
INTRODUCTION
In recent years, SETD2 has attracted a lot of interest as a gene whose inactivation is involved in tumor initiation and progression. However, Faber et al [1] had already identified a protein encoded by SETD2 in 1998 using a two-hybrid-based approach to search for proteins that interact with Huntingtin, the protein known to be associated with Huntington’s disease (HD). They identified several candidates, three of which contained a WW domain. One of these three proteins was Huntingtin Yeast Partner B (HYPB). Around the same time Mao et al [2] and Zhang et al [3] identified and analyzed a large set of transcripts from human umbilical cord CD34+ hematopoietic stem/progenitor cells. One of these transcripts, HSPC069, had a sequence identical to HYPB and represented the same gene. A few years later, HSPC069 was shown to contain an AWS-SET-PostSET domain and to possess histone methyl transferase activity specific for lysine 36 of histone 3 (H3K36) [4]. In a study focusing on proteins that interact with a DNA-binding motif in the E1A promoter, a transcript identical to HYPB was identified and named HBP231 [5]. The associated gene is ubiquitously expressed in all tissues and cell lines tested, including many cancer-derived cell lines.
Edmunds et al [6] introduced the gene symbol SETD2 in 2008, and made a more detailed analysis of the global and transcription-dependent distribution of tri-methylated histone H3 lysine 36 (H3K36me3) in mammalian cells. This was in line with the role of the Saccharomyces cerevisiae homologue of SETD2, ySET2, which had been identified in 2002 [7]. An important step in understanding the biology of ySET2 was its interaction with the serine2 phosphorylated C-terminal domain (CTD) of RNA polymerase II (RNA Pol II), linking ySET2 to the transcription elongation process [8]. A similar interaction was later confirmed for mammalian SETD2 [4, 9]. It was, however, not just its role in regulating transcription that attracted the interest of researchers over the years. The presence of inactivating mutations in a range of tumor types, most notably in clear cell renal cell cancer (ccRCC), sparked an additional focus of research: exploring the role of SETD2 in cancer development.
In this review the domains and functions of SETD2 in normal biology will be discussed in more detail. In the final part of the review, we focus on how loss of SETD2 function can contribute to cancer development.
THE FUNCTIONAL DOMAINS OF SETD2
The human SETD2 gene is located at the cytogenetic band p21.31 of chromosome 3, a region frequently targeted by copy number loss in various tumors [10]. SETD2 encompasses a genomic region of 147Kb, and the 21 exons encode an 8,452nt transcript. The SETD2 protein consists of 2,564 amino acids and has a molecular weight of 287.5 KD. Three conserved functional domains have been identified in the SETD2 protein: the triplicate AWS-SET-PostSET domains, a WW domain and a Set2 Rpb1 interacting (SRI) domain.
AWS-SET-PostSET domain
The human SET domain is a motif of 130 amino acids that is evolutionarily conserved from mammals to yeast and even in some bacteria and viruses [11, 12]. The SET domain was identified by comparison of the protein sequence of the Drosophila position-effect variegation suppressor gene, Su(var)3-9, with the protein sequence of several other genes [13]. The acronym SET stands for “Suppressor of Variegation, Enhancer of zeste and Trithorax”, which are the three genes that led to the discovery of this domain.
The SET domain is usually present as part of a multi-domain, flanked by an AWS (Associated with SET) and a PostSET domain. Generally, SET-domain-containing proteins transfer one or several methyl groups from S-adenosyl-L-methionine to the amino group of a lysine or an arginine residue of histones or other proteins [14]. This transfer is dependent on the flanking AWS and PostSET regions, which contain several conserved cysteine residues. In contrast to other methyltransferases, SET-domain-containing methyltransferases have a β-sheet structure that facilitates multiple rounds of methylation without substrate disassociation [15].
WW domain
The term “WW domain” was originally described in 1995 by Sudol et al [16] and refers to the presence of two conserved tryptophan (W) residues spaced 20-22 amino acids apart. Binding assays showed that the WW domain preferentially binds to proline-rich segments, mediating protein-protein interactions to participate in a variety of molecular processes [17]. The WW domain recognizes motifs like Proline-Proline-x-Tyrosine (PPxY) [18], phospho-Serine-Proline (p-SP) or phospho-Threonine-Proline (p-ST) [19], and mediates protein binding [20]. Aberrant expression of WW-domain-containing genes has been associated with different diseases such as HD [21], Alzheimer’s disease [22], and multiple cancer subtypes [23, 24]. The WW domain in the C-terminal region of SETD2 interacts with the Huntingtin protein via its proline-rich segment, regardless of the length of the HD-associated polyglutamine track [1], and may also interact with TP53 [25]. Gao et al [26] performed a detailed nuclear magnetic resonance study on the interaction of SETD2 with Huntingtin. SETD2 contains a proline-rich stretch that precedes the WW domain. This proline-rich stretch functions as an intramolecular WW-interacting domain that can block the WW domain of SETD2 from interacting with the proline-rich stretch of Huntingtin, and most likely of other proteins as well.
SRI domain
By analyzing a series of SET2-deletion-mutants, Kizer et al [27] identified a novel domain that specifically interacted with the hyperphosphorylated C-terminal domain (CTD) of Rpb1, the largest subunit of RNA Pol II. This Set2 Rpb1 Interacting (SRI) domain is conserved from yeast to human [27]. In human, the primary C-terminal domain-docking site of RNA Pol II is located at the first and second helices of SETD2 [9]. This domain directs the activity of SETD2 towards actively transcribed genes. Yeast experiments by Li et al [8] revealed a high affinity of ySET2 to the Ser2-phosphorylated CTD of RNA Pol II that is present only when transcription is well under way. ySET2 binds to the Ser5-phosphorylated CTD with intermediate affinity, while it has no affinity to the unphosphorylated CTD [28]. This interaction is dependent on the activity of the RNA Pol II CTD kinase CTK1, the enzyme responsible for the phosphorylation of Ser2 [29].
FROM PROTEIN STRUCTURE TO BIOLOGICAL FUNCTION
The above-mentioned functional domains define the biological function of SETD2. By virtue of its AWS-SET-PostSET domains, SETD2 mediates trimethylation of H3K36 [4]. In vitro, human SETD2 can carry out mono-, di-, and tri-methylation of H3K36 [30], but in vivo the scenario is different. While ySET2 catalyzes all methylation levels of H3K36 [7], SETD2 only modulates H3K36me3 in mammals. Knockdown of SETD2 induces a complete absence of H3K36me3 without disturbing the levels of H3K36me1 and H3K36me2 [6]. In human, trimethylation of H3K36 is carried out by a complex, of which SETD2 and Heterogeneous Nuclear Ribonucleoprotein L (hnRNPL) are the major subunits [31]. Based on these studies, it has become evident that SETD2 is solely responsible for this modification. Catalyzing H3K36 trimethylation is now regarded as the main function of SETD2. H3K36me3 is recognized by so-called readers, effector proteins that are recruited by specific histone modifications and determine the functional outcome of those modifications [32] (Table 1). A schematic representation of how SETD2-mediated-trimethylation of H3K36 is involved in various biological processes is shown in Figure 1.
Table 1: Overview of currently known H3K36me3 readers and their interacting domains.
Gene symbol |
binding domain |
Function |
Ref. |
BRPF1/2 |
PWWP |
Histone acetylation |
|
DNMT3A/B |
PWWP |
DNA methylation |
[35] |
GLYR1 |
PWWP |
Histone methylation |
[36] |
HDGF |
PWWP |
DNA binding |
[77] |
IWS1 |
PWWP |
Transcription elongation, splicing, mRNA export |
[78] |
MORF4L1 |
Chromo |
Alternative splicing |
|
MSH6 |
PWWP |
DNA mismatch repair |
|
MTF2 |
Tudor |
Histone methylation |
|
MSL3 |
Chromo |
Histone acetylation |
[80] |
MUM1 |
PWWP |
DNA damage repair |
|
NSD1 |
PWWP |
Histone methylation |
|
PHF1/19 |
Tudor |
Histone methylation |
|
PSIP1 |
PWWP |
Splicing and HR repair |
|
SPT16H |
PWWP |
Facilitate transcription and repress cryptic transcription |
[52] |
WHSC1/L1 |
PWWP |
Histone methylation |
|
ZMYND11 |
PWWP |
Transcription elongation |
[83] |
Note: BRPF1/2, Bromodomain And PHD Finger Containing 1 and 2; GLYR1, Glyoxylate Reductase 1 Homolog; HDGF, Hepatoma-Derived Growth Factor; MSH6, MutS Homolog 6; MTF2, Metal Response Element Binding Transcription Factor 2; MSL3, Male-Specific Lethal 3 Homolog; MUM1, Melanoma Associated Antigen 1;NSD1, nuclear receptor binding SET domain protein 1; PHD1/19, PHD Finger Protein 1/19; WHSC1, Wolf-Hirschhorn Syndrome Candidate 1; WHSC1L1, Wolf-Hirschhorn Syndrome Candidate 1-Like 1; ZMYND11, Zinc Finger MYND-Type Containing 11.
The most prominent function of SETD2 is thus indirectly determined by the factors that target SETD2 to specific nucleosomes to be trimethylated on the one hand, and the so-called readers of this modification on the other. Vezzoli et al [33] showed that BRPF1 (Bromodomain And PHD Finger Containing 1) interacts with H3K36me3 through its PWWP domain, a finding later corroborated by a study of Wu et al [34]. Subsequently, several other “readers” were identified that interact with H3K36me3 by virtue of their PWWP domain [35, 36, 37]. More recently, additional proteins were identified that interact with H3K36me3 through their tudor domain [38] or chromodomain [39].
In addition to its role in histone modification, SETD2 may also interact directly with other proteins, most likely through its WW domain. The BioGRID database (http://thebiogrid.org) lists multiple proteins that directly interact with SETD2. Co-immunoprecipitation assays showed that the C-terminal domain of SETD2 interacts with the N-terminal domain of TP53 [25]. Binding of SETD2 to TP53 modulates the expression of a specific set of TP53 downstream target genes, including the apoptosis related genes puma, noxa, and p53AIP1. However, because no follow-up studies have corroborated these findings, the importance of the SETD2-TP53 interaction remains to be established. Given the well-known role of TP53 in cancer development, exploring its interactions with SETD2 may be relevant to elucidating the role of SETD2 mutations in cancer. To date, most studies have focused on the SETD2-dependent trimethylation of H3K36.
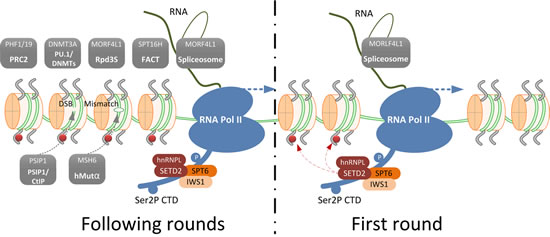
Figure 1: Schematic representation of SETD2-mediated trimethylation of H3K36 and an overview of the H3K36me3 readers that define its role in various biological processes. During the first round of transcription, the transcription elongation factor and histone chaperone SPT6-IWS1 are recruited to Ser2P CTD tail of RNA Pol II. This results in the recruitment of the SETD2-hnRNPL complex that trimethylates H3K36. This mark is preserved on the histones in the following rounds of transcription and serves as a signal beacon to recruit H3K36me3 readers (shown in grey boxes). Facilitates Chromatin Transcription (FACT) complex, Histone deacetylase (HDAC) complex, PU.1 (also known as Spi-1 proto-oncogene, SPI1)/ DNA (cytosine-5-)-methyltransferase (DNMTs) complex and Polycomb Repressive Complex 2 (PCR2) complex are recruited for chromatin structure remodeling to facilitate transcription elongation and to prevent cryptic transcription initiation. The spliceosome is recruited through MORF4L1 for splicing selection; PSIP1/CtIP complex is recruited through PSIP1 for homologous recombination (HR) repair of double strand breaks (DSBs) and hMutα complex is recruited through MSH6 for DNA mismatch repair.
Distribution of H3K36me3
Krogan et al [29] were the first to report a specific distribution of H3K36me3 over the yeast genome, with enrichment of H3K36me3 in actively transcribed coding regions. In C. elegans, actively transcribed genes also have much higher levels of H3K36me3 than transcriptionally repressed genes [40]. The same pattern is observed in higher eukaryotes, with high H3K36me3 levels downstream of the first exon of actively transcribed genes and across the whole gene body with a peak near the 3’ end [41, 42].
In both human and mouse, intron-containing genes showed relatively higher levels of H3K36me3 than intron-less genes, irrespective of transcriptional activity [43]. Along the gene body, H3K36me3 enrichment also appears to be discrete, co-localizing to exons rather than introns, and with higher levels of H3K36me3 at constitutively included exons as compared to alternatively spliced exons [40]. The distribution pattern of H3K36me3 indicates a role for SETD2 in modulating splicing events by marking exonic and intronic regions.
It should be noted that H3K36me3 is not confined to actively transcribed genes. A study by Chantalat et al [44] showed a high level of H3K36me3 at the silenced Snurf-Snrpn region in mice, a well-known facultative heterochromatin domain. Pericentromeric regions, which consist mainly of constitutive heterochromatin, are also enriched for H3K36me3 [44]. In these regions the H3K36me3 mark is apparently not correlated with transcriptional events. In the remainder of this review we will discuss how the loss of or decrease in H3K36me3 caused by functional loss of SETD2 could contribute to cancer development.
H3K36me3-MEDIATED BIOLOGICAL FUNCTIONS
H3K36me3 participates in transcription elongation and splicing selection
Deletion of the SRI domain of SETD2 not only abolishes its interaction with RNA Pol II but also leads to a defect in trimethylation of H3K36, suggesting that H3K36 trimethylation and transcription elongation are coupled processes [27, 28]. Splicing and transcription are also coupled processes regulated by many factors, including chromatin remodeling complexes [45], RNA Pol II elongation rate [46], RNA binding elements [47] and histone modifications [48]. Direct evidence to support participation of SETD2 in splicing came from studies on alternative splicing of the human fibroblast growth factor receptor 2 (FGFR2) gene [49]. FGFR2 is spliced into two mutually exclusive and tissue-specific isoforms: FGFR2-IIIb (exon IIIb is included) and FGFR2-IIIc (exon IIIc is included). Alternative splicing is modulated by polypyrimidine tract binding protein 1 (PTBP1, also known as PTB). PTBP1 is recruited by histone tail-binding protein Mortality Factor 4 like 1 (MORF4L1, also known as Eaf3 and MRG15), which recognizes H3K36me3 through its chromo domain [39, 50]. Overexpression of ySET2 leads to a global increase of H3K36me3 and a decreased inclusion of exon IIIb, whereas siRNA-mediated knockdown of human SETD2 resulted in inclusion of the PTB1-repressed exon IIIb [49].
This “chromatin affects splicing” model does not, however, explain by what mechanism chromatin is modified to direct splicing. Subsequently a “splicing affects chromatin” model was proposed [43]. Inhibiting splicing, either by knockdown of splicing factor Sin3A-associated protein (SAP130) or D-ribofuranosyl-benzimidazole (DRB) treatment, leads to a decreased recruitment of SETD2 and reduced H3K36me3 levels [43]. Thus, the splicing machinery itself may play a role in the recruitment of SETD2 by RNA Pol II and the subsequent trimethylation of H3K36. DRB-treatment of HeLa cells reduced the H3K36me3 levels on internal exons to a level that remained higher than the level in intergenic regions, even though both regions have a comparable RNA Pol II occupancy. This indicates that, although splicing is not required for trimethylation, it does modulate H3K36me3 levels [43]. Kim et al [51] showed that introducing mutations that prevent splicing, or interfere with the splicing machinery using splicing inhibitor spliceostatinA (SSA), led to a redistribution of H3K36me3 with a shift towards the 3’ region, again indicating a direct causal relationship between splicing and H3K36me3.
H3K36me3 prevents spurious transcription
Modification of nucleosomes plays an important role in protecting genomic DNA and regulating its accessibility. A compact nucleosome structure of the gene body is needed to prevent spurious transcription initiation from cryptic promoters. Removal of this barrier during transcription elongation upon passage of RNA pol II results in a more accessible chromatin. Reconstitution of completely evicted nucleosomes with acetylated nucleosomes from the soluble pool after passage of RNA pol II could result in a more accessible chromatin structure of transcribed genes. This would allow intergenic transcription initiation from cryptic promoter sequences. Trimethylation of H3K36 during transcription elongation by RNA pol II-bound SETD2 is thought to prevent spurious transcription from cryptic promoters. H3K36me3 recruits Facilitates Chromatin Transcription complex (FACT) [52] and Polycomb repressive complex 2 (PRC2) [38, 53] to restore the repressed chromatin structure after elongation. The FACT complex disassembles the H2A-H2B dimer from the nucleosomes. After passage of RNA Pol II, the same complex promotes the replacement of the H2A-H2B dimers. This allows the transcription elongation complex to pass without the need to remove histone H4 and H3 [54]. Thus, the H3K36 trimethylated nucleosomes are kept on their position. The IWS1:SPT6:CTD complex is needed for the recruitment of SETD2 to RNA Pol II for trimethylation of H3K36 [55]. SPT6 was already known to enhance the elongation rate by displacing the nucleosomes in front of RNA pol II [56]. However, several studies have indicated that SPT6 also enhances the elongation rate in the absence of nucleosomes [57-59]. Experiments in S. cerevisiae have shown that inactivation of SPT6 or the FACT subunit Suppressor Of Ty 16 Homolog (SPT16H, also known as SPT16) resulted in intragenic transcription initiation events from cryptic promoters [52, 60, 61]. Taken together, the prevention of spurious intragenic transcription initiation is an important function of H3K36me3 and thus, indirectly, of SETD2.
H3K36me3 maintains genomic integrity and stability
The enriched level of H3K36me3 in transcribed regions not only serves to restore chromatin structure after transcription but also functions in maintaining genomic integrity. H3K36me3 is a crucial factor in the repair of DNA damage in transcribed regions by modulating two different pathways: (i) the DNA Mismatch Repair (MMR) pathway responsible for the repair of nucleotide mismatches and small insertion/deletion loops of simple repeat sequences and (ii) the homologous recombination (HR) repair of DNA double strand breaks (DSBs).
DNA MMR is a mechanism for correcting base-base mismatches and insertion/deletion loops produced during replication. The most abundant machinery responsible for DNA MMR is the hMutSα (MSH2-MSH6) complex. Li et al showed that the binding of hMutSα to chromatin is H3K36me3-dependent as its subunit MSH6 reads the H3K36me3 signal by virtue of its PWWP domain [62]. Depletion of SETD2 abolished the localization of hMutSα, which led to a DNA-MMR-deficient mutator phenotype. The DNA MMR defect in SETD2-deficient UOK143 cells could be restored by enforced expression of ySET2. This demonstrates the crucial role of H3K36me3 in recruiting the DNA MMR repairing machinery.
DNA MMR predominantly occurs during the S-phase of the cell cycle, whereas HR repair preferentially takes place in the late S/G2 phase. H3K36me3 consistently peaks in the late G1/early S phase and disappears in the late S/G2 phase [63] (reviewed by Li et al [64]). This is additional proof that H3K36me3-modification enables a safe transition from the G1 to the S phase by recruiting repairing machineries to correct the errors produced during replication. When H3K36me3 is abolished due to SETD2 inactivation, the repair machinery cannot localize to damaged sites, resulting in an accumulation of errors and genomic instability, a hallmark of tumorigenesis.
H3K36me3 also serves as a signal to recruit proteins to DNA double strand breaks (DSBs) to initiate repair. An accurate repair of DSBs relies on HR. The PWWP domain of PC4 And SFRS1 Interacting Protein 1 (PSIP1, also known as Lens Epithelium-Derived Growth Factor, LEDGF) is the basis of this HR process, and H3K36me3 plays a key role through the recruitment of PSIP1 [65-67]. This is consistent with the finding that SETD2 is required for ATM-activation upon DSBs [68] and the notion that SETD2-deficient cells fail to activate a proper DNA damage response, including activation of TP53 [68]. SETD2 inactivation abolishes H3K36me3 and consequently the binding of PSIP1 to the damage sites. To compensate for the HR deficiency, cells have to use alternative mechanisms to repair the DSBs, such as nonhomologous end-joining and/or microhomology-mediated end-joining [68]. These approaches are error prone and may lead to deletions [69]. Although the HR repair machinery in SETD2-inactivated cells is still competent [67, 68], these cells are not capable of recruiting the DNA repair components to the damaged sites due to loss of the H3K36me3 signal.
H3K36me3 and DNA methylation
Several publications have indicated that actively transcribed genes are extensively methylated at the gene body [70-73]. This has raised the question of whether H3K36 trimethylation is associated with gene body DNA methylation. Hahn et al [74] carried out a detailed study of the association of a number of epigenetic markers in human bronchial epithelial cells and colorectal cancer cell line HCT116, focusing on chromosome 19 genes. Of the expressed genes, 74% had a high level of both gene body DNA methylation and H3K36me3. DNA methylation and H3K36me3 have been linked in both yeast and mouse [75]. In addition, a group of genes, mostly Zinc Finger genes, were identified in which H3K36me3 occurred in combination with the repressive intragenic H3K9me3 mark [75]. On average these genes were expressed at a low level and had a relatively low number of intragenic CpG dinucleotides that were largely unmethylated. By analyzing cells that were either made defective in H3K36 trimethylation or in CpG methylation, Hahn et al [74] further showed that the levels of these two epigenetic markers are established independently. However, Dhalayan et al demonstrated a high affinity of DNA (cytosine-5)-methyltransferase 3A (DNMT3A) to H3K36me3 in vitro [35]. DNMT3A is targeted to H3K36me3-containing nucleosomes, e.g. in heterochromatic regions as well as gene bodies, by virtue of its PWWP domain. DNMT3A/B interacts with PU.1 to form a complex for de novo site-specific methylation [76]. This indicates that the H3K36me3 mark could recruit DNMT3A/B to establish DNA methylation.
SETD2 knock-out mouse
In mice, SETD2-/- knockout is embryonic lethal in E10.5 to E11.5 due to defects in angiogenesis in the yolk sac and placenta [84]. Expression profiling of SETD2-/- and wild-type yolk sacs revealed significantly altered expression levels of genes involved in vascular remodeling. Both SETD2-/- embryonic bodies derived from embryonic stem cells and from cultured human endothelial cells treated with siRNAs-directed against SETD2 showed defects in cell migration and invasion [84, 85]. Thus, SETD2 appears to be crucial for a proper embryonic development although many cancer cells appear to function well without SETD2. In the literature, no clues can be found of heterozygous SETD2 knockout mice being predisposed to any kind of disease or cancer.
SETD2 IN DISEASE
Luscan et al [86] identified a missense and a nonsense SETD2 mutation in 2 out of 11 patients with Sotos syndrome, an overgrowth syndrome first described by Sotos et al [87]. It is unknown if these mutations were present in the germline and there is no direct functional evidence that links these mutations to SOTOS. However, it is remarkable that the gene most frequently mutated in SOTOS is the PWWP-domain-containing Nuclear Receptor Binding SET Domain Protein 1 (NSD1, also known as KMT3B) gene [88] responsible for mono- and di-methylation of H3K36 [30, 89]. We are not aware of any reports that link SETD2 germline mutations to an inherited syndrome in humans.
SETD2 IN CANCER
The first report on SETD2 mutations in cancer dates from 2010 when Dalgliesh et al identified inactivation mutations in ccRCC [90]. At the same time, using a “Gene Identification by Nonsense-mediated mRNA decay Inhibition (GINI)” strategy, our group identified inactivating SETD2 mutations in 5 out of 10 ccRCC-derived cell lines [91]. All cell lines showed copy number loss for most of the short arm of chromosome 3, indicating complete functional loss of SETD2 in these cell lines. Subsequent targeted sequencing of the SETD2 coding regions revealed SETD2 mutations in 2 out of 10 primary ccRCC tumors [92]. This bi-allelic inactivation of SETD2 was the first clue that the gene might be a tumor suppressor gene. Two large cohort studies revealed an overall frequency of SETD2 mutations of approximately 11% in ccRCC [93, 94]. The fraction of truncating mutations in ccRCC was more than 50% in the study of Hakimi et al [95] and 57% in COSMIC, which is significantly higher than the fraction of truncating mutations in non-ccRCC tumors (32%, COSMIC). Still, whole-exome sequencing studies did reveal somatic SETD2 mutations in various types of cancer (Table 2), and this can be seen as an indication that SETD2 inactivation plays a role in the development of other tumors, albeit with low frequencies in most of them (COSMIC [96], Tumorportal [97] and cBIOPortal [98, 99], accessed in January 2016). It should be noted that in many studies it is not clear if the mutation resulted in a bi-allelic inactivation of SETD2. Moreover, the majority of somatic SETD2 mutations were missense mutations for which the functional consequences are often unclear (Table 2). This is illustrated by the study of Zhu et al [100] of 241 cases of leukemia (134x acute myeloid leukemia (AML) and 107x acute lymphcytic leukemia (ALL)) in which only 8 of the 19 somatic SETD2 mutations identified in 15 patients were truncating. Bi-allelic mutations were detected in only 4 patients. It cannot be excluded that in ALL, and possibly in other tumors as well, SETD2 haploinsufficiency does lead to a disease phenotype. SETD2 mutations appeared to be most frequent in leukemias that carried a MLL gene rearrangement [100].
Table 2: Overview of SETD2 mutation frequencies in a selection of tumors based on the COSMIC database (Feb 2016)*.
Tissue/tumour subtype |
Percentage of samples with mutation |
cases tested |
|
truncating |
missense |
||
Kidney |
4.19 |
3.10 |
2197 |
ccRCC |
5.43 |
4.14 |
1473 |
Lung |
1.26 |
1.42 |
1826 |
Adenocarcinoma |
3.51 |
3.51 |
550 |
Skin |
1.08 |
2.65 |
1017 |
Liver |
0.74 |
1.55 |
1611 |
Hepatocellular carcinoma |
0.78 |
1.12 |
893 |
Soft tissue |
0.70 |
4.67 |
428 |
Biliary tract |
0.66 |
0.66 |
152 |
Adenocarcinoma |
0.67 |
0.67 |
150 |
Endometrium |
0.63 |
3.49 |
631 |
Endometrioid carcinoma |
0.74 |
4.08 |
539 |
Large intestine |
0.59 |
3.05 |
1345 |
Adenocarcinoma |
0.62 |
3.10 |
1298 |
Breast |
0.58 |
0.94 |
1378 |
Central nervous system |
0.47 |
0.38 |
2128 |
Pancreas |
0.46 |
0.33 |
1521 |
Ductal carcinoma |
0.40 |
0.57 |
1240 |
Stomach |
0.34 |
2.04 |
587 |
Urinary tract |
0.30 |
0.90 |
666 |
Haematopoietic and lymphoid |
0.24 |
0.87 |
2519 |
Acute lymphoblastic B cell leukaemia |
1.54 |
2.32 |
258 |
Acute lymphoblastic T cell leukaemia |
0.97 |
0.97 |
207 |
Diffuse large B cell lymphoma |
0.00 |
3.20 |
250 |
Ovary |
0.24 |
0.59 |
843 |
Serous carcinoma |
0.31 |
0.78 |
641 |
Bone |
0.20 |
0.60 |
496 |
Prostate |
0.10 |
0.88 |
1019 |
Adenocarcinoma |
0.12 |
0.48 |
827 |
* Tumor subtypes with a sample size less than 100 cases have been excluded.
In ccRCC, SETD2 is ranked in the top-5 most commonly mutated genes (COSMIC, rank 4), indicating its specific role in this tumor type. In Tumorportal, SETD2 mutations are indicated as “highly significant” in ccRCC and glioblastoma multiform and indicated as “near significant” in bladder cancer. In all cancers combined, there is a slight clustering of SETD2 missense mutations in an approximately 200 amino acid segment (p.M1468 up to p.Q1668) that overlaps with the SET domain (Figure 2). The same region is relatively devoid of missense variants in the normal population (ExAC database, http://exac.broadinstitute.org, accessed January 2016, and Figure 2), indicating that missense mutations in this domain might be more often damaging. SETD2 nonsense mutations leading to loss-of-function can be located throughout the entire gene (Figure 2). Further studies on the potential functional consequences of SETD2 missense mutations are required to establish their role in tumor development and/or progression.
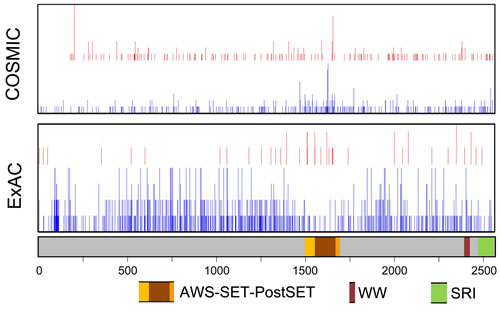
Figure 2: Schematic representation of SETD2 with the location of functional domains and nonsynonymous mutations and variants. The location of nonsynonymous mutations was obtained from ExAC (Germline variants in ~120000 alleles; January 2016) and COSMIC (somatic variants in 23,249 cases; January 2016). Intronic regions and 3’- and 5’-untranslated regions are not shown. Red, position of inactivating variants; Blue, position of missense variants. For the COSMIC data, the height of the bar is relative to the number of mutations. For the ExAC data, the height of the bars indicate 1, 2-5, 6-10, or >10 variants per triplet.
Pena-Llopis et al [101] collected data on 924 primary ccRCC of which 300 cases had a PBRM1 mutation and 66 cases had a SETD2 mutation, while 33 cases had a mutation in both genes. This number was shown to be significantly higher than the expected number of cases with mutations in both genes (n = 21, Fisher exact test, p = 0.003). This suggests that mutations of PBMR1 and SETD2 may have a synergistic effect in ccRCC, possibly by disrupting different pathways. Moreover, the Cancer Genome Atlas database (TCGA) reveals co-mutation of PBRM1 and SETD2 in multiple tumors despite the low mutation frequency of both genes in these cancers. Thus, having both SETD2 and PBRM1 mutations might strengthen their oncogenic potential, and the underlying mechanism deserves exploration. Sato et al [94] found that SETD2 mutations predominantly occurred in tumors with pre-existing VHL mutations, again indicating a role in tumor progression. However, in other studies SETD2 mutations were also identified in ccRCC cases with wild type VHL [95, 102].
The high frequency of inactivating SETD2 mutations in ccRCC points to a tumor-suppressor-like function of this gene. Additional proof for a tumor suppressor role of SETD2 came from Sleeping Beauty transposon experiments. This approach is based on the assumption that commonly observed transposon insertion sites can harbor tumor-driver genes. These studies revealed transposon integration sites in SETD2 in various tumors such as leukemia’s [103] and colorectal cancer [104], albeit at a low frequency.
Correlation with clinical data
Al Sarakbi et al [105] found a negative association of SETD2 expression levels with increasing tumor stage in breast cancers. In gliomas, SETD2 mutations were predominantly seen in high-grade (16 out of 178 cases) but not in low-grade cases (0 out of 45 cases) [106]. ccRCC patients with somatic SETD2 mutations had a higher relapse rate compared to cases with wild-type SETD2, but no effect was observed on overall survival. In a study including 185 ccRCC patients, SETD2 mutations were significantly associated with advanced tumor stage (P = 0.02) [95]. In the TCGA, SETD2 mutations were found to be associated with worse cancer-specific survival (P = 0.036; HR 1.68; 95% CI 1.04-2.73), and the presence of SETD2 mutations was a predictor of ccRCC recurrence in an univariant analysis (P = 0.002; HR 2.5; 95% CI 1.38-4.5) [107]. Further evidence supporting a role of SETD2 inactivation in progression of tumors comes from a recent study performed by Ho et al [108]. Using immuno-histochemical approaches, Ho et al (108) observed a decrease in H3K36me3 levels in metastatic ccRCC as compared to primary ccRCC. Either acquired SETD2 mutations or alternate mechanisms may be the cause of this, suggesting that a decreased level of H3K36me3 is correlated with progression. They also noted that loss of one allele of SETD2, a common event due to the widespread copy number loss of the short arm of chromosome 3 in ccRCC, did not result in a reduced level of H3K36me3. Thus, SETD2 haploinsufficiency does not cause a H3K36me3-related phenotype in ccRCC. In addition, intra-tumor heterogeneity studies have indicated that SETD2-inactivation may be a late event in cancer development. Gerlinger et al [109] carried out a genomic analysis of multiple regions of four primary ccRCC tumors and detected intratumor heterogeneity in every case. Using whole exome sequencing and H3K36me3-staining of tissue sections, they identified different SETD2 mutations in different regions of the same primary tumor in three cases. This suggested that loss of SETD2 can be a late event that provides a selective advantage to tumor cells [109]. Lentiviral-mediated knockdown of SETD2 in pre-leukemic cells carrying a MLL fusion-gene increased both the colony-forming capacity and the growth rate of these cells [100]. This indicates that loss of functional SETD2 facilitates initiation as well as progression of leukemias. Thus, it appears that SETD2-inactivation may function not only in driving the development of tumors, but also in promoting progression of the disease.
SETD2 functional studies in cancer
Alternative splicing is considered as a major impetus driving proteome diversity and promoting progression of cancer [110]. SETD2-mutated ccRCC tumors showed an altered chromatin accessibility in the H3K36me3 marked regions, which led to widespread defects in transcript processing, including intron retention, exon utilization and different transcriptional start and stop site usage, especially in highly expressed genes [111]. A specific set of transcripts showed an increased retention of introns in H3K36me3-deficient tumors, and several of the affected genes, including PTEN, TP53, ATR, RAD50, POLN, XRCC1, CCNB1, and CCND3, are important in tumor development. Since intron retention could lead to loss of function of the protein product, SETD2-inactivation will probably also have an impact on the functionality of these genes. Additionally, in the study of Ho et al [108], decreased levels of H3K36me3 in ccRCC, most likely due to SETD2-inactivating mutations, resulted in alternative exon usage for a selection of genes [108]. Li et al [112] carried out a detailed study on the splicing of CDH1 in gastric cancer cell lines in comparison to the human gastric mucosal epithelial cell line GES-1. In all samples, the wild type product and a transcript that lacks part of exon 8 were identified. A higher level of H3K36me3 appeared to favor the use of the splice donor site within exon 8. Attempts to influence the ratio between the two transcript variants were most successful using siRNA directed against SETD2 and, to a lesser extent, using an HDAC inhibitor.
HR repair and DNA MMR defects have been observed in SETD2-inactivated tumor cell lines, although the repair machineries themselves are not abolished in these cells [62, 67]. The SETD2-deficient ccRCC-derived cell line UOK143 showed insufficient MutSα-mediated DNA MMR in S phase. In contrast, in the SETD2-proficient ccRCC cell line UOK12, abundant MSH6 foci were formed during S phase and most of those loci co-localized with the H3K36me3 signal. SETD2-inactivated ccRCC cell lines RCC-MF and RCC-FG2 showed defects in DSB repair [68]. These studies indicated that SETD2 is important to maintain the genomic integrity in ccRCC.
Additional factors modulating H3K36me3 levels
When examining several databases, it becomes clear that SETD2 is ubiquitously expressed in most if not all tissues. This is not surprising given its function as the sole gene responsible for the trimethylation of H3K36. However, two factors have been identified in cancer-related studies that can modulate the level of SETD2 in cancer cells, and may also do so in non-cancerous cells (Figure 3). A recent study on liver cancer demonstrated a negative correlation between expression of SETD2 and the HOX transcript antisense RNA (HOTAIR) [116]. HOTAIR expression has been associated with several cancers and is shown to be an oncogenic long noncoding RNA [117]. HOTAIR suppressed the transcription of SETD2, and reduced the level of H3K36me3. Thus, HOTAIR overexpression is linked to various cellular processes mediated by H3K36me3 readers.
Xiang et al [118] showed that miR-106b-5p could bind to, and inhibit translation of, the SETD2 mRNA transcript in ccRCC. SETD2 levels increased by inhibiting miR-106b-5p and this resulted in suppression of cell proliferation and a G0/G1 cell cycle arrest.
A number of genes other than SETD2 can influence H3K36me3 levels. KDM4A, -B and -C are known to demethylate H3k36me3 [113]. Overexpression of these genes, which is a relative common event in various types of cancer [114], may thus interfere with all processes that involve H3K36me3 readers. As an example, it was recently shown that an enhanced expression of KDM4A-C promotes genomic instability [115]. By demethylating H3K36me3 the recruitment of MSH6 is prevented.
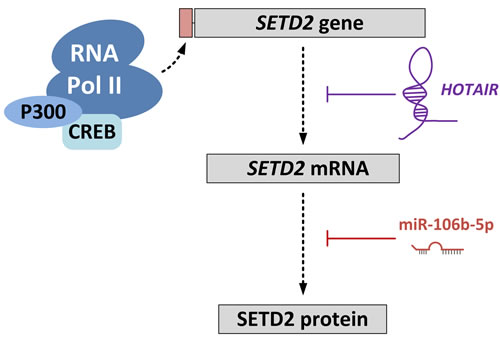
Figure 3: Regulation of SETD2 expression. The long non-coding RNA HOTAIR regulates SETD2 expression at the transcriptional level by competitively blocking loading of CREB-P300-RNA Pol II complex to the SETD2 promoter. MicroRNA-106-5p (miR-106-5p) regulates SETD2 expression at the translational level by binding to the 3’-UTR of the SETD2 mRNA transcript.
EMERGING THERAPEUTIC OPPORTUNITIES
Now that it is evident that SETD2-inactivation can be an important factor in tumor development and progression, especially in ccRCC, understanding the SETD2-inactivation-related pathways may offer new targets for therapy. The Genomics of Drug Sensitivity in Cancer database [119] lists four chemical compounds with a selective inhibitory capacity for SETD2-/- cell lines. Two of these components target P13Kbeta. Feng et al [120] further analyzed the effects of AZD6482 on SETD2-/- ccRCC cell lines and showed that tumor cells were selectively inhibited. This represents the first indication that novel compounds targeting SETD2-/- tumors might become feasible treatment for ccRCC patients. In recent years many studies have focused on the ability of small molecules to target specific histone modifications, which could eventually be used in targeted therapies. A recent study shows that the combination of WEE1-inactivation by the AZD1775 inhibitor and H3K36me3-deficiency is lethal for cultured human cells [121]. These results were then validated in xenograft models of two tumor-derived SETD2-/- cell lines. The underlying mechanism appears to be inhibition of the replication process. These recent developments may open the doors that allow for the development of targeted therapies for H3K36me3-deficient tumors in combination with WEE1 inhibitors. The WEE1 inhibitor is currently being tested in several phase II clinical trials (http://www.clinicaltrials.gov).
CONCLUDING REMARKS
SETD2 is responsible for the trimethylation of H3K36 in the gene body of actively transcribed genes and its inactivation interferes with the function of readers of this specific histone modification. The role of H3K36me3 on specific cellular functions is becoming more and more clear. Loss of one allele of SETD2, most likely a common event in many tumors due to widespread and frequent 3p copy number loss, may not be enough to cause a significant change in H3K36me3. On the other hand, biallelic inactivation of SETD2 is not the only mechanism that may cause loss of H3K36me3. Loss of SETD2 may also cause regional genomic instability, RNA processing defects and intragenic transcription initiations. Both genomic instability and alternative splicing are known as hallmarks of cancer. The former is a key force in carcinogenesis. The latter is an important mechanism for driving proteome diversity, which contributes to cancer development. In combination with the presence of SETD2-inactivating mutations in a substantial proportion of ccRCC, this clearly demonstrates SETD2’s role as a suppressor of both tumor initiation and progression.
Our knowledge on SETD2-regulated signaling pathways is quite limited, especially in the context of SETD2 binding proteins. Recent studies have indicated that SETD2 may interact with multiple proteins [122-124]. The challenge will be to unravel novel SETD2 functionalities that are independent of its function as trimethylator of H3K36. Conditional, and/or tissue-specific, SETD2 knockout mice may be of help to identify the crucial pathways that are affected upon inactivation of SETD2. Loss of SETD2 appears to play an essential role in a substantial subset of ccRCC. However, the specific effect of SETD2 inactivation on ccRCC precursor cells, kidney primary tubular epithelial cells, is still unknown. As SETD2 mutations are also seen in other cancer types, understanding the role of SETD2 in ccRCC will contribute to our understanding of these tumors.
ACKNOWLEDGMENTS
We are grateful to Kate McIntyre for critically editing the manuscript.
FUNDING
JL was supported by a China Scholarship Council of Research fellowship.
CONFLICTs OF INTEREST
There is no conflict of interest for any of the authors.
REFERENCES
1. Faber PW, Barnes GT, Srinidhi J, Chen J, Gusella JF, MacDonald ME. Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet. 1998; 7: 1463-74.
2. Mao M, Fu G, Wu JS, Zhang QH, Zhou J, Kan LX, Huang QH, He KL, Gu BW, Han ZG, Shen Y, Gu J, Yu YP, et al. Identification of genes expressed in human CD34 hematopoietic stem/progenitor cells by expressed sequence tags and efficient full-length cDNA cloning. Proc Natl Acad Sci USA. 1998; 95: 8175-80.
3. Zhang QH, Ye M, Wu XY, Ren SX, Zhao M, Zhao CJ, Fu G, Shen Y, Fan HY, Lu G, Zhong M, Xu XR, Han ZG, et al. Cloning and functional analysis of cDNAs with open reading frames for 300 previously undefined genes expressed in CD34 hematopoietic stem/progenitor cells. Genome Res. 2000; 10: 1546-60.
4. Sun X, Wei J, Wu X, Hu M, Wang L, Wang H, Zhang Q, Chen S, Huang Q, Chen Z. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem. 2005; 280: 35261-71.
5. Rega S, Stiewe T, Chang D, Pollmeier B, Esche H, Bardenheuer W, Marquitan G, Pützer BM. Identification of the full-length huntingtin-interacting protein p231HBP/HYPB as a DNA-binding factor. Mol Cell Neurosci. 2001; 18: 68-79.
6. Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008; 27: 406-20.
7. Strahl BD, Grant PA, Briggs SD, Sun Z, Bone JR, Caldwell JA, Mollah S, Cook RG, Shabanowitz J, Hunt DF. Allis CD. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol. 2002; 22: 1298-1306.
8. Li J, Moazed D, Gygi SP. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J Biol Chem. 2002; 277: 49383-49388.
9. Li M, Phatnani HP, Guan Z, Sage H, Greenleaf AL, Zhou P. Solution structure of the Set2-Rpb1 interacting domain of human Set2 and its interaction with the hyperphosphorylated C-terminal domain of Rpb1. Proc Natl Acad Sci U S A. 2005; 102: 17636-41.
10. Kok K, Naylor SL, Buys CH. Deletions of the short arm of chromosome 3 in solid tumors and the search for suppressor genes. Adv Cancer Res. 1997; 71: 27-92.
11. Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000; 406: 593-9.
12. Tschiersch B, Hofmann A, Krauss V, Dorn R, Korge G, Reuter G. The protein encoded by the Drosophila position-effect variegation suppressor gene Su (var) 3-9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J. 1994; 13: 3822-31.
13. Jenuwein T, Laible G, Dorn R, Reuter G. SET domain proteins modulate chromatin domains in eu-and heterochromatin. Cell Mol Life Sci. 1998; 54: 80-93.
14. Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005; 6: 227.
15. Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X. Structural basis for the product specificity of histone lysine methyltransferases. Mol Cell. 2003; 12: 177-85.
16. Sudol M, Chen HI, Bougeret C, Einbond A, Bork P. Characterization of a novel protein-binding module—the WW domain. FEBS letters. 1995; 369: 67-71.
17. Ingham RJ, Colwill K, Howard C, Dettwiler S, Lim CS, Yu J, Hersi K, Raaijmakers J, Gish G, Mbamalu G, Taylor L, Yeung B, Vassilovski G, et al. WW domains provide a platform for the assembly of multiprotein networks. Mol Cell Biol. 2005; 25: 7092-106.
18. Macias MJ, Hyvönen M, Baraldi E, Schultz J, Sudol M, Saraste M, Oschkinat H. Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature. 1996; 382: 646-9.
19. Lu P, Zhou XZ, Shen M, Lu KP. Function of WW domains as phosphoserine-or phosphothreonine-binding modules. Science. 1999; 283: 1325-8.
20. Sudol M, Hunter T. NeW wrinkles for an old domain. Cell. 2000; 103: 1001-4.
21. Passani LA, Bedford MT, Faber PW, McGinnis KM, Sharp AH, Gusella JF, Vonsattel J, MacDonald ME. Huntingtin’s WW domain partners in Huntington’s disease post-mortem brain fulfill genetic criteria for direct involvement in Huntington’s disease pathogenesis. Hum Mol Genet. 2000; 9: 2175-82.
22. Sze CI, Su M, Pugazhenthi S, Jambal P, Hsu LJ, Heath J, Schultz L, Chang NS. Down-regulation of WW Domain-containing Oxidoreductase Induces Tau Phosphorylation in Vitro. A potential role in Alzheimer’s disease. J Biol Chem. 2004; 279: 30498-506.
23. Bednarek AK, Laflin KJ, Daniel RL, Liao Q, Hawkins KA, Aldaz CM. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23. 3-24.1, a region frequently affected in breast cancer. Cancer Res. 2000; 60: 2140-45.
24. Yendamuri S, Kuroki T, Trapasso F, Henry AC, Dumon KR, Huebner K, Williams NN, Kaiser LR, Croce CM. WW domain containing oxidoreductase gene expression is altered in non-small cell lung cancer. Cancer Res. 2003; 63: 878-81.
25. Xie P, Tian C, An L, Nie J, Lu K, Xing G, Zhang L, He F. Histone methyltransferase protein SETD2 interacts with p53 and selectively regulates its downstream genes. Cell Signal. 2008; 20: 1671-8.
26. Gao YG, Yang H, Zhao J, Jiang YJ, Hu HY. Autoinhibitory Structure of the WW Domain of HYPB/SETD2 Regulates Its Interaction with the Proline-Rich Region of Huntingtin. Structure. 2014; 22: 378-86.
27. Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005; 25: 3305-16.
28. Li B, Howe L, Anderson S, Yates JR 3rd, Workman JL. The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. J Biol Chem. 2003; 278: 8897-903.
29. Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, Richards DP, Beattie BK, Emili A, Boone C, Shilatifard A, Buratowski S, Greenblatt J. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2003; 23: 4207-18.
30. Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012; 13: 115-26.
31. Yuan W, Xie J, Long C, Erdjument-Bromage H, Ding X, Zheng Y, Tempst P, Chen S, Zhu B, Reinberg D. Heterogeneous nuclear ribonucleoprotein L Is a subunit of human KMT3a/Set2 complex required for H3 Lys-36 trimethylation activity in vivo. J Biol Chem. 2009; 284: 15701-07.
32. Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Res. 2011; 21: 564-78.
33. Vezzoli A, Bonadies N, Allen MD, Freund SM, Santiveri CM, Kvinlaug BT, Huntly BJ, Göttgens B, Bycroft M. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol. 2010; 17: 617-9.
34. Wu H, Zeng H, Lam R, Tempel W, Amaya MF, Xu C, Dombrovski L, Qiu W, Wang Y, Min J. Structural and histone binding ability characterizations of human PWWP domains. PLoS One. 2011; 6: e18919.
35. Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. 2010; 285: 26114-20.
36. Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann, M. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010; 142: 967-80.
37. Qin S, Min J. Structure and function of the nucleosome-binding PWWP domain. Trends Biochem Sci. 2014; 39: 536-47.
38. Cai L, Rothbart SB, Lu R, Xu B, Chen WY, Tripathy A, Rockowitz S, Zheng D, Patel DJ, Allis CD, Strahl BD, Song J, Wang GG. An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol Cell. 2013; 49: 571-82.
39. Sun B, Hong J, Zhang P, Dong X, Shen X, Lin D, Ding J. Molecular basis of the interaction of Saccharomyces cerevisiae Eaf3 chromo domain with methylated H3K36. J Biol Chem. 2008; 283: 36504-12.
40. Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet. 2009; 41: 376-81.
41. Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Spatial distribution of di-and tri-methyl lysine 36 of histone H3 at active genes. J Biol Chem. 2005; 280: 17732-36.
42. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129: 823-37.
43. De Almeida SF, Grosso AR, Koch F, Fenouil R, Carvalho S, Andrade J, Levezinho H, Gut M, Eick D, Gut I, Andrau JC, Ferrier P, Carmo-Fonseca M. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat Struct Mol Biol. 2011; 18: 977-83.
44. Chantalat S, Depaux A, Héry P, Barral S, Thuret JY, Dimitrov S, Gérard M. Histone H3 trimethylation at lysine 36 is associated with constitutive and facultative heterochromatin. Genome Res. 2011; 21: 1426-37.
45. Batsché E, Yaniv M, Muchardt C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol. 2006; 13: 22-9.
46. Ip JY, Schmidt D, Pan Q, Ramani AK, Fraser AG, Odom DT, Blencowe BJ. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res. 2011; 21: 390-401.
47. Fu X, Ares Jr M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014; 15: 689-701.
48. Zhou HL, Luo G, Wise JA, Lou H. Regulation of alternative splicing by local histone modifications: potential roles for RNA-guided mechanisms. Nucleic Acids Res. 2014; 42: 701-13.
49. Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010; 327: 996-1000.
50. Zhang P, Du J, Sun B, Dong X, Xu G, Zhou J, Huang Q, Liu Q, Hao Q, Ding J. Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic Acids Res. 2006; 34: 6621-8.
51. Kim S, Kim H, Fong N, Erickson B, Bentley DL. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc Natl Acad Sci U S A. 2011; 108: 13564-9.
52. Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, Carmo-Fonseca M, de Almeida SF. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res. 2013; 41: 2881-93.
53. Qin S, Guo Y, Xu C, Bian C, Fu M, Gong S, Min J. Tudor domains of the PRC2 components PHF1 and PHF19 selectively bind to histone H3K36me3. Biochem Biophys Res Commun. 2013; 430: 547-53.
54. Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003; 301: 1090-3.
55. Yoh SM, Lucas JS, Jones KA. The Iws1: Spt6: CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev. 2008; 22: 3422-34.
56. Bortvin A, Winston F. Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science. 1996; 272: 1473-76.
57. Endoh M, Zhu W, Hasegawa J, Watanabe H, Kim DK, Aida M, Inukai N, Narita T, Yamada T, Furuya A, Sato H, Yamaguchi Y, Mandal SS, et al. Human Spt6 stimulates transcription elongation by RNA polymerase II in vitro. Mol Cell Biol. 2004; 24: 3324-36.
58. Ardehali MB, Yao J, Adelman K, Fuda NJ, Petesch SJ, Webb WW, Lis JT. Spt6 enhances the elongation rate of RNA polymerase II in vivo. EMBO J. 2009; 28: 1067-77.
59. Kwak H, Lis JT. Control of transcriptional elongation. Annu Rev Genet. 2013; 47: 483-508.
60. Mason PB, Struhl K. The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo. Mol Cell Biol. 2003; 23: 8323-33.
61. Kaplan CD, Laprade L, Winston F. Transcription elongation factors repress transcription initiation from cryptic sites. Science. 2003; 301: 1096-9.
62. Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM. The Histone Mark H3K36me3 Regulates Human DNA Mismatch Repair through Its Interaction with MutS. Cell. 2013; 153: 590-600.
63. Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010; 20: 761-70.
64. Li F, Ortega J, Gu L, Li GM. Regulation of mismatch repair by histone code and posttranslational modifications in eukaryotic cells. DNA Repair. 2016; 38: 68-74.
65. Eidahl JO, Crowe BL, North JA, McKee CJ, Shkriabai N, Feng L, Plumb M, Graham RL, Gorelick RJ, Hess S, Poirier MG, Foster MP, Kvaratskhelia M. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013; 41: 3924-36.
66. Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012; 8: e1002717.
67. Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ, Helleday T, Legube G, La Thangue NB, Porter AC, Humphrey TC. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014; 7: 2006-18.
68. Carvalho S, Vitor AC, Sridhara SC, Martins FB, Raposo AC, Desterro JM, Ferreira J, de Almeida SF. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife. 2014; 3: e02482.
69. Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011; 45: 247-71.
70. Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007; 315: 1141-3.
71. Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q, Edsall L, Antosiewicz-Bourget J, Stewart R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009; 462: 315-22.
72. Jjingo D, Conley AB, Soojin VY, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012; 3: 462-74. doi: 10.18632/oncotarget.497.
73. Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, Absher DM, Wold BJ, Myers RM. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013; 23: 555-67.
74. Hahn MA, Wu X, Li AX, Hahn T, Pfeifer GP. Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS One. 2011; 6: e18844.
75. Morselli M, Pastor WA, Montanini B, Nee K, Ferrari R, Fu K, Bonora G, Rubbi L, Clark AT, Ottonello S, Jacobsen SE, Pellegrini M. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. Elife. 2015; 4: e06205.
76. Suzuki M, Yamada T, Kihara-Negishi F, Sakurai T, Hara E, Tenen DG, Hozumi N, Oikawa T. Site-specific DNA methylation by a complex of PU. 1 and Dnmt3a/b. Oncogene. 2006; 25: 2477-88.
77. Lukasik SM, Cierpicki T, Borloz M, Grembecka J, Everett A, Bushweller JH. High resolution structure of the HDGF PWWP domain: a potential DNA binding domain. Protein Sci. 2006; 15: 314-23.
78. Maltby VE, Martin BJ, Schulze JM, Johnson I, Hentrich T, Sharma A, Kobor MS, Howe L. Histone H3 lysine 36 methylation targets the Isw1b remodeling complex to chromatin. Mol Cell Biol. 2012; 32: 3479-85.
79. Xu C, Cui G, Botuyan MV, Mer G. Structural basis for the recognition of methylated histone H3K36 by the Eaf3 subunit of histone deacetylase complex Rpd3S. Structure. 2008; 16: 1740-50.
80. Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, Yang P, Workman JL, Park PJ, Kuroda MI. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol Cell. 2007; 28: 121-33.
81. Huen MS, Huang J, Leung JW, Sy SM, Leung KM, Ching YP, Tsao SW, Chen J. Regulation of chromatin architecture by the PWWP domain-containing DNA damage-responsive factor EXPAND1/MUM1. Mol Cell. 2010; 37: 854-64.
82. Musselman CA, Avvakumov N, Watanabe R, Abraham CG, Lalonde ME, Hong Z, Allen C, Roy S, Nuñez JK, Nickoloff J. Kulesza CA, Yasui A, Côté J, et al. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat Struct Mol Biol. 2012; 19: 1266-72.
83. Wang J, Qin S, Li F, Li S, Zhang W, Peng J, Zhang Z, Gong Q, Wu J, Shi Y. Crystal structure of human BS69 Bromo-ZnF-PWWP reveals its role in H3K36me3 nucleosome binding. Cell Res. 2014; 24: 890-3.
84. Hu M, Sun XJ, Zhang YL, Kuang Y, Hu CQ, Wu WL, Shen SH, Du TT, Li H, He F, Xiao HS, Wang ZG, Liu TX, et al. Histone H3 lysine 36 methyltransferase Hypb/Setd2 is required for embryonic vascular remodeling. Proc Natl Acad Sci U S A. 2010; 107: 2956-61.
85. Zhang Y, Xie S, Zhou Y, Xie Y, Liu P, Sun M, Xiao H, Jin Y, Sun X, Chen Z, Huang Q, Chen S. H3K36 histone methyltransferase Setd2 is required for murine embryonic stem cell differentiation toward endoderm. Cell Rep. 2014; 8: 1989-2002.
86. Luscan A, Laurendeau I, Malan V, Francannet C, Odent S, Giuliano F, Lacombe D, Touraine R, Vidaud M, Pasmant E, Cormier-Daire V. Mutations in SETD2 cause a novel overgrowth condition. J Med Genet. 2014; 51: 512-7.
87. Sotos JF, Dodge PR, Muirhead D, Crawford JD, Talbot NB. Cerebral gigantism in childhood: a syndrome of excessively rapid growth with acromegalic features and a nonprogressive neurologic disorder. New Eng. J. Med. 1964; 271: 109-16.
88. Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, Ohashi H, Naritomi K, Tsukahara M, Makita Y, Sugimoto T, Sonoda T, Hasegawa T. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet. 2002; 30: 365-6.
89. Li Y, Trojer P, Xu CF, Cheung P, Kuo A, Drury WJ,3rd, Qiao Q, Neubert TA, Xu RM, Gozani O, Reinberg D. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem. 2009; 284: 34283-95.
90. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010; 463: 360-3.
91. Duns G, van den Berg E, van Duivenbode I, Osinga J, Hollema H, Hofstra RM, Kok K. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 2010; 70: 4287-91.
92. Duns G, Hofstra RM, Sietzema JG, Hollema H, van Duivenbode I, Kuik A, Giezen C, Jan O, Bergsma JJ, Bijnen H. van der Vlies P, van den Berg E, Kok K. Targeted exome sequencing in clear cell renal cell carcinoma tumors suggests aberrant chromatin regulation as a crucial step in ccRCC development. Hum Mutat. 2012; 33: 1059-62.
93. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013; 499: 43-9.
94. Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki H, Nagata Y, Yoshida K, Kon A, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013; 45: 860-7.
95. Hakimi AA, Chen YB, Wren J, Gonen M, Abdel-Wahab O, Heguy A, Liu H, Takeda S, Tickoo SK, Reuter VE, Voss MH, Motzer RJ, Coleman JA, et al. Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma Eur Urol. 2013; 63: 848-54.
96. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43: D805-11.
97. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505: 495-501.
98. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6: pl1.
99. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2: 401-4.
100. Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, Yan X, Wei W, Pang Y, Cheng H, Hua C, Zhang Y, Yang X. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014; 46: 287-293.
101. Pena-Llopis S, Christie A, Xie XJ, Brugarolas J. Cooperation and antagonism among cancer genes: the renal cancer paradigm. Cancer Res. 2013; 73: 4173-9.
102. Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin M, Teague J, Bignell G, Butler A, Cho J, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011; 469: 539-42.
103. Berquam-Vrieze KE, Nannapaneni K, Brett BT, Holmfeldt L, Ma J, Zagorodna O, Jenkins NA, Copeland NG, Meyerholz DK, Knudson CM, Mullighan CG, Scheetz TE, Dupuy AJ. Cell of origin strongly influences genetic selection in a mouse model of T-ALL. Blood. 2011; 118: 4646-56.
104. March HN, Rust AG, Wright NA, ten Hoeve J, de Ridder J, Eldridge M, van der Weyden L, Berns A, Gadiot J, Uren A. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet. 2011; 43: 1202-9.
105. Al Sarakbi W, Sasi W, Jiang W, Roberts T, Newbold R, Mokbel K. The mRNA expression of SETD2 in human breast cancer: correlation with clinico-pathological parameters. BMC cancer. 2009; 9: 290.
106. Fontebasso AM, Schwartzentruber J, Khuong-Quang D, Liu X, Sturm D, Korshunov A, Jones DT, Witt H, Kool M, Albrecht S. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013; 125: 659-69.
107. Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen Y, Gonen M, Liu H, Takeda S, Voss MH, Tickoo SK, Reuter VE, Russo P, Cheng EH, et al. Adverse Outcomes in Clear Cell Renal Cell Carcinoma with Mutations of 3p21 Epigenetic Regulators BAP1 and SETD2: A Report by MSKCC and the KIRC TCGA Research Network. Clin Cancer Res. 2013; 19: 3259-67.
108. Ho TH, Park IY, Zhao H, Tong P, Champion M, Yan H, Monzon F, Hoang A, Tamboli P, Parker AS, Joseph RW, Qiao W, Dykema K, et al. High-resolution profiling of histone H3 lysine 36 trimethylation in metastatic renal cell carcinoma. Oncogene. 2015; 35: 1565-74.
109. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366: 883-92.
110. Oltean S, Bates D. Hallmarks of alternative splicing in cancer. Oncogene. 2014; 33: 5311-8.
111. Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, Weiser M, Ho TH, Kuan PF, Jonasch E, Furey TS, Prins JF, Lieb JD, Rathmell WK, et al. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res. 2014; 24: 241-50.
112. Li X, Shi B, Yang Q, Wu J, Wu H, Wang Y, Wu Z, Fan Y, Wang Y. Epigenetic regulation of CDH1 exon 8 alternative splicing in gastric cancer. BMC Cancer. 2015; 15: 954.
113. Labbé RM, Holowatyj A, Yang ZQ. Histone lysine demethylase (KDM) subfamily 4: structures, functions and therapeutic potential. Am J Transl Res. 2013; 6: 1-15.
114. Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013; 73: 2936-42.
115. Awwad SW, Ayoub N. Overexpression of KDM4 lysine demethylases disrupts the integrity of the DNA mismatch repair pathway. Biol Open. 2015; 4: 498-504.
116. Li H, An J, Wu M, Zheng Q, Gui X, Li T, Pu H, Lu D. LncRNA HOTAIR promotes human liver cancer stem cell malignant growth through downregulation of SETD2. Oncotarget. 2015; 6: 27847-64. doi: 10.18632/oncotarget.4443.
117. Tang L, Zhang W, Su B, Yu B. Long noncoding RNA HOTAIR is associated with motility, invasion, and metastatic potential of metastatic melanoma. Biomed Res Int. 2013; 2013: 251098.
118. Xiang W, He J, Huang C, Chen L, Tao D, Wu X, Wang M, Luo G, Xiao X, Zeng F, Jiang G. miR-106b-5p targets tumor suppressor gene SETD2 to inactive its function in clear cell renal cell carcinoma. Oncotarget. 2015; 6: 4066-79. doi: 10.18632/oncotarget.2926.
119. Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, Ramaswamy S, Futreal PA, Haber DA, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013; 41: D955-61.
120. Feng C, Sun Y, Ding G, Wu Z, Jiang H, Wang L, Ding Q, Wen H. PI3K Inhibitor TGX221 Selectively Inhibits Renal Cell Carcinoma Cells with Both VHL and SETD2 mutations and Links Multiple Pathways. Sci Rep. 2015; 5: 9465.
121. Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G, Mavrommati I, Pai C, Zalmas L, Drobnitzky N, Dianov GL, Verrill C, Macaulay VM, et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell. 2015; 28: 557-68.
122. Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, et al. The BioPlex network: a systematic exploration of the human interactome. Cell. 2015; 162: 425-40.
123. Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015; 163: 712-3.
124. Kirli K, Karaca S, Dehne HJ, Samwer M, Pan KT, Lenz C, Urlaub H, Görlich D. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. Elife. 2015; 4. pii: e11466.