORs from NMDAR-HINT1 complexes and disrupt the cross-regulation between these receptors. Pharmacologically we can take advantage of σ1R antagonists as adjuvants of opioid antinociception with a view to reducing the development of opioid tolerance [60].
In contrast to what is observed for the MOR, the CB1R hinders the activity of NMDARs. As witnessed for the MOR, the CB1R also forms CB1R-HINT1-σ1R complexes with non-phosphorylated NMDARs [10, 39]. However, there is no activation of the NMDAR in the CB1R environment and the σ1R remains at the GPCR allowing endocannabinoids to stabilize the weak activity of NMDARs. As observed for MORs in their interaction with NMDARs, in the absence of σ1R ligands or in the presence of σ1R agonists, the HINT1-σ1R switch enables CB1Rs to associate with inactive NMDARs, the ON situation. By contrast, σ1R antagonists promote the shift of HINT1 from the CB1R to the NMDAR NR1 subunit disconnecting both receptors, the OFF state, thereby preventing cannabinoids from producing NMDAR hypoactivity [39, 10].
In the absence of GPCR or NMDAR activity, binding of GPCR-associated HINT1 proteins to resting NMDARs is blocked by sumoylated RGS-Rz proteins, mostly RGS17 and RGS20 [42, 63, 3]. It is the activity of MOR-activated PKCγ or of NMDAR-activated CaMKII that disrupts the HINT1 interaction with the RGS-Rz barrier [63, 9, 25], thereby allowing the MOR/CB1R-HINT1 complex to associate with the NMDAR NR1 subunits (Figures 2B & 3A). Thus, the HINT1-σ1R tandem sustains the association between these GPCRs and NMDARs, and in the absence of HINT1 proteins or of σ1Rs their relationship is disrupted [37, 9, 39]. Indeed, in HINT1-/- or σ1R-/- mice, the MOR and the NMDAR are physically and functionally uncoupled, and thus, morphine does not recruit NMDARs nor do NMDARs dampen opioid antinociception [63, 9]. Similarly, in these mice cannabinoids fail to reduce NMDAR calcium influx and the subsequent release of endogenous zinc, and they also provide no protection against NMDAR excitotoxicity [25, 4].
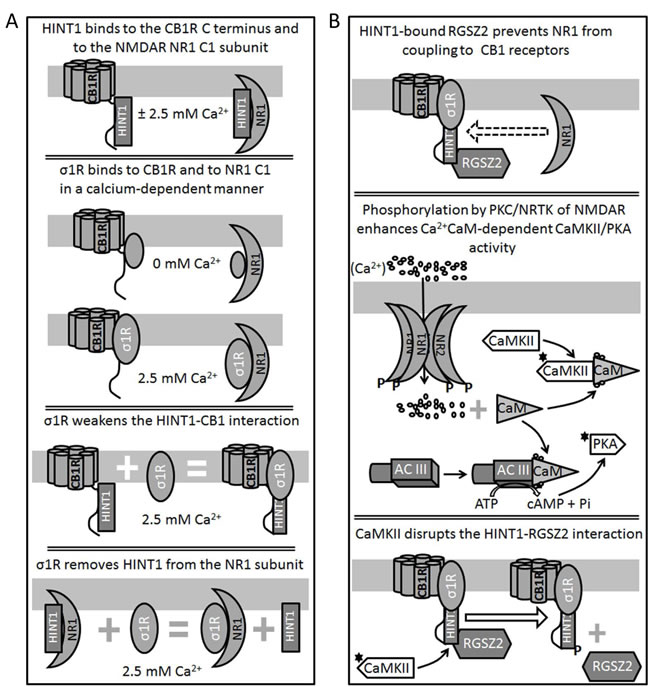
Figure 2: Diagram showing the relationship between HINT1 proteins and σ1Rs in their association with the NMDAR NR1 C1 subunits and CB1Rs. A. Binding of HINT1 and σ1R to NR1 subunits and CB1Rs. Their interaction is unidirectional and while HINT1 does not dampen σ1R binding to CB1R or NR1 subunits, the σ1R dissociates HINT1 from the NR1 and weakens its interaction with the CB1R. B. CB1Rs bind to the NMDAR NR1 subunits via HINT1 proteins. The binding of RGS-Rz proteins to HINT1 prevents the formation of the CB1R-NMDAR complex and, NMDAR-activated CaMKII removes this barrier to make their coupling and cross-regulation possible.
IV.2. Functional aspects
If the activity of NMDARs reaches a given threshold, excitatory signals recruit the negative control of the endocannabinoid system via CB1Rs [34]. Thus, the NMDAR-induced release of endocannabinoids [64] provokes the stabilization of CB1R-HINT1 complexes along with silent NMDARs [25, 39], thereby reducing the pool of NMDARs that can be potentially activated (Figure 3A). Since exocannabinoids internalize CB1Rs better than endocannabinoids [38], they promote the co-internalization of the CB1R-HINT1 complexes bound to NR1 subunits and probably, to surface NMDAR NR2 subunits as well [65, 25]. Thus, exocannabinoids better disassemble and inactivate CB1R-associated NMDARs efficiently reducing the risk of the excitotoxicity mediated by NMDAR calcium influx. Notwithstanding, the absence of σ1Rs disrupts the control cannabinoids exert on NMDAR excitatory signaling. In these mice, CB1Rs are separated from the NR1 subunits and the HINT1 proteins switch to the NR1 C1 subunits [9, 39].
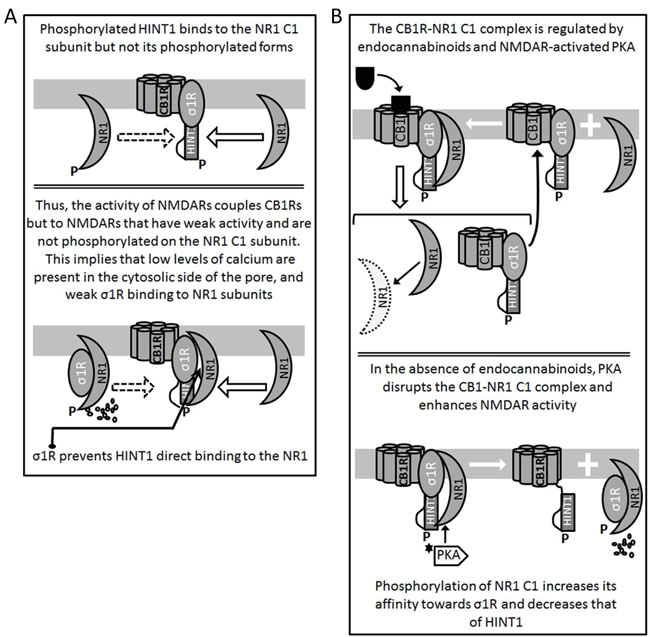
Figure 3: Formation and regulation of the CB1R-NMDAR complex. A. CB1R-coupled HINT1 proteins freed of RGS-Rz proteins bind to the C1 region of NR1 subunits. PKC/PKA phosphorylation, such as that of activated NMDARs weakens the HINT1-NR1 C1 association (P-S890, 896, 897) while P-T879 abolishes it [9]. B. Cannabinoids and PKA determine the fate of the CB1R-NMDAR complex. When cannabinoids bind to CB1Rs they dampen the activity of the coupled NMDARs and they may even provoke the co-internalization of NR1 subunits. As a result, overall NMDAR activity diminishes. However, in the absence of CB1R-bound cannabinoids, PKA acts on the NR1 C1 segment and disrupts its association with HINT1 in the CB1R complex.
V. The CB1R-NMDAR complex
V.1. Functional relevance
Abnormally high spiking activity can damage neurons and the endogenous cannabinoid system provides on-demand protection against acute excitotoxicity. A series of studies suggest that the endocannabinoid system controls NMDAR activity intracellularly through signaling pathways that converge on those triggered by the glutamate receptor [66, 67, 68, 69], although other studies indicate that this control is the result of direct physical coupling between CB1Rs and NMDAR NR1 subunits [25, 39]. In this respect, the absence of CB1Rs abrogates the control that endocannabinoids exert on NMDAR activity, whilst the pharmacological antagonism of NMDARs decreases cannabinoid CB1R mRNA expression [67, 34] [67, 34]. The CB1R is one of the most abundant GPCRs in the nervous system and although it is mostly localized at the pre-synapse, it is also present in the somata and dendrites [70, 71]. Moreover, there is immunocytochemical and ultrastructural evidence that CB1Rs exist in the post-synapse, both at the spinal [72, 68, 73] and supraspinal level [74, 71], co-localizing with NMDARs and PSD95 proteins [75, 25].
The presence of NMDARs in the pre-synapse [76, 77] makes the physical association between CB1Rs and NR1 subunits possible at both sides of the synaptic cleft. As such, pre-synaptic CB1Rs could reduce the release of glutamate into the cleft, contributing to NMDAR hypofunction [78], whereas post-synaptic CB1Rs might interfere with intracellular NMDAR signaling [66], thereby negatively regulating the activity of glutamate by directly inhibiting calcium influx [66, 79]. This latter possibility is also supported by whole-cell patch clamp recordings [78]. Thus, besides interfering with NMDAR signaling, cannabinoids can also directly diminish NMDAR mediated calcium flux channel. In this respect, the control exerted by cannabinoids on NMDAR calcium influx, zinc metabolism and excitotoxicity requires CB1Rs, HINT1 and σ1R proteins. In the absence of the σ1Rs or HINT1 proteins, cannabinoids cannot control NMDARs yet the expression of these proteins in HINT1 and σ1R deficient mice restores the cross-regulation between CB1Rs and NMDARs [4, 9]. These observations bring to the fore the role of HINT1 and σ1R proteins in the restraint that endocannabinoids exert on NMDAR function through CB1Rs.
The HINT1-σ1R machinery couples the CB1R to the NMDAR and controls its capacity to promote oxidative stress, a regulatory event in which PKA plays an essential role. Thus, NMDAR activity augments the formation of the Ca2+-CaM that regulates adenylyl cyclase activity, primarily that of types I and VIII but to a lesser extent that of type III, increasing cAMP levels and consequently PKA activity [80, 81]. PKA phosphorylates the protein inhibitor-1 and inhibits the PP1 responsible for dephosphorylating P-Thr286 and inhibiting CaMKII [18, 19]. CaMKII displaces RGS-Rz proteins from HINT1 proteins associated to CB1Rs, an event that is necessary for endocannabinoids to promote and stabilize the inhibitory association of CB1Rs with NMDARs [25] (Figure 3B). Notwithstanding, PKA also favors NMDAR stimulated Ca2+ currents, disrupting the CB1R-NMDAR complexes not affected by endocannabinoids, and thereby preserving glutamate function [25] (Figure 3B). Hence, the formation of the CB1R-NMDAR complex requires HINT1 and σ1R but also, endocannabinoids and NMDAR-activated PKA, exerting bidirectional control on this mechanism.
When NMDAR activity decreases, the calcium concentration falls, as does the strength of Ca2+-CaM/AC/cAMP/PKA/CaMKII signaling. In these circumstances, the formation of CB1R-NMDAR complexes diminishes, and the NMDARs in the existing complexes display little or no activity. Thus, both receptors should be disconnected to prevent undesirable glutamate hypofunction, and in conditions of low calcium/low PKA activity, this regulation is achieved by transferring of HINT1 proteins from CB1Rs to the NMDAR NR1 C1 subunits (Figure 4). The σ1R and its endogenous regulators, probably neurosteroids, apparently play an essential role in this physiological process, which releases NMDAR activity from the negative control of cannabinoids [39, 9].
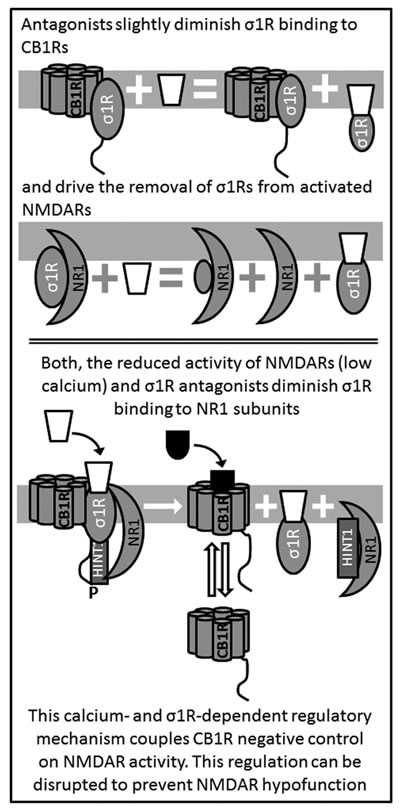
Figure 4: Antagonists of σ1R release NMDAR from the negative control of CB1Rs.
V.2. Implications in neural disturbances
Our current understanding of glutamate NMDAR neurotransmission enables us to better define the benefits and risks of its pharmacological manipulation. Whilst, excessive NMDAR activity can be excitotoxic, compromising cell viability, too little activity dysregulates the coordination between GPCRs and NMDARs to set synaptic tone [82]. In order to prevent these anomalies, the endogenous cannabinoid system collaborates with NMDARs to maintain their activity within physiological limits.
V.2.1 Psychosis/Schizophrenia - NMDAR hypofunction
The prolific amount of data being generated through studies into psychosis/schizophrenia suggests that both GPCRs and NMDARs participate in the pathophysiology of these mental illnesses. Alterations to GPCRs, like dopamine and GABA receptors, concur with a decrease in NMDAR activity in patients suffering psychosis/schizophrenia [83, 84, 85]. The relationship between these GPCRs and NMDARs is bidirectional, and this cross-regulation could account for the disturbances observed between NMDAR transmission and dopamine receptors in schizophrenia. Indeed, experimental NMDAR hypofunction (induced by its antagonists) causes glutamate metabotropic hyperfunction and dopaminergic hypofunction in the prefrontal cortex, as well as inducing psychotic symptoms and neurocognitive disturbances similar to schizophrenia [83, 16]. Hence, NMDAR dysfunction appears to lie at the crux of the hierarchy of events provoking schizophrenia.
Several clinical and neurobiological findings suggest that endocannabinoids are implicated in NMDAR dysfunction and thus, in the pathophysiology of schizophrenia [86, 87]. The CB1R gene (CNR1) maps to chromosome 6q14-15 and linkage studies have suggested a schizophrenia-susceptibility locus lies in this region [88, 89]. A variety of CNR1 polymorphisms have been studied for associations with schizophrenia, with mixed results [90, 91, 92, 93]. Post-mortem studies carried out on the brains of patients with schizophrenia have demonstrated alterations to the CB1R, such as reduced levels of its mRNA and protein expression in the dorsolateral prefrontal cortex [94], or increased CB1R binding in the corticolimbic areas implicated in this disorder [95, 96, 97]. Accordingly, it is commonly accepted that prolonged cannabis consumption precipitates symptoms of psychosis in vulnerable subjects [98, 99, 100], as well as triggering the relapse of psychotic symptoms in schizophrenic patients and worsening other symptoms of schizophrenia [87, 101].
The endogenous cannabinoid anandamide is involved in regulating pain, mood and cognition [102], and its content in cerebrospinal fluid and plasma augments in patients with schizophrenia, although these levels are negatively correlated with the intensity of the symptoms experienced by these subjects [103, 104, 105]. Pharmacological blockade of anandamide degradation in rodents appears to attenuate certain psychotic-like behaviors induced by amphetamine and phencyclidine [106]. Conversely, the psychotic symptoms induced by Δ9-tetrahydrocannabinol and other cannabinoid agonists in healthy volunteers [107, 108] and schizophrenic patients [109] suggest that hyperactivity of the endocannabinoid system contributes to the psychotic state. Thus, it is unclear whether endocannabinoids protect against or intensify schizophrenia [110]. The evidence suggests that exocannabinoids more effectively precipitate psychotic symptoms than endocannabinoids, and that they may even play opposite roles in the expression of this mental illness. The effect of CB1R antagonism in schizophrenia has been evaluated in pre-clinical and clinical studies yielded promising although not definite results [111, 112, 113]. Unfortunately, few studies are available on the therapeutic use of cannabinoids in psychosis and schizophrenia.
The functional relationship between CB1Rs and silent NMDARs depends on the HINT1-σ1R switch, and it is stabilized by endocannabinoids. The σ1R is a calcium sensor [49] that associates with the CB1R-NMDAR complex and when calcium levels are reduced, σ1R antagonists release inactive NMDARs from their association with CB1Rs through the transfer of HINT1 proteins. The freed NMDAR can then be activated, preventing endocannabinoids from producing glutamate hypofunction [39] (Figure 5A). Delays in operating this molecular switch would promote NMDAR hypofunction and the persistence of such a situation could bring about symptoms of psychosis, possibly even precipitating schizophrenia. Similarly, if the endocannabinoid system applies a disproportionate negative control on NMDAR activity (i.e.: there is early recruitment of endocannabinoids and/or an increased number of functional CB1Rs), HINT1 swaps to NMDARs, disconnecting both receptors and preventing glutamatergic hypofunction. Notwithstanding, this early and inopportune recruitment of the endocannabinoid system prevents the HINT1-primed pool of NMDARs from associating with GPCR-HINT1 complexes, thereby reducing the influence of the GPCR-NMDAR system on synaptic tone (Figure 5B). These situations may be exaggerated when exocannabinoids co-internalize CB1Rs and NMDAR subunits, accelerating the onset and duration of NMDAR hypofunction. In such circumstances, antagonists of CB1Rs or an increase in endocannabinoids could counteract the negative actions of exocannabinoids.
Hence, the endocannabinoid system as a target of exocannabinoids is a candidate to produce schizophrenia by inducing NMDAR hypofunction and/or altering NMDAR-GPCR cross-regulation [3, 39], while in humans the HINT1 and σ1R genes have also been implicated in schizophrenia [114, 115, 116]. Mice lacking the HINT1 protein display altered dopamine transmission that might favor drug abuse [117], or antidepressant and anxiolytic-like behaviors [118]. Notably, σ1R ligands induce antidepressant and anxiolytic-like behaviors in mice [119, 53], effects that could derive from the regulatory role of σ1Rs on the HINT1 protein in the GPCR-NMDAR complex.
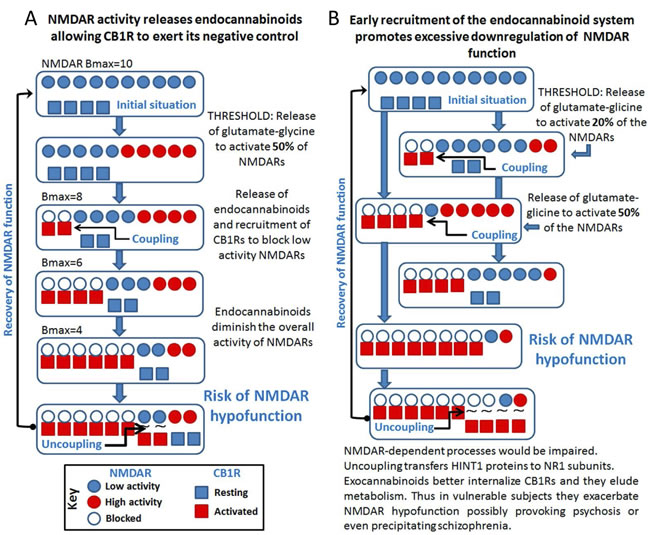
Figure 5: Cross-regulation between excitatory glutamate NMDAR signaling and the endocannabinoid system: Implications in psychosis and schizophrenia. A. The activity of NMDARs demands endocannabinoid control via CB1Rs. B. Disproportionate endocannabinoid control could cause NMDAR hypofunction.
V.2.2 Convulsive disorders - NMDAR hyperfunction
Epilepsy is a chronic disorder suffered by approximately 50 million people worldwide (WHO, Fact sheet N° 999, May 2015). It is well established that altered central inhibitory (e.g., γ-aminobutyric acid or GABA) and excitatory (e.g., glutamate) neurotransmission plays a pivotal role in the etiology of epilepsy, with excess glutamatergic transmission and the ensuing overactivation of glutamate receptors being particularly relevant to its clinical manifestations [20, 120, 121]. Many basic and clinical studies have focused over the past two decades on NMDARs, showing how blocking or suppressing NMDAR activity can prevent, and in some cases reverse, certain pathological effects associated with neurological diseases, including epilepsy [122, 123, 82].
In epilepsy it would appear that NMDAR stimulation escapes from the physiological controls responsible for maintaining excitatory activity within tolerable limits (Figure 6A). Different strategies have been explored to alleviate convulsive disorders in which NMDARs are implicated. Both competitive and non-competitive NMDAR channel blockers provoke potent anti-convulsant activity [124, 125, 126], although treatment of epilepsy with chronic selective NMDAR antagonists has mostly disappointed in clinical trials [127]. The side-effects of NMDAR antagonists, pose significant problems, as they include memory dysfunction, learning deficits, psychotomimetic effects and motor disturbances [122]. High-doses of the low-affinity and non-competitive NMDAR antagonist memantine (e.g., 20 mg/kg) induce spontaneous motor seizures in amygdala-kindled rats [124]. Yet, at an adequate dose memantine has anticonvulsant effects against maximal electroshock seizures [128, 129] and seizures induced by different chemoconvulsants [130, 131, 126, 32]. Similarly, there is evidence that another blocker of NMDAR channels, ketamine, may also be useful to treat refractory status epilepticus [132]. Notably, these antagonists show preference for highly activated NMDARs, with the unblocked receptors functioning normally.
The drugs currently used to treat epilepsy (antiepileptic drugs -AEDs) mostly decrease electrical activity in the brain by: i) preventing neuronal depolarization by blocking excitatory sodium or calcium channels; ii) enhancing the depressor function of potassium channels; iii) inhibiting the excitatory action of glutamate; or iv) inhibiting neuronal excitability by GABA [133]. The efficacy of these medications varies in function of etiology. Despite the relatively large number of AEDs available to treat convulsive syndromes, up to 30% of patients are resistant to the pharmacotherapies currently available [134, 135, 136] and some are not candidates for surgery. Therefore, therapeutic interventions are still sought for such epilepsies unresponsive to the available treatments. Dravet and Lennox-Gastaut syndromes are examples where pharmacoresistant epilepsy not only responds poorly to conventional AEDs, but some AEDs may even worsen the patient’s condition.
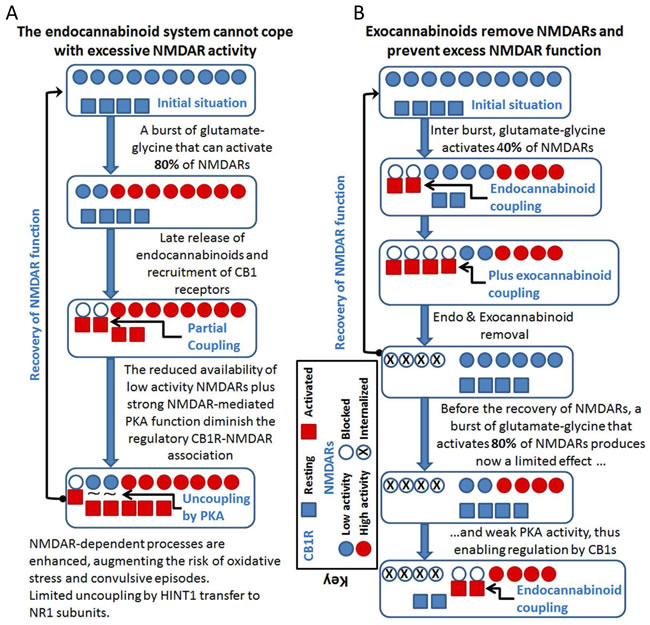
Figure 6: Cannabinoids can prevent the overactivation of glutamate NMDARs and reduce the incidence of convulsive episodes. A. If the endocannabinoid system fails to control excess NMDAR activation, PKA activity increases disrupting CB1R-NMDAR complexes before they can be acted on by endocannabinoids. B. Exocannabinoids acting through NMDAR-recruited CB1Rs would reduce glutamate excitatory signaling. The exocannabinoids acting on the preformed CB1R-NMDAR complexes prevent PKA from disrupting these complexes (indicated as “plus exocannabinoid coupling”).
V.2.2.1. The cannabinoid system as an anti-convulsant
Intensive ongoing research with cannabinoids has produced some promising results in terms of the treatment of pediatric epilepsy and there is evidence that the endocannabinoid system plays a key role in regulating seizure activity in brain [137, 138, 139, 140]. NMDAR hyperactivity might be implicated in the manifestation of these convulsive syndromes and thus, interest has grown regarding the role of endocannabinoids as antiepileptic agents [141, 142]. In some preclinical models of seizures, Δ9-tetrahydrocannabinol (Δ9-THC) and synthetic CB1R agonists reduced seizure frequency or severity. However, no such effect or even potentiation of convulsive episodes has been reported elsewhere [143]. Thus, activation of CB1Rs by exogenous substances has an anticonvulsant effect in various models of experimental epilepsy, such as the maximal electroshock model of grand-mal seizure [142, 144], the rat pilocarpine model of acquired epilepsy [145, 138, 140], the in vitro hippocampal neuronal culture models of acquired epilepsy and status epilepticus [146, 137], the pentylenetetrazole (PTZ) model of myoclonic seizures in mice [147, 148], and the penicillin-induced model of epileptiform activity in rats [149]. Since exogenous activators of CB1Rs alleviate these epileptogenic syndromes, the endogenous cannabinoid receptors must be operative but their control on NMDARs is overridden by glutamatergic dysfunction (Figure 6B).
V.2.2.2. Cannabis sativa
For thousands of years, humans have used the Cannabis sativa plant for its sedative/hypnotic, antidepressant, analgesic, anticonvulsant, antiemetic, anti-inflammatory, anti-spasmodic and appetite-stimulating effects [86]. Thus, it is not surprising that current medicine should take advantage of the anti-epileptic potential of cannabis [150]. However, the use of cannabis by individuals to treat their epilepsy may precipitate a re-emergence of convulsive seizures when it is no longer used, while resuming cannabis consumption again controls epilepsy. Therefore, it has been complicated to obtain consistent data regarding the benefits of cannabis consumption as an anticonvulsant. In a recent informal interview of >215 patients with active epilepsy who have used recreational cannabis intermittently or regularly, more than 90% of them failed to appreciate any benefits of cannabis in seizure control. Only 7% believed that their seizures were better controlled while the remainder felt that their seizures were worse due to cannabis use [151]. In a 1976 study, 29% of patients with epilepsy reported self-medication with cannabis for their condition, of whom one reported that cannabis provoked seizures and another patient indicated an improvement with cannabis use [152]. In a more recent Canadian study, 28/165 patients with epilepsy were active users of cannabis, of whom 68% reported improvements in seizure severity and 54% in seizure frequency [153]. A careful analysis showed that cannabis use in men (but not in women) protected against new-onset unprovoked seizures and against new-onset provoked seizures when used within 90 days of seizure presentation. Yet overall, there is insufficient epidemiological data to reach hard conclusions [154], even though cannabis may protect patients from new-onset seizures and it may help patients with established epilepsy control their seizures.
V.2.3 Phytocannabinoids in schizophrenia and convulsive disorders
Cannabis contains several substances with unknown effects on psychosis/schizophrenia and epilepsy, including phytocannabinoids and non-cannabinoid compounds. Indeed, this plant has a complex mixture of chemicals that includes phytocannabinoids, terpenoids, flavonoids, steroids and enzymes [155] with Δ9-THC, cannabidiol (CBD), and cannabinol (CBN) constituting the major cannabinoids in marijuana. Despite the fact that the potential benefits remains unclear, interest in the therapeutic potential of compounds derived from Cannabis sativa has resurged in recent years. Thus, well-designed retrospective and prospective studies should be carried out to investigate the various cannabis preparations, strengths and compositions that have been studied. The principal psychoactive component of marijuana is Δ9-THC, which is a partial agonist of the CB1Rs that are primarily located in the brain (on inhibitory GABAergic and excitatory glutamatergic neurons) [156]. Δ9-THC is also a partial agonist of the CB2Rs that are mainly located on immune and hematopoietic cells, yet also to some extent on neural cells.
CBD is probably the most promising non-psychoactive anti-convulsant and anti-psychotic phytocannabinoid investigated to date. CBD has been seen to exert anti-convulsant effects in animal models and humans [157, 158, 159, 142], and it prevents some of the psychotic-like effects produced by Δ9-THC [160]. Such effects are promoted through mechanisms that remain unknown but that probably do not involve direct binding to the CB1R. In this respect, CBD only weakly competes with 3[H] CP55940 at both CB1Rs and CB2Rs, and at concentrations in the micromolar range [161, 162]. Despite its low affinity for CB receptors, CBD can produce effects at reasonably low concentrations and in fact, in the low nanomolar range CBD it alters the binding of agonists to the CB1/CB2 receptors [162]. However, while the role of CBD at CBRs remains controversial, its influence on endocannabinoid signaling appears convincing. Indeed, CBD potentiates such signaling, increasing anandamide levels by inhibiting its reuptake and degradation, the latter involving a dampening of FAAH expression and activity (fatty acid amide hydrolase - the enzyme involved in anandamide breakdown) [163, 164]. However, the concentrations of CBD required to inhibit anandamide reuptake and hydrolysis are quite high (>20 μM) [163].
It should be noted that CBD exerts other effects that could also contribute to its antiepileptic/antipsychotic activities. These include the modulation of the equilibrative nucleoside transporter, the orphan G-protein-coupled receptor 55 and the transient receptor potential of melastatin type 8 channel [165]. CBD modifies intracellular calcium concentrations and it inhibits T-type calcium channels [166]. At higher concentrations, CBD activates the nuclear peroxisome proliferator-activated receptor-γ (PPAR-γ) and the transient receptor potential of vanilloid type 1 (TRPV1) and TRPV2 channels [167]. In addition, CBD has anti-apoptotic, neuroprotective, and anti-inflammatory effects [168]. CBD also displays some agonist activity at α3 and α1 glycine receptors and at the transient receptor potential of ankyrin type 1 [169]. At present, there is no convincing information as to the precise molecular mechanisms by which CBD produces its antipsychotic or anticonvulsant effects. Thus, the relationship between CBD and NMDARs is apparently indirect, and it probably resides in the context of its influence on CB1Rs. Since our aim is to analyze these neural dysfunctions from a new perspective, that of the control exerted by the CB1R via HINT1/σ1R on NMDAR function, we will describe just a few promising findings regarding the effects of CBD on schizophrenia, and the effects of CBD combined with cannabinoids in the treatment of NMDAR convulsive episodes.
In healthy volunteers CBD attenuates the impairment of time production tasks and the euphoria induced by Δ9-THC [170, 171]. In humans, CBD significantly reduces psychotic symptoms in acute schizophrenia with potency similar to that of the antipsychotic amisulpride [167, 8, 172]. Notably, schizophrenic patients treated with CBD present higher anandamide serum levels than those receiving amisulpride, and in the CBD group there was a significant association between anandamide levels and improvement of psychotic symptoms [167]. Indeed, CBD inhibits FAAH activity at a concentration that does not interact with receptors commonly associated with schizophrenia, such as dopamine, GABA, serotonin and glutamate receptors. CBD appears to have pharmacological profile similar to that of atypical antipsychotic drugs in preventing human experimental psychosis, and it is apparently effective in open case reports and clinical trials in patients with schizophrenia [173]. Several mechanisms were proposed to explain the antipsychotic properties of CBD, such as the activation of CB1Rs via increased levels of anandamide or the activation of TRPV1 channels facilitating the pre-synaptic release of glutamate that would counteract NMDAR hypofunction. Facilitation of CB1R-mediated neurotransmission by CBD increases adult hippocampal neurogenesis, a mechanism that could improve the cognitive deficits seen in schizophrenic patients. Amongst these possible mechanisms, CBD facilitation of 5-HT1A mediated neurotransmission could account for its anti-psychotic. Besides the CB1R, certain serotonin receptors are also negatively coupled to NMDAR activity [4] and CBD displays some affinity for 5-HT1A receptors [169], providing another possible explanation for its effects that is worthy of consideration.
Alternatively, drugs like fenfluramine that acts on 5HT1R [174] can control seizures in patients with Dravet syndrome [175]. Phase 3 clinical trials with ZX008 are currently ongoing in the US and Europe (Zogenix: low-dose fenfluramine-orphan drug designation granted for ZX008 in Dravet syndrome by FDA). Therefore, it is feasible that continuous seizures (status epilepticus) can be controlled by acting on GPCRs like CB1R or 5HT1A receptors, with the HINT1-σ1R tandem coupling their function to that of over activated NMDARs. There are conflicting reports on the possible role of neurosteroids in convulsive disorders [176, 177] although the pro-convulsant effects of progesterone appear not to be related to σ1Rs given that σ1R ligands have mostly anti-convulsant effects [178, 179]. As such, recent data on the involvement of σ1R in rare CNS diseases highlights the potential of the ANAVEX 2-73 sigma ligand to treat other CNS disorders, including epilepsy [180]. Additional studies with highly selective σ1R ligands would definitely shed some light on their therapeutic potential as anti-convulsive agents.
Many of the pharmacological activities of CBD have only been established in vivo and hence, some of them may be due to CBD metabolites. Like most cannabinoids, CBD is metabolized extensively by the liver, where it is hydroxylated to 7-hydroxyl-CBD by cytochrome P450 (CYP) enzymes, predominantly isozymes of the CYP3A (2/4) and CYP2C (8/9/19) families [181]. These metabolites then undergo further metabolism in the liver, and their products are excreted in the feces and secreted in the urine [182]. Recent in vitro studies show that CBD is a potent inhibitor of multiple cytochrome P450 enzymes including CYP1A2, CYP2B6, CYP2C9, CYP2D6 and CYP3A4 [183, 184, 185, 186]. Consequently, CBD metabolism could influence the pharmacokinetics of other pharmacological agents, although there is currently little data available in this regard. In some studies, CBD has been shown to mildly augment the levels of Δ9-THC (metabolized by CYP2C9, CYP2C19 and CYP3A4) by reducing its conversion to 11-hydroxy-THC [187, 188]. Moreover, CBD reduces the potency of some anti-convulsants and it enhances that of others, even though it is uncertain whether this effect is a pharmacokinetic activity [189, 190].
V.2.4 Clinical studies
Early clinical studies to evaluate the possible efficacy of CBD and other cannabinoids in epilepsy had important methodological limitations. A recent Cochrane review identified four studies published between 1978 and 1990 that were randomized, controlled trials, blind (single or double) or unblind, and that included 9 to 15 patients [191]. These studies failed to provide evidence of cannabinoid efficacy in treating epilepsy and the main conclusion was that short term CBD treatment (in the 200-300 mg/day range) is usually well tolerated in adults [192, 158, 193, 194]. In another study, an improvement was reported in children with grand mal epilepsy that were administered isomeric homologues of TCH [195].
The use of cannabinoids in children with refractory epilepsy was assessed recently by surveying an internet-based (Facebook) group of approximately 150 children with various types of medication-resistant epilepsies, including Dravet and Lennox-Gastaut syndromes [196]. In 19 cases, 12 of whom had Dravet syndrome, parents had explored the use of CBD enriched cannabis to manage pediatric treatment-resistant epilepsy. Of the parents surveyed, 53% reported a >80% reduction in seizure frequency, with 11% of children remaining seizure free during a 3-month period. The parents also often reported better mood, improved alertness and better sleep, with no severe side effects. Interestingly, orphan drug designation has been granted by the FDA to treat Dravet and Lennox-Gastaut syndromes with the low-THC/high-CBD product Epidiolex(R) (GW Pharmaceuticals). Evidence for its efficacy has come from the first pivotal Phase 3 study on Dravet syndrome where Epidiolex was compared to a placebo, measuring the frequency of convulsive seizures during the 14-week treatment relative to the 4-week basal observation period (http://www.gwpharm.com/news.aspx; March 2016). Patients receiving Epidiolex achieved a highly significant 39% median monthly reduction in convulsive seizures compared with a 13% reduction in those receiving a placebo (p=0.01). This difference between Epidiolex and the placebo emerged during the first month of treatment and was sustained throughout the entire treatment period. Sensitivity analyses of this primary endpoint confirmed the robustness of this result.
The complex composition of the Cannabis sativa plant itself makes it a challenge to understand why it apparently has contradictory effects in epilepsy. It has 489 known constituents [155] only 70 of which are cannabinoids, with the remainder including potentially neuroactive substances such as terpenes, hydrocarbons, ketones, aldehydes and other small hydrophobic compounds capable of crossing the blood-brain barrier. Strain-specific variability in the ratio of the most common cannabinoid, THC, and the second most common cannabinoid, CBD, offers further complexity when using whole cannabis as an antiepileptic agent, although most marijuana strains used to treat epilepsy are thought to have a high CBD/THC ratio. The extraction method is also critical, as the conditions and solvents used to separate these phytocompounds may alter them.
There are multiple potential routes of administration for CBD/THC. The inhaled route is the most common delivery form as a constituent of smoked cannabis used for recreational or medicinal purposes. Delivery through aerosols or vaporization using specialized devices has been examined, reporting rapid peak plasma concentrations (<10 min) and bioavailability of ~31% [197]. Combinations of low THC/high CBD have been delivered orally in an oil-based capsule in some human trials, although poor water solubility and erratic gastrointestinal absorption leads to variable pharmacokinetics. Bioavailability from oral delivery has been estimated at 6% due to significant first-pass metabolism in the liver [198]. Oral-mucosal/sublingual delivery through sprays/lozenges has similar bioavailability to the oral route but less variable (Guy and Robson, 2004: A Phase I, Open Label, Four-Way Crossover Study to Compare the Pharmacokinetic Profiles of a Single Dose of 20 mg of a Cannabis Based Medicine Extract -CBME- GWPK0112). Transdermal approaches have also been investigated but due to the strong lipophilicity, special ethosomal delivery systems are needed to prevent drug accumulation in the skin, making this approach impractical and costly [199]. The bioavailability of oral and smoked cannabis in humans was found to be around 6% and 31%, respectively, further support for a substantial first-pass effect [200, 181, 197, 201]. The adverse effects of cannabis are likely to be minimized by using active principles rather than the whole plant in medicine. Moreover, the therapeutic bioavailability of these active principles can be controlled using the adequate route of administration and dosage.
VI. Concluding remarks
The glutamate NMDAR is implicated in certain neurological disorders and whilst psychosis/schizophrenia concurs with reduced NMDAR activity, these receptors are hyperactive in convulsive disorders like epilepsy. The use of agonists or antagonists of NMDARs to treat such conditions is commonly ineffective in clinical trials on humans as directly altering synaptic NMDAR transmission compromises neuronal survival [202]. Therefore, approaches that indirectly modulate NMDAR activity are currently being developed and validated clinically [12, 203]. Subjects with schizophrenia and bipolar disorder experience alterations to neuroactive steroids like pregnenolone, dehydroepiandrosterone and allopregnanolone [204]. These alterations might reinforce the HINT1-σ1R switch and rescue GPCR-induced activation of NMDARs, indicating that regulators of σ1Rs and HINT1 proteins [205, 9] could represent more reliable and effective therapies. In fact, preliminary clinical trials with pregnenolone highlight its potential to alleviate symptoms of schizophrenia [206], and synthetic σ1R antagonists have completed phase I safety and pharmacokinetic evaluations in humans [207]. Similarly, NMDAR-induced continuous seizures (status epilepticus) can be controlled by CB1R and here, CBD and regulators of HINT1-σ1R activity seem to be promising agents. Although the precise mechanism of action of CBD in this particular context remains unknown, its therapeutic potential may stem from endogenous compensatory systems, such as the endocannabinoid system. Research in this field is particularly relevant to treat severe seizures in pediatric epilepsy. Thus, palliative treatments for psychosis/schizophrenia and convulsive syndromes that directly focus on NMDAR or GPCR activity could be complemented or even substituted with others that modify the GPCR/NMDAR interactions.
Funding
The work was supported by Spanish Ministerio de Sanidad, Servicios Sociales e Igualdad, Plan de Drogas (grant number 2014-012) and the Ministerio de Economía y Competividad (grant number SAF 2015-65420R).
Conflicts of Interest
The authors have no conflicts of interest to declare.
References
1. Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nature Neuroscience. 1999; 2: 331-338.
2. Rojas A, Dingledine R. Ionotropic glutamate receptors: regulation by G-protein-coupled receptors. Molecular Pharmacology. 2013; 83: 746-752.
3. Sánchez-Blázquez P, Rodríguez-Muñoz M, Garzón J. The cannabinoid receptor 1 associates with NMDA receptors to produce glutamatergic hypofunction: implications in psychosis and schizophrenia. Frontiers in Pharmacology. 2014; 4: 169.
4. Vicente-Sánchez A, Sánchez-Blázquez P, Rodríguez-Muñoz M, Garzón J. HINT1 protein cooperates with cannabinoid 1 receptor to negatively regulate glutamate NMDA receptor activity. Molecular Brain. 2013; 6: 42.
5. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nature Reviews Neuroscience. 2013; 14: 383-400.
6. Regan MC, Romero-Hernandez A, Furukawa H. A structural biology perspective on NMDA receptor pharmacology and function. Current Opinion in Structural Biology. 2015; 33: 68-75.
7. Lu HC, Mackie K. An Introduction to the Endogenous Cannabinoid System. Biological Psychiatry. 2016; 79: 516-525.
8. Leweke FM, Mueller JK, Lange B, Rohleder C. Therapeutic Potential of Cannabinoids in Psychosis. Biological Psychiatry. 2016; 79: 604-612.
9. Rodríguez-Muñoz M, Sánchez-Blázquez P, Herrero-Labrador R, Martínez-Murillo R, Merlos M, Vela JM, Garzón J. The sigma1 receptor engages the redox-regulated HINT1 protein to bring opioid analgesia under NMDA receptor negative control. Antioxidants & Redox Signaling. 2015; 22: 799-818.
10. Rodríguez-Muñoz M, Cortés-Montero E, Pozo-Rodrigálvarez A, Sánchez-Blázquez P, Garzón-Niño J. The ON:OFF switch, sigma1R-HINT1 protein, controls GPCR-NMDA receptor cross-regulation: Implications in neurological disorders. Oncotarget. 2015; 6: 35458-35477. doi: 10.18632/oncotarget.6064.
11. Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. Journal of Nutrition. 2000; 130: 1007S-1015S.
12. Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nature Reviews Drug Discovery. 2006; 5: 160-170.
13. Chapman V, Haley JE, Dickenson AH. Electrophysiologic analysis of preemptive effects of spinal opioids on N-methyl-D-aspartate receptor-mediated events. Anesthesiology. 1994; 81: 1429-1435.
14. Sigtermans MJ, van Hilten JJ, Bauer MC, Arbous MS, Marinus J, Sarton EY, Dahan A. Ketamine produces effective and long-term pain relief in patients with Complex Regional Pain Syndrome Type 1. Pain. 2009; 145: 304-311.
15. Maeng S, Zarate CA, Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Current Psychiatry Reports. 2007; 9: 467-474.
16. Mechri A, Saoud M, Khiari G, d’Amato T, Dalery J, Gaha L. Glutaminergic hypothesis of schizophrenia: clinical research studies with ketamine. Encephale. 2001; 27: 53-59.
17. Mori H, Mishina M. Structure and function of the NMDA receptor channel. Neuropharmacology. 1995; 34: 1219-1237.
18. Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998; 280: 1940-1942.
19. Makhinson M, Chotiner JK, Watson JB, O’Dell TJ. Adenylyl cyclase activation modulates activity-dependent changes in synaptic strength and Ca2+/calmodulin-dependent kinase II autophosphorylation. Journal of Neuroscience. 1999; 19: 2500-2510.
20. Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacological Reviews. 1999; 51: 7-61.
21. Grishin AA, Benquet P, Gerber U. Muscarinic receptor stimulation reduces NMDA responses in CA3 hippocampal pyramidal cells via Ca2+-dependent activation of tyrosine phosphatase. Neuropharmacology. 2005; 49: 328-337.
22. Yuen EY, Jiang Q, Chen P, Gu Z, Feng J, Yan Z. Serotonin 5-HT1A receptors regulate NMDA receptor channels through a microtubule-dependent mechanism. Journal of Neuroscience. 2005; 25: 5488-5501.
23. Liu W, Yuen EY, Allen PB, Feng J, Greengard P, Yan Z. Adrenergic modulation of NMDA receptors in prefrontal cortex is differentially regulated by RGS proteins and spinophilin. Proceedings of the National Academy of Sciences USA. 2006; 103: 18338-18343.
24. Gu Z, Liu W, Wei J, Yan Z. Regulation of N-methyl-D-aspartic acid (NMDA) receptors by metabotropic glutamate receptor 7. Journal of Biological Chemistry. 2012; 287: 10265-10275.
25. Sánchez-Blázquez P, Rodríguez-Muñoz M, Vicente-Sánchez A, Garzón J. Cannabinoid receptors couple to NMDA receptors to reduce the production of NO and the mobilization of zinc induced by glutamate. Antioxidants & Redox Signaling. 2013; 19: 1766-1782.
26. Grosshans DR, Browning MD. Protein kinase C activation induces tyrosine phosphorylation of the NR2A and NR2B subunits of the NMDA receptor. Journal of Neurochemistry. 2001; 76: 737-744.
27. Martin G, Nie Z, Siggins GR. Mu-opioid receptors modulate NMDA receptor-mediated responses in nucleus accumbens neurons. Journal of Neuroscience. 1997; 17: 11-22.
28. Greengard P. The neurobiology of slow synaptic transmission. Science. 2001; 294: 1024-1030.
29. Guo W, Wei F, Zou S, Robbins MT, Sugiyo S, Ikeda T, Tu JC, Worley PF, Dubner R, Ren K. Group I metabotropic glutamate receptor NMDA receptor coupling and signaling cascade mediate spinal dorsal horn NMDA receptor 2B tyrosine phosphorylation associated with inflammatory hyperalgesia. Journal of Neuroscience. 2004; 24: 9161-9173.
30. Tyszkiewicz JP, Gu Z, Wang X, Cai X, Yan Z. Group II metabotropic glutamate receptors enhance NMDA receptor currents via a protein kinase C-dependent mechanism in pyramidal neurones of rat prefrontal cortex. Journal of Physiology. 2004; 554: 765-777.
31. Yuen EY, Jiang Q, Chen P, Feng J, Yan Z. Activation of 5-HT2A/C receptors counteracts 5-HT1A regulation of N-methyl-D-aspartate receptor channels in pyramidal neurons of prefrontal cortex. Journal of Biological Chemistry. 2008; 283: 17194-17204.
32. Pasternak GW, Kolesnikov YA, Babey AM. Perspectives on the N-methyl-D-aspartate/nitric oxide cascade and opioid tolerance. Neuropsyschopharmacology. 1995; 13: 309-313.
33. Rodríguez-Muñoz M, Garzón J. Nitric Oxide and Zinc-Mediated Protein Assemblies Involved in Mu Opioid Receptor Signaling. Molecular Neurobiology. 2013; 48: 769-782.
34. Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, Lopez-Rodriguez ML, Casanova E, Schutz G et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003; 302: 84-88.
35. Fiorentini C, Gardoni F, Spano P, Di LM, Missale C. Regulation of dopamine D1 receptor trafficking and desensitization by oligomerization with glutamate N-methyl-D-aspartate receptors. Journal of Biological Chemistry. 2003; 278: 20196-20202.
36. Perroy J, Raynaud F, Homburger V, Rousset MC, Telley L, Bockaert J, Fagni L. Direct interaction enables cross-talk between ionotropic and group I metabotropic glutamate receptors. Journal of Biological Chemistry. 2008; 283: 6799-6805.
37. Rodríguez-Muñoz M, Sánchez-Blázquez P, Vicente-Sánchez A, Berrocoso E, Garzón J. The Mu-Opioid Receptor and the NMDA Receptor Associate in PAG Neurons: Implications in Pain Control. Neuropsychopharmacology. 2012; 37: 338-349.
38. Garzón J, Torre-Madrid E, Rodríguez-Muñoz M, Vicente-Sánchez A, Sánchez-Blázquez P. Gz mediates the long-lasting desensitization of brain CB1 receptors and is essential for cross-tolerance with morphine. Molecular Pain. 2009; 5: 11.
39. Sánchez-Blázquez P, Rodríguez-Muñoz M, Herrero-Labrador R, Burgueño J, Zamanillo D, Garzón J. The calcium-sensitive Sigma-1 receptor prevents cannabinoids from provoking glutamate NMDA receptor hypofunction: implications in antinociception and psychotic diseases. International Journal of Neuropsychopharmacology. 2014; 17: 1943-1955.
40. Sánchez-Blázquez P, Rodríguez-Muñoz M, Bailón C, Garzón J. GPCRs promote the release of zinc ions mediated by nNOS/NO and the Redox transducer RGSZ2 protein. Antioxidants & Redox Signaling. 2012; 17: 1163-1177.
41. Rodríguez-Muñoz M, Torre-Madrid E, Sánchez-Blázquez P, Wang JB, Garzón J. NMDAR-nNOS generated zinc recruits PKCgamma to the HINT1-RGS17 complex bound to the C terminus of Mu-opioid receptors. Cellular Signalling. 2008; 20: 1855-1864.
42. Ajit SK, Ramineni S, Edris W, Hunt RA, Hum WT, Hepler JR, Young KH. RGSZ1 interacts with protein kinase C interacting protein PKCI-1 and modulates mu opioid receptor signaling. Cellular Signalling. 2007; 19: 723-730.
43. Rodríguez-Muñoz M, Torre-Madrid E, Sánchez-Blázquez P, Garzón J. NO-released zinc supports the simultaneous binding of Raf-1 and PKCgamma cysteine-rich domains to HINT1 protein at the mu-opioid receptor. Antioxidants & Redox Signaling. 2011; 14: 2413-2425.
44. Kitaichi K, Chabot JG, Moebius FF, Flandorfer A, Glossmann H, Quirion R. Expression of the purported sigma1 (s1) receptor in the mammalian brain and its possible relevance in deficits induced by antagonism of the NMDA receptor complex as revealed using an antisense strategy. Journal of Chemical Neuroanatomy. 2000; 20: 375-387.
45. Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. Journal of Pharmacology and Experimental Therapeutics. 1976; 197: 517-532.
46. Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. The Proceedings of the National Academy of Sciences USA. 1996; 93: 8072-8077.
47. Schmidt HR, Zheng S, Gurpinar E, Koehl A, Manglik A, Kruse AC. Crystal structure of the human sigma1 receptor. Nature. 2016; 532: 527-530.
48. Balasuriya D, Stewart AP, Edwardson JM. The sigma-1 receptor interacts directly with GluN1 but not GluN2A in the GluN1/GluN2A NMDA receptor. Journal of Neuroscience. 2013; 33: 18219-18224.
49. Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007; 131: 596-610.
50. Kourrich S, Hayashi T, Chuang JY, Tsai SY, Su TP, Bonci A. Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell. 2013; 152: 236-247.
51. Aydar E, Palmer CP, Klyachko VA, Jackson MB. The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron. 2002; 34: 399-410.
52. Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends in Pharmacological Sciences. 2010; 31: 557-566.
53. Hayashi T, Tsai SY, Mori T, Fujimoto M, Su TP. Targeting ligand-operated chaperone sigma-1 receptors in the treatment of neuropsychiatric disorders. Expert Opinion on Therapeutic Targets. 2011; 15: 557-577.
54. Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacology & Therapeutics. 2009; 124: 195-206.
55. Kourrich S, Su TP, Fujimoto M, Bonci A. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends in Neurosciences. 2012; 35: 762-771.
56. Robson MJ, Noorbakhsh B, Seminerio MJ, Matsumoto RR. Sigma-1 receptors: potential targets for the treatment of substance abuse. Current Pharmaceutical Design. 2012; 18: 902-919.
57. Diaz JL, Zamanillo D, Corbera J, Baeyens JM, Maldonado R, Pericas MA, Vela JM, Torrens A. Selective sigma-1 (s1) receptor antagonists: emerging target for the treatment of neuropathic pain. Central Nervous System Agents in Medicinal Chemistry. 2009; 9: 172-183.
58. Romero L, Zamanillo D, Nadal X, Sanchez-Arroyos R, Rivera-Arconada I, Dordal A, Montero A, Muro A, Bura A, Segales C, Laloya M, Hernandez E, Portillo-Salido E et al. Pharmacological properties of S1RA, a new sigma-1 receptor antagonist that inhibits neuropathic pain and activity-induced spinal sensitization. British Journal of Pharmacology. 2012; 166: 2289-2306.
59. Kim FJ, Kovalyshyn I, Burgman M, Neilan C, Chien CC, Pasternak GW. Sigma 1 receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Molecular Pharmacology. 2010; 77: 695-703.
60. Vidal-Torres A, de la Puente B, Rocasalbas M, Tourino C, Andreea BS, Fernandez-Pastor B, Romero L, Codony X, Zamanillo D, Buschmann H, Merlos M, Manuel BJ, Maldonado R et al. Sigma-1 receptor antagonism as opioid adjuvant strategy: Enhancement of opioid antinociception without increasing adverse effects. European Journal of Pharmacology. 2013; 711: 63-72.
61. Monnet FP, Debonnel G, Junien JL, De MC. N-methyl-D-aspartate-induced neuronal activation is selectively modulated by sigma receptors. European Journal of Pharmacology. 1990; 179: 441-445.
62. Ortega-Roldan JL, Ossa F, Schnell JR. Characterization of the human sigma-1 receptor chaperone domain structure and binding immunoglobulin protein (BiP) interactions. Journal of Biological Chemistry. 2013; 288: 21448-21457.
63. Rodríguez-Muñoz M, Sánchez-Blázquez P, Vicente-Sánchez A, Bailón C, Martín-Aznar B, Garzón J. The histidine triad nucleotide-binding protein 1 supports mu-opioid receptor-glutamate NMDA receptor cross-regulation. Cellular and Molecular Life Sciences. 2011; 68: 2933-2949.
64. Hashimotodani Y, Ohno-Shosaku T, Watanabe M, Kano M. Roles of phospholipase Cbeta and NMDA receptor in activity-dependent endocannabinoid release. Journal of Physiology. 2007; 584: 373-380.
65. Fan N, Yang H, Zhang J, Chen C. Reduced expression of glutamate receptors and phosphorylation of CREB are responsible for in vivo Delta9-THC exposure-impaired hippocampal synaptic plasticity. Journal of Neurochemistry. 2010; 112: 691-702.
66. Liu Q, Bhat M, Bowen WD, Cheng J. Signaling pathways from cannabinoid receptor-1 activation to inhibition of N-methyl-D-aspartic acid mediated calcium influx and neurotoxicity in dorsal root ganglion neurons. Journal of Pharmacology and Experimental Therapeutics. 2009; 331: 1062-1070.
67. Mailleux P, Vanderhaeghen JJ. Glutamatergic regulation of cannabinoid receptor gene expression in the caudate-putamen. European Journal of Pharmacology. 1994; 266: 193-196.
68. Ong WY, Mackie K. A light and electron microscopic study of the CB1 cannabinoid receptor in primate brain. Neuroscience. 1999; 92: 1177-1191.
69. Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. Journal of Neuroscience. 2001; 21: 109-116.
70. Pickel VM, Chan J, Kearn CS, Mackie K. Targeting dopamine D2 and cannabinoid-1 (CB1) receptors in rat nucleus accumbens. The Journal of Comparative Neurology. 2006; 495: 299-313.
71. Rodriguez JJ, Mackie K, Pickel VM. Ultrastructural localization of the CB1 cannabinoid receptor in mu-opioid receptor patches of the rat Caudate putamen nucleus. Journal of Neuroscience. 2001; 21: 823-833.
72. Hohmann AG, Briley EM, Herkenham M. Pre- and postsynaptic distribution of cannabinoid and mu opioid receptors in rat spinal cord. Brain Research. 1999; 822: 17-25.
73. Salio C, Fischer J, Franzoni MF, Conrath M. Pre- and postsynaptic localizations of the CB1 cannabinoid receptor in the dorsal horn of the rat spinal cord. Neuroscience. 2002; 110: 755-764.
74. Kofalvi A, Rodrigues RJ, Ledent C, Mackie K, Vizi ES, Cunha RA, Sperlagh B. Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: a combined immunochemical and pharmacological analysis. Journal of Neuroscience. 2005; 25: 2874-2884.
75. Marchalant Y, Cerbai F, Brothers HM, Wenk GL. Cannabinoid receptor stimulation is anti-inflammatory and improves memory in old rats. Neurobiology of Aging. 2008; 29: 1894-1901.
76. Conti F, Minelli A, DeBiasi S, Melone M. Neuronal and glial localization of NMDA receptors in the cerebral cortex. Molecular Neurobiology. 1997; 14: 1-18.
77. Corlew R, Brasier DJ, Feldman DE, Philpot BD. Presynaptic NMDA receptors: newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist. 2008; 14: 609-625.
78. Liu XJ, Salter MW. Glutamate receptor phosphorylation and trafficking in pain plasticity in spinal cord dorsal horn. European Journal of Neuroscience. 2010; 32: 278-289.
79. Zhuang SY, Bridges D, Grigorenko E, McCloud S, Boon A, Hampson RE, Deadwyler SA. Cannabinoids produce neuroprotection by reducing intracellular calcium release from ryanodine-sensitive stores. Neuropharmacology. 2005; 48: 1086-1096.
80. Chetkovich DM, Sweatt JD. NMDA receptor activation increases cyclic AMP in area CA1 of the hippocampus via calcium/calmodulin stimulation of adenylyl cyclase. Journal of Neurochemistry. 1993; 61: 1933-1942.
81. Cooper DM. Regulation and organization of adenylyl cyclases and cAMP. Biochemical Journal. 2003; 375: 517-529.
82. Parsons CG, Stoffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system—too little activation is bad, too much is even worse. Neuropharmacology. 2007; 53: 699-723.
83. Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. International Review of Neurobiology. 2007; 78: 69-108.
84. Javitt DC. Glutamatergic theories of schizophrenia. The Israel Journal of Psychiatry and Related Sciences. 2010; 47: 4-16.
85. Poels EM, Kegeles LS, Kantrowitz JT, Slifstein M, Javitt DC, Lieberman JA, Abi-Dargham A, Girgis RR. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Molecular Psychiatry. 2014; 19: 20-29.
86. Zuardi AW. History of cannabis as a medicine: a review. Revista Brasileira de Psiquiatria. 2006; 28: 153-157.
87. Leweke FM, Koethe D. Cannabis and psychiatric disorders: it is not only addiction. Addiction Biology. 2008; 13: 264-275.
88. Hoehe MR, Caenazzo L, Martinez MM, Hsieh WT, Modi WS, Gershon ES, Bonner TI. Genetic and physical mapping of the human cannabinoid receptor gene to chromosome 6q14-q15. New Biology. 1991; 3: 880-885.
89. Cao Q, Martinez M, Zhang J, Sanders AR, Badner JA, Cravchik A, Markey CJ, Beshah E, Guroff JJ, Maxwell ME, Kazuba DM, Whiten R, Goldin LR et al. Suggestive evidence for a schizophrenia susceptibility locus on chromosome 6q and a confirmation in an independent series of pedigrees. Genomics. 1997; 43: 1-8.
90. Leroy S, Griffon N, Bourdel MC, Olie JP, Poirier MF, Krebs MO. Schizophrenia and the cannabinoid receptor type 1 (CB1): association study using a single-base polymorphism in coding exon 1. American Journal of Medical Genetics. 2001; 105: 749-752.
91. Ujike H, Takaki M, Nakata K, Tanaka Y, Takeda T, Kodama M, Fujiwara Y, Sakai A, Kuroda S. CNR1, central cannabinoid receptor gene, associated with susceptibility to hebephrenic schizophrenia. Molecular Psychiatry. 2002; 7: 515-518.
92. Martinez-Gras I, Hoenicka J, Ponce G, Rodriguez-Jimenez R, Jimenez-Arriero MA, Perez-Hernandez E, Ampuero I, Ramos-Atance JA, Palomo T, Rubio G. (AAT)n repeat in the cannabinoid receptor gene, CNR1: association with schizophrenia in a Spanish population. European Archives of Psychiatry and Clinical Neuroscience. 2006; 256: 437-441.
93. Chavarria-Siles I, Contreras-Rojas J, Hare E, Walss-Bass C, Quezada P, Dassori A, Contreras S, Medina R, Ramirez M, Salazar R, Raventos H, Escamilla MA. Cannabinoid receptor 1 gene (CNR1) and susceptibility to a quantitative phenotype for hebephrenic schizophrenia. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2008; 147: 279-284.
94. Eggan SM, Hashimoto T, Lewis DA. Reduced cortical cannabinoid 1 receptor messenger RNA and protein expression in schizophrenia. Archives of General Psychiatry. 2008; 65: 772-784.
95. Dean B, Sundram S, Bradbury R, Scarr E, Copolov D. Studies on [3H]CP-55940 binding in the human central nervous system: regional specific changes in density of cannabinoid-1 receptors associated with schizophrenia and cannabis use. Neuroscience. 2001; 103: 9-15.
96. Zavitsanou K, Garrick T, Huang XF. Selective antagonist [3H]SR141716A binding to cannabinoid CB1 receptors is increased in the anterior cingulate cortex in schizophrenia. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 2004; 28: 355-360.
97. Newell KA, Deng C, Huang XF. Increased cannabinoid receptor density in the posterior cingulate cortex in schizophrenia. Experimental Brain Research. 2006; 172: 556-560.
98. Stefanis NC, Delespaul P, Henquet C, Bakoula C, Stefanis CN, van OJ. Early adolescent cannabis exposure and positive and negative dimensions of psychosis. Addiction. 2004; 99: 1333-1341.
99. Henquet C, Murray R, Linszen D, van OJ. The environment and schizophrenia: the role of cannabis use. Schizophrenia Bulletin. 2005; 31: 608-612.
100. Moore TH, Zammit S, Lingford-Hughes A, Barnes TR, Jones PB, Burke M, Lewis G. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007; 370: 319-328.
101. Sewell RA, Skosnik PD, Garcia-Sosa I, Ranganathan M, D’Souza DC. [Behavioral, cognitive and psychophysiological effects of cannabinoids: relevance to psychosis and schizophrenia]. Revista Brasileira de Psiquiatria. 2010; 32 Suppl 1: S15-S30.
102. Di M, V, Petrosino S. Endocannabinoids and the regulation of their levels in health and disease. Current Opinion in Lipidology. 2007; 18: 129-140.
103. Leweke FM, Giuffrida A, Wurster U, Emrich HM, Piomelli D. Elevated endogenous cannabinoids in schizophrenia. Neuroreport. 1999; 10: 1665-1669.
104. De MN, De PL, Orlando P, Daniele F, Fezza F, Di M, V. Endocannabinoid signalling in the blood of patients with schizophrenia. Lipids in Health and Disease. 2003; 2: 5.
105. Giuffrida A, Leweke FM, Gerth CW, Schreiber D, Koethe D, Faulhaber J, Klosterkotter J, Piomelli D. Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacology. 2004; 29: 2108-2114.
106. Seillier A, Advani T, Cassano T, Hensler JG, Giuffrida A. Inhibition of fatty-acid amide hydrolase and CB1 receptor antagonism differentially affect behavioural responses in normal and PCP-treated rats. International Journal of Neuropsychopharmacology. 2010; 13: 373-386.
107. D’Souza DC, Perry E, MacDougall L, Ammerman Y, Cooper T, Wu YT, Braley G, Gueorguieva R, Krystal JH. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: implications for psychosis. Neuropsychopharmacology. 2004; 29: 1558-1572.
108. Morrison PD, Nottage J, Stone JM, Bhattacharyya S, Tunstall N, Brenneisen R, Holt D, Wilson D, Sumich A, McGuire P, Murray RM, Kapur S, Ffytche DH. Disruption of frontal theta coherence by Delta9-tetrahydrocannabinol is associated with positive psychotic symptoms. Neuropsychopharmacology. 2011; 36: 827-836.
109. D’Souza DC, Abi-Saab WM, Madonick S, Forselius-Bielen K, Doersch A, Braley G, Gueorguieva R, Cooper TB, Krystal JH. Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis, and addiction. Biological Psychiatry. 2005; 57: 594-608.
110. Emrich HM, Leweke FM, Schneider U. Towards a cannabinoid hypothesis of schizophrenia: cognitive impairments due to dysregulation of the endogenous cannabinoid system. Pharmacology, Biochemistry and Behavior. 1997; 56: 803-807.
111. Scatton B, Sanger DJ. Pharmacological and molecular targets in the search for novel antipsychotics. Behavioural Pharmacology. 2000; 11: 243-256.
112. Meltzer HY, Arvanitis L, Bauer D, Rein W. Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. American Journal of Psychiatry. 2004; 161: 975-984.
113. Black MD, Stevens RJ, Rogacki N, Featherstone RE, Senyah Y, Giardino O, Borowsky B, Stemmelin J, Cohen C, Pichat P, Arad M, Barak S, De LA et al. AVE1625, a cannabinoid CB1 receptor antagonist, as a co-treatment with antipsychotics for schizophrenia: improvement in cognitive function and reduction of antipsychotic-side effects in rodents. Psychopharmacology (Berl). 2011; 215: 149-163.
114. Altar CA, Vawter MP, Ginsberg SD. Target identification for CNS diseases by transcriptional profiling. Neuropsychopharmacology. 2009; 34: 18-54.
115. Chen Q, Wang X, O’Neill FA, Walsh D, Kendler KS, Chen X. Is the histidine triad nucleotide-binding protein 1 (HINT1) gene a candidate for schizophrenia? Schizophrenia Research. 2008; 106: 200-207.
116. Watanabe Y, Nunokawa A, Kaneko N, Shibuya M, Egawa J, Someya T. Supportive evidence for the association between the Gln2Pro polymorphism in the SIGMAR1 gene and schizophrenia in the Japanese population: a case-control study and an updated meta-analysis. Schizophrenia Research. 2012; 141: 279-280.
117. Jackson KJ, Wang JB, Barbier E, Damaj MI, Chen X. The histidine triad nucleotide binding 1 protein is involved in nicotine reward and physical nicotine withdrawal in mice. Neuroscience Letters. 2013; 550: 129-133.
118. Barbier E, Wang JB. Anti-depressant and anxiolytic like behaviors in PKCI/HINT1 knockout mice associated with elevated plasma corticosterone level. BMC Neuroscience. 2009; 10: 132.
119. Fishback JA, Robson MJ, Xu YT, Matsumoto RR. Sigma receptors: potential targets for a new class of antidepressant drug. Pharmacology & Therapeutics. 2010; 127: 271-282.
120. McNamara JO. Cellular and molecular basis of epilepsy. Journal of Neuroscience. 1994; 14: 3413-3425.
121. Urbanska EM, Czuczwar SJ, Kleinrok Z, Turski WA. Excitatory amino acids in epilepsy. Restorative Neurology and Neuroscience. 1998; 13: 25-39.
122. Ghasemi M, Schachter SC. The NMDA receptor complex as a therapeutic target in epilepsy: a review. Epilepsy and Behavior. 2011; 22: 617-640.
123. Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist—a review of preclinical data. Neuropharmacology. 1999; 38: 735-767.
124. Löscher W, Hönack D. High doses of memantine (1-amino-3,5-dimethyladamantane) induce seizures in kindled but not in non-kindled rats. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1990; 341: 476-481.
125. Löscher W. Pharmacology of glutamate receptor antagonists in the kindling model of epilepsy. Progress in Neurobiology. 1998; 54: 721-741.
126. Meldrum BS, Turski L, Schwarz M, Czuczwar SJ, Sontag KH. Anticonvulsant action of 1,3-dimethyl-5-aminoadamantane. Pharmacological studies in rodents and baboon, Papio papio. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1986; 332: 93-97.
127. Rogawski MA. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Currents. 2011; 11: 56-63.
128. Parsons CG, Quack G, Bresink I, Baran L, Przegalinski E, Kostowski W, Krzascik P, Hartmann S, Danysz W. Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology. 1995; 34: 1239-1258.
129. Urbanska E, Dziki M, Czuczwar SJ, Kleinrok Z, Turski WA. Antiparkinsonian drugs memantine and trihexyphenidyl potentiate the anticonvulsant activity of valproate against maximal electroshock-induced seizures. Neuropharmacology. 1992; 31: 1021-1026.
130. Geter-Douglass B, Witkin JM. Behavioral effects and anticonvulsant efficacies of low-affinity, uncompetitive NMDA antagonists in mice. Psychopharmacology. 1999; 146: 280-289.
131. Mares P, Mikulecka A. Different effects of two N-methyl-D-aspartate receptor antagonists on seizures, spontaneous behavior, and motor performance in immature rats. Epilepsy and Behavior. 2009; 14: 32-39.
132. Dorandeu F, Dhote F, Barbier L, Baccus B, Testylier G. Treatment of status epilepticus with ketamine, are we there yet? CNS Neuroscience and Therapeutics. 2013; 19: 411-427.
133. Lason W, Chlebicka M, Rejdak K. Research advances in basic mechanisms of seizures and antiepileptic drug action. Pharmacological Reports. 2013; 65: 787-801.
134. Hao X, Goldberg D, Kelly K, Stephen L, Kwan P, Brodie MJ. Uncontrolled epilepsy is not necessarily the same as drug-resistant epilepsy: differences between populations with newly diagnosed epilepsy and chronic epilepsy. Epilepsy and Behavior. 2013; 29: 4-6.
135. Kwan P, Brodie MJ. Early identification of refractory epilepsy. The New England Journal of Medicine. 2000; 342: 314-319.
136. Kwan P, Schachter SC, Brodie MJ. Drug-resistant epilepsy. The New England Journal of Medicine. 2011; 365: 919-926.
137. Deshpande LS, Sombati S, Blair RE, Carter DS, Martin BR, DeLorenzo RJ. Cannabinoid CB1 receptor antagonists cause status epilepticus-like activity in the hippocampal neuronal culture model of acquired epilepsy. Neuroscience Letters. 2007; 411: 11-16.
138. Falenski KW, Carter DS, Harrison AJ, Martin BR, Blair RE, DeLorenzo RJ. Temporal characterization of changes in hippocampal cannabinoid CB(1) receptor expression following pilocarpine-induced status epilepticus. Brain Research. 2009; 1262: 64-72.
139. Luszczki JJ, Czuczwar P, Cioczek-Czuczwar A, Czuczwar SJ. Arachidonyl-2’-chloroethylamide, a highly selective cannabinoid CB1 receptor agonist, enhances the anticonvulsant action of valproate in the mouse maximal electroshock-induced seizure model. European Journal of Pharmacology. 2006; 547: 65-74.
140. Wallace MJ, Blair RE, Falenski KW, Martin BR, DeLorenzo RJ. The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. Journal of Pharmacology and Experimental Therapeutics. 2003; 307: 129-137.
141. Hofmann ME, Frazier CJ. Marijuana, endocannabinoids, and epilepsy: potential and challenges for improved therapeutic intervention. Experimental Neurology. 2013; 244: 43-50.
142. Wallace MJ, Wiley JL, Martin BR, DeLorenzo RJ. Assessment of the role of CB1 receptors in cannabinoid anticonvulsant effects. European Journal of Pharmacology. 2001; 428: 51-57.
143. dos Santos RG, Hallak JE, Leite JP, Zuardi AW, Crippa JA. Phytocannabinoids and epilepsy. Journal of Clinical Pharmacy and Therapeutics. 2015; 40: 135-143.
144. Wallace MJ, Martin BR, DeLorenzo RJ. Evidence for a physiological role of endocannabinoids in the modulation of seizure threshold and severity. European Journal of Pharmacology. 2002; 452: 295-301.
145. Falenski KW, Blair RE, Sim-Selley LJ, Martin BR, DeLorenzo RJ. Status epilepticus causes a long-lasting redistribution of hippocampal cannabinoid type 1 receptor expression and function in the rat pilocarpine model of acquired epilepsy. Neuroscience. 2007; 146: 1232-1244.
146. Blair RE, Deshpande LS, Sombati S, Falenski KW, Martin BR, DeLorenzo RJ. Activation of the cannabinoid type-1 receptor mediates the anticonvulsant properties of cannabinoids in the hippocampal neuronal culture models of acquired epilepsy and status epilepticus. Journal of Pharmacology and Experimental Therapeutics. 2006; 317: 1072-1078.
147. Gholizadeh S, Shafaroodi H, Ghasemi M, Bahremand A, Sharifzadeh M, Dehpour AR. Ultra-low dose cannabinoid antagonist AM251 enhances cannabinoid anticonvulsant effects in the pentylenetetrazole-induced seizure in mice. Neuropharmacology. 2007; 53: 763-770.
148. Shafaroodi H, Samini M, Moezi L, Homayoun H, Sadeghipour H, Tavakoli S, Hajrasouliha AR, Dehpour AR. The interaction of cannabinoids and opioids on pentylenetetrazole-induced seizure threshold in mice. Neuropharmacology. 2004; 47: 390-400.
149. Kozan R, Ayyildiz M, Agar E. The effects of intracerebroventricular AM-251, a CB1-receptor antagonist, and ACEA, a CB1-receptor agonist, on penicillin-induced epileptiform activity in rats. Epilepsia. 2009; 50: 1760-1767.
150. O’Shaughnessy WB. On the preparations of the Indian Hemp, or gunjah: Cannabis Indica their effects on the animal system in health, and their utility in the treatment of tetanus and other convulsive diseases. Provincial Medical Journal and Retrospect of the Medical Sciences. 1843; 5: 343-347.
151. Gordon E, Devinsky O. Alcohol and marijuana: effects on epilepsy and use by patients with epilepsy. Epilepsia. 2001; 42: 1266-1272.
152. Feeney DM. Letter: Marihuana use among epileptics. Journal of the American Medical Association. 1976; 235: 1105.
153. Gross DW, Hamm J, Ashworth NL, Quigley D. Marijuana use and epilepsy: prevalence in patients of a tertiary care epilepsy center. Neurology. 2004; 62: 2095-2097.
154. Hamerle M, Ghaeni L, Kowski A, Weissinger F, Holtkamp M. Cannabis and other illicit drug use in epilepsy patients. European Journal of Neurology. 2014; 21: 167-170.
155. Elsohly MA, Slade D. Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sciences. 2005; 78: 539-548.
156. Lutz B. On-demand activation of the endocannabinoid system in the control of neuronal excitability and epileptiform seizures. Biochemical Pharmacology. 2004; 68: 1691-1698.
157. Consroe P, Benedito MA, Leite JR, Carlini EA, Mechoulam R. Effects of cannabidiol on behavioral seizures caused by convulsant drugs or current in mice. European Journal of Pharmacology. 1982; 83: 293-298.
158. Cunha JM, Carlini EA, Pereira AE, Ramos OL, Pimentel C, Gagliardi R, Sanvito WL, Lander N, Mechoulam R. Chronic administration of cannabidiol to healthy volunteers and epileptic patients. Pharmacology. 1980; 21: 175-185.
159. Jones NA, Hill AJ, Smith I, Bevan SA, Williams CM, Whalley BJ, Stephens GJ. Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. Journal of Pharmacology and Experimental Therapeutics. 2010; 332: 569-577.
160. Karniol IG, Carlini EA. Pharmacological interaction between cannabidiol and delta 9-tetrahydrocannabinol. Psychopharmacologia. 1973; 33: 53-70.
161. Griffin G, Atkinson PJ, Showalter VM, Martin BR, Abood ME. Evaluation of cannabinoid receptor agonists and antagonists using the guanosine-5’-O-(3-[35S]thio)-triphosphate binding assay in rat cerebellar membranes. Journal of Pharmacology and Experimental Therapeutics. 1998; 285: 553-560.
162. Thomas BF, Gilliam AF, Burch DF, Roche MJ, Seltzman HH. Comparative receptor binding analyses of cannabinoid agonists and antagonists. Journal of Pharmacology and Experimental Therapeutics. 1998; 285: 285-292.
163. Bisogno T, Hanus L, De PL, Tchilibon S, Ponde DE, Brandi I, Moriello AS, Davis JB, Mechoulam R, Di M, V. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. British Journal of Pharmacology. 2001; 134: 845-852.
164. De Filippis D., Iuvone T, d’amico A, Esposito G, Steardo L, Herman AG, Pelckmans PA, de Winter BY, de Man JG. Effect of cannabidiol on sepsis-induced motility disturbances in mice: involvement of CB receptors and fatty acid amide hydrolase. Neurogastroenterology and Motility. 2008; 20: 919-927.
165. Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. British Journal of Pharmacology. 2008; 153: 199-215.
166. Ryan D, Drysdale AJ, Lafourcade C, Pertwee RG, Platt B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. Journal of Neuroscience. 2009; 29: 2053-2063.
167. Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, Klosterkotter J, Hellmich M, Koethe D. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Translational Psychiatry. 2012; 2: e94.
168. Izzo AA, Borrelli F, Capasso R, Di M, V, Mechoulam R. Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends in Pharmacological Sciences. 2009; 30: 515-527.
169. Devinsky O, Cilio MR, Cross H, Fernandez-Ruiz J, French J, Hill C, Katz R, Di M, V, Jutras-Aswad D, Notcutt WG, Martinez-Org, Robson PJ, Rohrback BG et al. Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia. 2014; 55: 791-802.
170. Karniol IG, Shirakawa I, Kasinski N, Pfeferman A, Carlini EA. Cannabidiol interferes with the effects of delta 9 - tetrahydrocannabinol in man. European Journal of Pharmacology. 1974; 28: 172-177.
171. Dalton WS, Martz R, Lemberger L, Rodda BE, Forney RB. Influence of cannabidiol on delta-9-tetrahydrocannabinol effects. Clinical Pharmacology & Therapeutics. 1976; 19: 300-309.
172. Zuardi AW, Morais SL, Guimaraes FS, Mechoulam R. Antipsychotic effect of cannabidiol. Journal of Clinical Psychiatry. 1995; 56: 485-486.
173. Zuardi AW, Crippa JA, Hallak JE, Bhattacharyya S, Atakan Z, Martin-Santos R, McGuire PK, Guimaraes FS. A critical review of the antipsychotic effects of cannabidiol: 30 years of a translational investigation. Current Pharmaceutical Design. 2012; 18: 5131-5140.
174. Raiteri M, Maura G, Barzizza A. Activation of presynaptic 5-hydroxytryptamine1-like receptors on glutamatergic terminals inhibits N-methyl-D-aspartate-induced cyclic GMP production in rat cerebellar slices. Journal of Pharmacology and Experimental Therapeutics. 1991; 257: 1184-1188.
175. Ceulemans B, Boel M, Leyssens K, Van RC, Neels P, Jorens PG, Lagae L. Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia. 2012; 53: 1131-1139.
176. Reddy DS, Mohan A. Development and persistence of limbic epileptogenesis are impaired in mice lacking progesterone receptors. Journal of Neuroscience. 2011; 31: 650-658.
177. Tuveri A, Paoletti AM, Orru M, Melis GB, Marotto MF, Zedda P, Marrosu F, Sogliano C, Marra C, Biggio G, Concas A. Reduced serum level of THDOC, an anticonvulsant steroid, in women with perimenstrual catamenial epilepsy. Epilepsia. 2008; 49: 1221-1229.
178. Tortella FC, Pellicano M, Bowery NG. Dextromethorphan and neuromodulation: old drug coughs up new activities. Trends In Pharmacological Sciences. 1989; 10: 501-507.
179. Thurgur C, Church J. The anticonvulsant actions of sigma receptor ligands in the Mg2+-free model of epileptiform activity in rat hippocampal slices. British Journal of Pharmacology. 1998; 124: 917-929.
180. Donnier-Marechal M, Larchanche PE, Le BD, Furman C, Carato P, Melnyk P. Carboline- and phenothiazine-derivated heterocycles as potent SIGMA-1 protein ligands. European Journal of Medicinal Chemistry. 2015; 89: 198-206.
181. Agurell S, Halldin M, Lindgren JE, Ohlsson A, Widman M, Gillespie H, Hollister L. Pharmacokinetics and metabolism of delta 1-tetrahydrocannabinol and other cannabinoids with emphasis on man. Pharmacological Reviews. 1986; 38: 21-43.
182. Harvey DJ, Mechoulam R. Metabolites of cannabidiol identified in human urine. Xenobiotica. 1990; 20: 303-320.
183. Yamaori S, Kushihara M, Yamamoto I, Watanabe K. Characterization of major phytocannabinoids, cannabidiol and cannabinol, as isoform-selective and potent inhibitors of human CYP1 enzymes. Biochemical Pharmacology. 2010; 79: 1691-1698.
184. Yamaori S, Ebisawa J, Okushima Y, Yamamoto I, Watanabe K. Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: role of phenolic hydroxyl groups in the resorcinol moiety. Life Sciences. 2011; 88: 730-736.
185. Yamaori S, Okamoto Y, Yamamoto I, Watanabe K. Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metabolism and Disposition. 2011; 39: 2049-2056.
186. Yamaori S, Koeda K, Kushihara M, Hada Y, Yamamoto I, Watanabe K. Comparison in the in vitro inhibitory effects of major phytocannabinoids and polycyclic aromatic hydrocarbons contained in marijuana smoke on cytochrome P450 2C9 activity. Drug Metabolism and Pharmacokinetics. 2012; 27: 294-300.
187. Nadulski T, Sporkert F, Schnelle M, Stadelmann AM, Roser P, Schefter T, Pragst F. Simultaneous and sensitive analysis of THC, 11-OH-THC, THC-COOH, CBD, and CBN by GC-MS in plasma after oral application of small doses of THC and cannabis extract. Journal of Analytical Toxicology. 2005; 29: 782-789.
188. Hunt CA, Jones RT, Herning RI, Bachman J. Evidence that cannabidiol does not significantly alter the pharmacokinetics of tetrahydrocannabinol in man. Journal of Pharmacokinetics and Biopharmaceuticals. 1981; 9: 245-260.
189. Turkanis SA, Cely W, Olsen DM, Karler R. Anticonvulsant properties of cannabidiol. Research Communications in Chemical Pathololy and Pharmacology. 1974; 8: 231-246.
190. Consroe P, Wolkin A. Cannabidiol—antiepileptic drug comparisons and interactions in experimentally induced seizures in rats. Journal of Pharmacology and Experimental Therapeutics. 1977; 201: 26-32.
191. Gloss D, Vickrey B. Cannabinoids for epilepsy. Cochrane Database of Systematic Reviews. 2014; 3: CD009270.
192. Ames FR, Cridland S. Anticonvulsant effect of cannabidiol. South African Medical Journal. 1986; 69: 14.
193. Trembly B, Sherman M. Double-blind clinical study of cannabidiol as a secondary anticonvulsant. International Association for Cannabinoid Medicines. Marijuana ‘90 International Conference on Cannabis and Cannabinoids, Kolympari, Crete. 1990; Section 2: p5.
194. Mechoulam R, Carlini EA. Toward drugs derived from cannabis. Naturwissenschaften. 1978; 65: 174-179.
195. Davis JP, Ramsey HH. Antiepileptic action of marijuana-active substances. Federation Proceedings. 1949; 8: 284-285.
196. Porter BE, Jacobson C. Report of a parent survey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy. Epilepsy and Behavior. 2013; 29: 574-577.
197. Ohlsson A, Lindgren JE, Andersson S, Agurell S, Gillespie H, Hollister LE. Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomedical & Environmental Mass Spectrometry. 1986; 13: 77-83.
198. Hawksworth G, McArdle K. Metabolism and pharmacokinetics of cannabinoids. The medicinal uses of cannabis and cannabinoids, Edited by G.W. Guy, B.A. Whittle, P.J. Robson. Pharmaceutical Press: London. 2004; pp: 205-228. ISBN:0-85369-517-2.
199. Lodzki M, Godin B, Rakou L, Mechoulam R, Gallily R, Touitou E. Cannabidiol-transdermal delivery and anti-inflammatory effect in a murine model. Journal of Controlled Release. 2003; 93: 377-387.
200. Consroe P, Laguna J, Allender J, Snider S, Stern L, Sandyk R, Kennedy K, Schram K. Controlled clinical trial of cannabidiol in Huntington’s disease. Pharmacology, Biochemistry and Behavior. 1991; 40: 701-708.
201. Huestis MA. Pharmacokinetics and metabolism of the plant cannabinoids, delta9-tetrahydrocannabinol, cannabidiol and cannabinol. Handbook of Experimental Pharmacology. 2005; 168: 657-690.
202. Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? The Lancet Neurology. 2002; 1: 383-386.
203. Dang YH, Ma XC, Zhang JC, Ren Q, Wu J, Gao CG, Hashimoto K. Targeting of NMDA receptors in the treatment of major depression. Current Pharmaceutical Design. 2014; 20: 5151-5159.
204. Marx CE, Stevens RD, Shampine LJ, Uzunova V, Trost WT, Butterfield MI, Massing MW, Hamer RM, Morrow AL, Lieberman JA. Neuroactive steroids are altered in schizophrenia and bipolar disorder: relevance to pathophysiology and therapeutics. Neuropsychopharmacology. 2006; 31: 1249-1263.
205. Garzón J, Herrero-Labrador R, Rodríguez-Muñoz M, Shah R, Vicente-Sánchez A, Wagner CR, Sánchez-Blázquez P. HINT1 protein: a new therapeutic target to enhance opioid antinociception and block mechanical allodynia. Neuropharmacology. 2015; 89: 412-423.
206. Ritsner MS, Gibel A, Shleifer T, Boguslavsky I, Zayed A, Maayan R, Weizman A, Lerner V. Pregnenolone and dehydroepiandrosterone as an adjunctive treatment in schizophrenia and schizoaffective disorder: an 8-week, double-blind, randomized, controlled, 2-center, parallel-group trial. Journal of Clinical Psychiatry. 2010; 71: 1351-1362.
207. Abadias M, Escriche M, Vaque A, Sust M, Encina G. Safety, tolerability and pharmacokinetics of single and multiple doses of a novel sigma-1 receptor antagonist in three randomized phase I studies. British Journal of Clinical Pharmacology. 2013; 75: 103-117.