
INTRODUCTION
Approximately 20% of HCCs show activation of Wnt/β-catenin and TGFβ pathways, carry wild-type β-catenin, lose the hepatocyte phenotype and overexpress Frizzled (FZD) receptors [1, 2]. Wnt/β-catenin pathway activation involves interaction of Wnt ligands with cell surface FZD receptors and LRP5/6 co-receptors, followed by disruption of the Adenomatous polyposis coli (APC)-axin platform, thus halting GSK3B-dependent phosphorylation of β-catenin at key residues in exon 3. Non-phosphorylated β-catenin is thus stabilized, accumulates in the cytoplasm and nucleus and interacts with T-cell factor (TCF) transcription factors of target gene expression. Conversely, in the absence of interaction of Wnt ligands with their cell surface receptors, phosphorylated β-catenin undergoes proteasomal degradation. β-catenin exon 3 mutations in tumors bypass the GSK3B gatekeeper, thus switching the pathway to the ON position constitutively [3].
It is widely accepted that Wnt pathway activation in HCCs may be driven by upregulation of TGFB and tyrosine-kinase receptor pathways [2, 4, 5], Wnt ligands, their cell surface frizzled receptors and/or epigenetic silencing of a family of endogenous Wnt inhibitors, i.e., the Secreted Frizzled-Related Proteins (SFRPs) [6]. SFRPs are soluble decoy receptors composed of a cysteine-rich frizzled ligand-binding domain (FZD_CRD), thereby quenching Wnt activity at the cell surface [7]. Thus, increasing the ratio of SFRP-like Wnt inhibitors over Wnt ligands or receptors controls tumor growth [8-11].
Most cases of HCC arise in a background of chronic inflammation and tissue remodeling, leading to stem/progenitor cell amplification [12]. In the context of experimental liver regeneration in response to tissue damage, Wnt signals expand transit-amplifying liver progenitor cells but impair hepatocyte differentiation [13].Thus, the aim of the present study was to investigate Wnt-specific molecular pathways, cell fates, phenotypes and clinical outcomes in HCC stem/progenitor cells. To this end, we fine-tuned Wnt/β-catenin signaling with the prototypical Wnt3a ligand [14, 15] and inhibited Wnt signaling using β-catenin siRNA knockdown or soluble FZD8_CRD in HepaRG liver progenitor cells. These bipotent progenitors carry wild-type Wnt/β-catenin pathway components [16] and can differentiate into hepatocyte- or biliary-like cells [17, 18].
We report that Wnt3a reprograms liver progenitors to replicating and invasive fibrogenic myofibroblast-like cells expressing stem cell markers. Analysis of HCCs from 70 patients revealed two tight gene networks associating stem/progenitor markers, cell surface Wnt signaling and fibrogenesis. Both networks were linked by Hyaluronan And Proteoglycan Link Protein 1 (HAPLN1), a mesenchymal matrix protein, which is essential in development [19]. HAPLN1 links proteoglycans with hyaluronic acid, thereby building growth factor binding platforms [20]. Whereas HAPLN1 appeared de novo in aggressive tumors expressing stem cell markers and in vitro models of epithelial-mesenchymal transition; HAPLN1 knockdown downregulated key markers of mesenchymal cells. We hypothesize that HAPLN1 can be hijacked by tumor evolution as a selective advantage for cancer progression.
RESULTS
Wnt signals lead to myofibroblastic differentiation of liver progenitor cells
As Wnt3a binds frizzled (FZD) receptors at the low nanomolar range [3], HepaRG hepatic progenitor cells were incubated with Wnt3a-conditioned medium, which contains <10 nM Wnt3a [9] or with 7 nM purified recombinant Wnt3a for 13 days (Figure 1A and Supplementary Figure 1A and 1B). Control media led differentiation to hepatocyte-like cells, as expected [18]. In contrast, Wnt3a-enriched media led to albumin¯ fibroblast-like cells, containing alpha-smooth-muscle-actin+ (α-SMA+) stress fibers, thus indicating myofibroblast differentiation (Figure 1B). These cells expressed the basement membrane collagen type IV, the mesenchymal markers N-cadherin (Figure 1B), Vimentin and Fibronectin, as well as cytoplasmic β-catenin (Supplementary Figure 1C). Gene expression kinetics showed increased MYC, SNAI1, TWIST1 and TGFB1 after 12 h, with a peak in Collagen IV and α-SMA mRNAs after 5 days of incubation of progenitor cells with purified Wnt3a. After 13 days, myofibroblast-like cells showed an important increase in LGR5 and BIRC5 (Survivin) and decrease in Aldolase B, GGT1, NOTCH2, Keratin 19 and SOX9 (Figure 1C). Consistently, after Wnt3a treatment, SNAI1 showed a seven-fold increase within the 1st h and CD44 increased within the first 8 h (Supplementary Figure 1D). Wnt3a promoted Matrigel invasion, [inhibited by 30 nM FZD7_CRD or FZD8_CRD (Figure 1D and 1E)], and cell proliferation throughout the 13-day assay (Figure 1F).
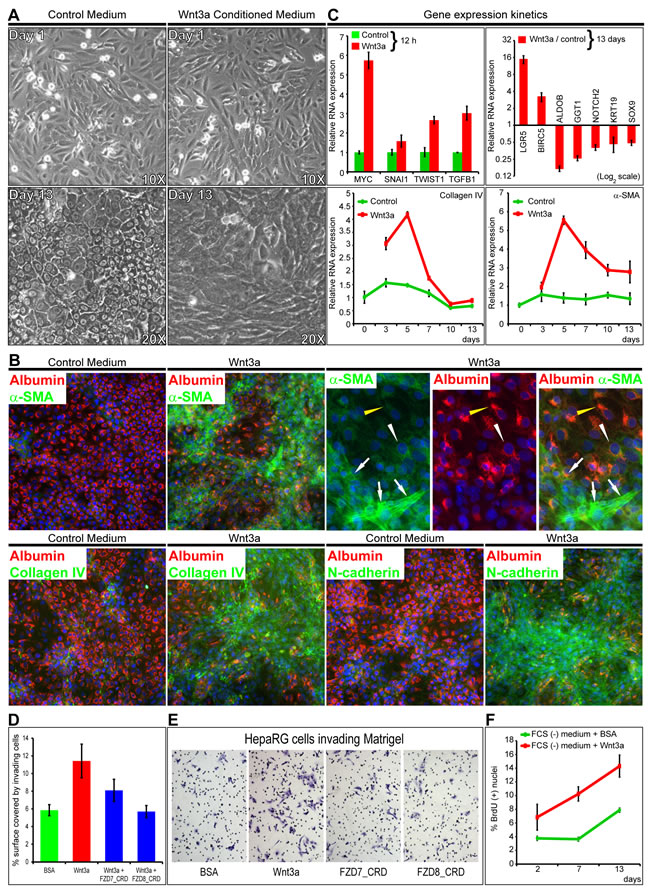
Figure 1: Wnt signals differentiate liver progenitors to myofibroblast-like cells invading Matrigel. A. and B. Incubation of progenitor HepaRG cells with control or Wnt3a-conditioned medium for 13 days. A. Controls are hepatocyte-like. Cells incubated with Wnt3a are fibroblast-like. B. Coimmunodetection of the indicated proteins. Alpha smooth muscle actin (α-SMA), collagen IV and N-cadherin: green (FITC); albumin, red (FluoProbes 594); nuclei, blue (DAPI). Images were acquired with a Cellomics station at 20X. Wnt3a-incubated cells show bridging fascicules of spindle α-SMA(+)/albumin(-) cells. Stress fibers typical of myofibroblasts (white arrows) embrace foci of transitional elongated α-SMA(+)/albumin(+) cells (white arrowheads), with granular and filamentous compartments (yellow arrowheads). C. Gene expression kinetics of HepaRG cells incubated with recombinant Wnt3a or control BSA and analyzed by real-time PCR. D. FZD7_CRD and FZD8_CRD inhibit Wnt3a-induced Matrigel invasion. Cells were incubated in Matrigel-coated Boyden chambers with FCS(-) medium plus BSA; 7 nM Wnt3a; Wnt3a plus 30 nM FZD7 or FZD8_CRD. E. HepaRG cells invading Matrigel. F. Wnt3a enhances cell proliferation. Cells were incubated in 96-well plates with FCS(-) medium plus BSA or Wnt3a. BrdU(+) nuclei were counted with a Cellomics station.
Wnt signals promote commitment of transit-amplifying liver progenitor cells to fibrogenic myofibroblast-like cells
To look at the gene networks leading to myofibroblastic differentiation of hepatic progenitors, we treated cells with 7 nM Wnt3a for three days. To identify β-catenin-dependent or FZD8_CRD sensitive gene networks, we transfected cells with β-catenin-targeting siRNA or treated them with the prototypical Wnt inhibitor FZD8_CRD. This strategy was validated by real-time PCR gene expression analysis of the β-catenin target gene LGR5 in HepaRG and Huh7 cells (Supplementary Figure 2A-2D). A whole transcriptome analysis identified 358 genes differentially regulated by Wnt3a in progenitor HepaRG cells (Supplementary Figure 2E-2H and Supplementary Table 3A). After filtering out 169/358 genes affected by control (scrambled) siRNA (Supplementary Table 3B), we obtained 189 genes, 142 (75%) of them were sensitive to β-catenin siRNA knockdown and 47 (25%) were not (Supplementary Table 3, C and D). Among the 358 genes modulated by Wnt3a in HepaRG cells, 233 (65%) of them were inhibited by 30 nM FZD8_CRD and 125 (35%) were not (Supplementary Table 3, E and F). Finally, 77 genes were both sensitive to both β-catenin knockdown and FZD8_CRD in independent experiments (Supplementary Table 3G).
Ingenuity Pathway Analysis revealed fibrogenesis and myofibroblast activation pathways (Figure 2A). Consistently, gene ontology analysis by FuncAssociate 2.0 [21] confirmed that Wnt signals induced highly significant and reproducible fibrogenic gene sets (Supplementary Table 4). Also, gene ontology analysis confirmed that the gene set affected by scrambled siRNA was only associated with actin crosslink formation, reflecting cytoskeletal rearrangements following electroporation [22]. Importantly, genes specifically silenced by β-catenin targeting siRNA and by FZD8_CRD were both associated with fibrogenesis.
To strengthen our observations, we compared the Wnt-induced signature in HepaRG cells with the whole genome mRNA expression profiles of epithelial 3P and fibroblast-like 3SP human HCC cell lines. These cells are derived from the same HCC, 3SP resulting from 3P cells that underwent spontaneous EMT [23]. Gene Set Enrichment Analysis (GSEA) revealed that the transcriptome of 3SP cells was significantly enriched in genes induced by Wnt3a in HepaRG progenitor cells (Figure 2B). By hierarchical clustering, fibroblast-like 3SP cells were unambiguously distinguished from epithelial 3P cells on the basis of the gene expression of Wnt Homepage targets (Figure 2C) and extracellular Wnt pathway components (Figure 2D) and associated functions (Supplementary Table 5). Moreover, comparison of the mRNA signature of transiently mesenchymal-competent cells induced by dedifferentiation of hepatocyte-like HepaRG cells [18] with our Wnt3a-induced signature confirmed that Wnt signals specifically promote differentiation of transit-amplifying liver progenitor cells to fibrogenic myofibroblast-like cells (Supplementary Table 6).
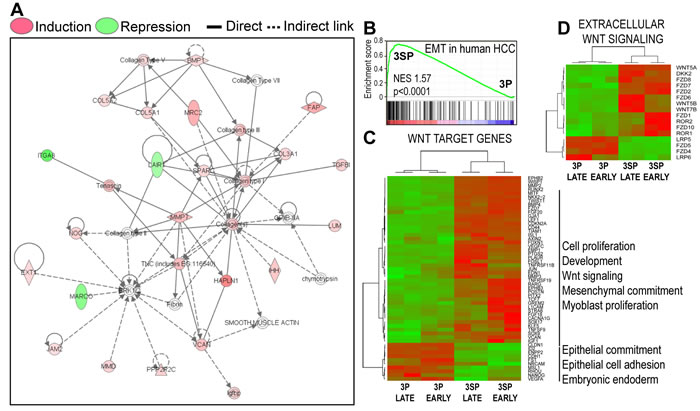
Figure 2: Wnt signals activate a fibrogenic gene expression program in liver progenitors. A. Gene networks applying Ingenuity pathway analysis on the 358 gene set differentially regulated by Wnt3a. B. Wnt-induced differentiation of hepatic progenitors into fibrogenic myofibroblast-like cells matches the transcriptome signature of spontaneously emerging EMT in human HCC (GSE26391). 3P and 3SP human HCC cell lines derive from the same tumor, mesenchymal 3SP resulting from epithelial 3P cells that underwent spontaneous EMT in a human HCC [23]. By gene set enrichment analysis, 3SP cells are significantly enriched in transcripts induced by Wnt3a in HepaRG cells. C. and D. Hierarchical clustering: 3SP and 3P cells are distinguished by C Wnt target genes from the Wnt Homepage (Supplementary Table 6A) and D Extracellular Wnt signaling (Wnt pathway ligands and receptors). Raw gene expression data were normalized by the Robust Multi-array Average (RMA) log2 algorithm and quantile filtered.
In vitro Wnt-induced fibrogenesis activates signaling networks that are functional in HCCs
Close analysis of Wnt-induced genes sensitive to both β-catenin targeting siRNA and to FZD8_CRD (Supplementary Table 3G) revealed crosstalks of Wnt with TGFB/BMP, Notch, Hedgehog and IGF signaling pathways [5]. To validate these crosstalks, β-catenin dependency of representative genes of these four signaling pathways was confirmed by real-time PCR (Figure 3A). Among these, the induction of the stem cell marker LGR5 by Wnt3a in Huh-7 or HepaRG cells was partially inhibited by the Hedgehog inhibitor KAAD-cyclopamine, but not by the inactive structural analog tomatidine (Figure 3B). Whereas LGR5 expression by myofibroblast-like cells suggested that they retained stem cell features (Figure 3C); silencing of LGR5 by specific siRNA upregulated the hepatocyte differentiation marker aldolase B (Figure 3D), suggesting that Wnt-induced LGR5 impairs hepatocyte differentiation.
We then sought to validate these findings in 79 HCCs, by assessing the mRNA expression of the genes shown in Figure 3A-3C, together with genes involved in cell surface Wnt signaling (GPC3, SFRP1, SFRP2, DKK1, FZD1, FZD7, WNT2) and mesenchymal commitment (ACTA2, LAMC1; COL4A1). Of note, SFRPs 1 and 2 also reflect mesenchymal commitment of stem cells [24]. Weighted Gene Co-expression Network Analysis revealed two tight networks associating stem/progenitor markers to cell surface Wnt signaling and mesenchymal commitment (Figure 3E and Supplementary Table 7). Both networks were linked by HAPLN1 and collagen IV (COL4A1). Hyaluronan And Proteoglycan Link Protein 1 (HAPLN1) is a cartilage gene that links proteoglycans to hyaluronic acid, thereby playing a key role in extracellular matrix assembly [20]. While its absence leads to dwarfism [19], tumors de novo expresses HAPLN1 [25]. Multiple correlation analyses confirmed that both SFRP1 and HAPLN1 were associated with ACTA2 (α-SMA) and collagen type IV (Figure 3F).
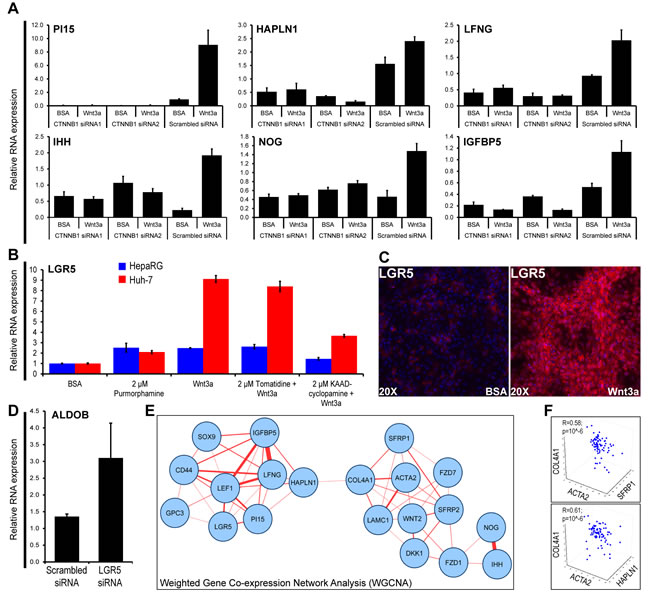
Figure 3: In vitro Wnt-induced fibrogenesis activates signaling networks that are functional in HCCs. A. RNA levels of the indicated genes in progenitor HepaRG HCC cells treated with Wnt3a after transfection with two β-catenin targeting siRNAs. B. LGR5 mRNA expression in HepaRG and Huh-7 cells treated with BSA (control), the Hedgehog pathway inducer Purmorphamine, Wnt3a and the Hedgehog inhibitor KAAD-cyclopamine. Tomatidine is a cyclopamine structural analog which does not inhibit the Hedgehog pathway. C. Immunodetection of LGR5 in HepaRG cells after 13 days of incubation with Wnt3a or BSA. LGR5, red (FluoProbes 594); nuclei, blue (DAPI). D. RNA expression of the hepatocyte differentiation marker ALDOB in HepaRG cells transfected with LGR5 siRNA. E. Gene networks generated by Weighted Correlation Network Analysis (WGCNA) [44] using mRNA expression data for the indicated genes in 79 HCCs. Networks with a correlation coefficient threshold ≥0.30 plotted with Cytoscape [45]. Closeness between nodes is proportional to the number of connections. Thickness of links is proportional to the correlation coefficients. F. 3D scatterplots showing the relationships between mRNA expression of the indicated genes in 79 HCCs. ACTA2, smooth muscle actin; COL4A1, procollagen, type IV, alpha 1 chain. Multiple regression coefficients (R) are indicated.
De novo expression of HAPLN1 hallmarks mesenchymal commitment
Wnt3a-induced upregulation of HAPLN1 was first detected at 12 h (~2.5 fold) reaching >30 folds at 13 days. At this time, HAPLN1 was deposited in the pericellular matrix of N-cadherin+ cells (Figure 4A and 4B). Generation of EPCAM¯/NCAM+ cells is the earliest stage of mesoderm commitment from undifferentiated human embryonic stem cells [26]. Analysis of this dataset (GSE21668) revealed de novo HAPLN1 expression at the earliest stages of mesoderm commitment (Figure 5A). Consistently, by real-time PCR, fibroblast-like HCC 3SP cells showed higher HAPLN1 mRNA levels than epithelial 3P cells (Figure 5B). Moreover, kinetics analysis of gene expression, upon dedifferentiation of hepatocyte-like HepaRG cells to transient mesenchyme-competent progenitors, revealed that HAPLN1 became detectable at 24 h and increased by 8 folds at 48 h post plating. Conversely, upon differentiation of these progenitors to hepatocyte-like cells, HAPLN1 decreased by >35 folds from day 4 to day 15 and was no longer detectable thereafter. This expression kinetics was similar to those of COL4A1 and LGR5 (Figure 5C). Furthermore, transfection of HepaRG progenitor cells with HAPLN1 siRNA downregulated the mesenchymal markers Snail, α-SMA, collagen type IV and the stem cell marker LGR5. Snail was also downregulated by β-catenin and LGR5-targeting siRNAs (Figure 6). Taken together, these findings indicate that HAPLN1 hallmarks mesenchymal commitment of hepatic progenitors.
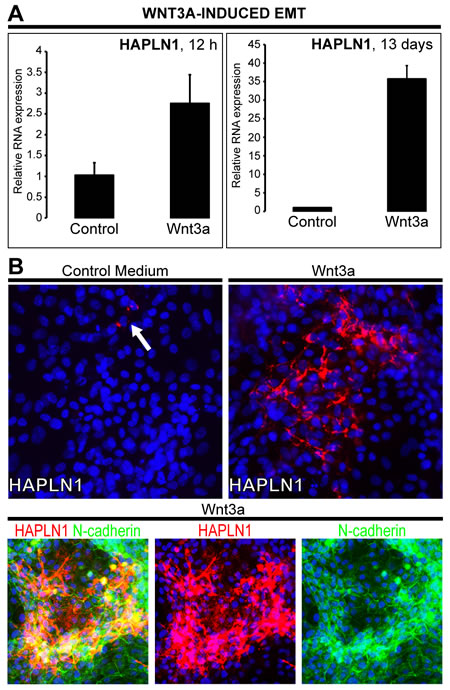
Figure 4: HAPLN1 upregulation by Wnt3a over 13 days. HepaRG progenitor cells were cultured with control FCS(-)/BSA(+) or FCS(-)/BSA(+)/Wnt3a. A. HAPLN1 mRNA levels 12 h and 13 days upon Wnt3a-induction. B. Immunodetection of HAPLN1, red (Cy5); N-cadherin, green (FITC). Nuclei, blue (DAPI). Images acquired at 20X. Control (BSA) shows very rare cells expressing HAPLN1 (arrow). HAPLN1 labels the pericellular matrix and colocalizes with N-cadherin.
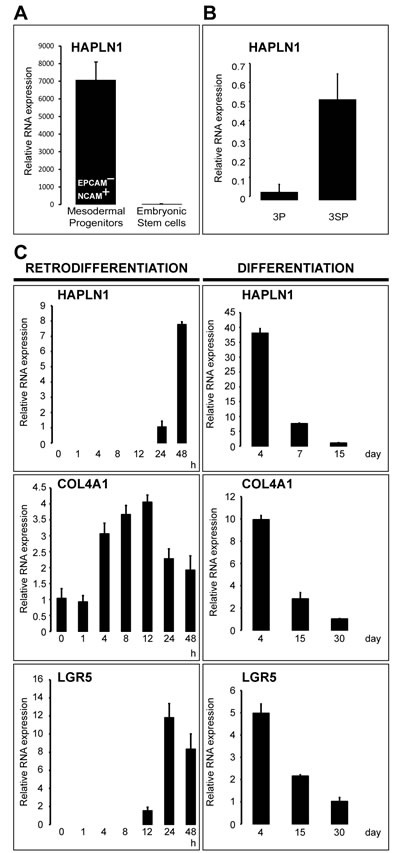
Figure 5: HAPLN1 hallmarks early mesoderm commitment of pluripotent hES cells and spontaneous EMT. A. Normalized mRNA data from GSE21668 [26] showing de novo HAPLN1 expression in early EPCAM¯/NCAM+ mesoderm-committed progenitors generated from undifferentiated human embryonic stem cells. B. HAPLN1 expression analyzed by real-time PCR in the 3P→3SP human HCC cell lines derived from the same tumor. Mesenchymal 3SP result from epithelial 3P cells that underwent spontaneous EMT [23]. C. Kinetics of mRNA expression analyzed by real-time PCR in HepaRG cells along the retrodifferentiation of hepatocytes into liver progenitors and the differentiation of liver progenitors to hepatocytes. Time points 0 h and 30 days (COL4A1 and LGR5) or 15 days (HAPLN1) were used as calibrators. HAPLN1 was not detectable at 30 days in differentiated hepatocyte-like cells.
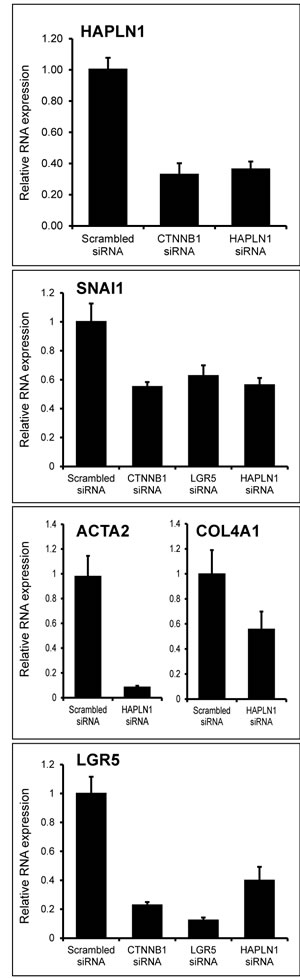
Figure 6: HAPLN1 silencing leads to mesenchymal and stem cell marker loss. Real-time-PCR-assessed mRNA expression of the indicated genes in HepaRG progenitor cells transfected with the indicated siRNAs. HAPLN1 silencing downregulates SNAI1; ACTA2; COL4A1 and LGR5 expression.
HAPLN1 is expressed at high levels in a subset of HCCs with bad outcome after curative resection.
HAPLN1 mRNA levels were measured by real-time PCR in 82 HCCs from 70 patients, 5 histologically normal liver controls, 11 FNHs and 66 matching non-tumor liver samples. HAPLN1 was detected at much higher levels in HCCs than in non-tumor livers in our cohort (Figure 7A), as well as in 80 HCCs with respect to 307 non-tumor hepatitis/cirrhosis livers in a public DASL microarray dataset (GSE10143) [12] (Figure 7B).
The clinical and biological features of the HCC patients in our cohort and the flowchart of inclusion criteria for survival analysis are detailed in Supplementary Table 8, A and B. A binary HAPLN1 expression score was generated from our real-time PCR and TMA immunohistochemistry data. HAPLN HIGH and LOW HCCs showed significantly different HAPLN1 mRNA levels and TMA immunohistochemistry scores (Figure 7C and 7D). HAPLN HIGH patients presented lower disease-free and overall survival (Figure 7E and 7F). Univariate Cox’s analysis (Supplementary Table 9) confirmed the association of HAPLN1 with poor survival after curative resection. Multivariate analysis after adjustment for alpha-fetoprotein, vascular invasion, number of resected liver segments and capsular infiltration by the HCC showed that HAPLN1 was independently associated with survival after curative resection, with a mean increase in relative risk of 4.6 folds (Figure 7G). Of note, HAPLN1 relationship with survival was not dependent on β-catenin exon 3 mutations (Figure 7H) assessed by Sanger sequencing (Supplementary Table 10). Indeed, HAPLN1 expression was not driven by β-catenin activating mutations (Supplementary Figure 3). This contrasts with LGR5 (Supplementary Figure 3), which expression may be governed by both β-catenin mutations [1, 2] and by Wnt-driven stem cell amplification [13].
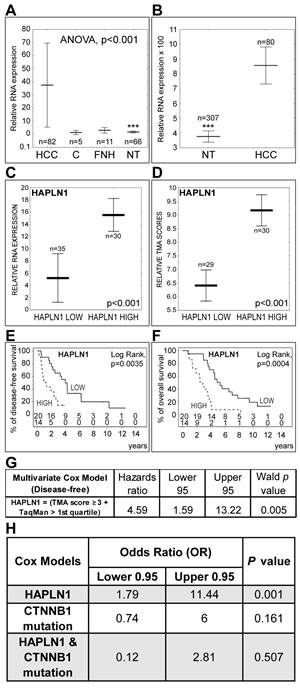
Figure 7: High HAPLN1 expression indicates bad outcome after curative HCC resection. A. Real-time PCR analysis of HAPLN1 mRNA expression in HCC, histologically normal liver controls C, focal nodular hyperplasias FNH and matching non-tumor livers NT. Controls were used as calibrator. Asterisks (***) indicate P<0.001 for NT versus HCC. B. Comparison of HAPLN1 mRNA levels in HCC versus NT from a public DASL microarray dataset (GSE10143) [12] normalized by the cubic spline algorithm and filtered by fold change (up/down>3). C. and D. HAPLN1 LOW and HAPLN HIGH classes display different mRNA and protein levels by real-time PCR and by TMA-based immunohistochemistry scoring. E. and F. Disease-free and overall survival after curative HCC resection. The number of patients at risk at each time point is shown (Upper row, HAPLN LOW; lower row, HAPLN1 HIGH). G. Multivariate Cox’s proportional hazards model after adjustment for alpha-fetoprotein, vascular invasion, number of resected liver segments and capsular infiltration. The model indicates that HAPLN1 is independently associated with survival. H. Cox’s model integrating β-catenin (CTNNB1) exon 3 mutations shows that HAPLN1 is not dependent on β-catenin mutations.
HAPLN1 emerges in aggressive HCCs expressing markers of tumor stem/progenitor cells
HCCs showed co-expression of HAPLN1 with other genes found to be induced by Wnt3a in vitro. HAPLN1 and the tumor progenitor cell markers CD44 and LGR5 were detected in tumor cells at the tumor-stroma interface and in cells arranged in an “Indian file” pattern (Figure 8A). HAPLN1 correlated with laminin ɣ1, α-SMA, CD44, as well as with other genes induced by Wnt3a, such as LFNG, IGFBP5, LEF1 and PI15 (Figure 8B). HCCs with microscopic vascular invasion had high mRNA levels of HAPLN1, LGR5, LEF1, SOX9, CD44, Glypican 3 and larger tumor size (Figure 8C). Serum AFP levels were higher in HAPLN1 HIGH class patients and, similarly, patients with high AFP levels displayed high immunohistochemistry scores for HAPLN1. Likewise, high HAPLN1 expression correlated with the stem cell marker PROM1 and with cytoplasmic β-catenin (Figure 8D). Consistently, cytoplasmic β-catenin staining is increased in HCCs showing enhanced Wnt signaling without β-catenin gene mutations [1, 2]. Moreover, a cluster of biomarkers induced by Wnt3a in HepaRG cells reflecting progenitor cells with mesenchymal commitment was associated with high Edmondson-Steiner’s score in HCCs, indicating poor tumor differentiation (Supplementary Figure 4). Taken together, these findings indicate that HAPLN1 emerges in mesenchymal-committed tumor progenitor cells driving tumor aggressiveness and poor outcome in HCC.
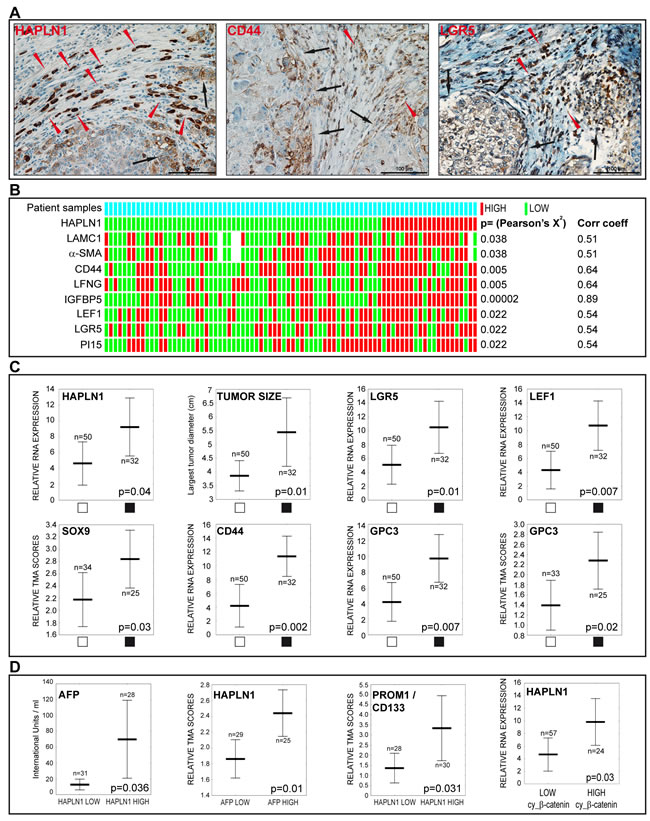
Figure 8: HAPLN1 is detected in HCCs featuring stem cell markers, high alpha-fetoprotein levels, vascular invasion and β-catenin activation. A. Immunodetection of HAPLN1, CD44 and LGR5 in formalin-fixed, paraffin embedded HCCs. Brown color, specific signal; blue, hematoxylin counterstaining. The three proteins are detected in tumor cells at the tumor-stroma interface (black arrows) and in cells arranged in “Indian files” (red arrowheads). B. Heatmap showing associations of HAPLN1 mRNA expression with the indicated genes. White bars indicate missing data (n=8/729 spots). Pearson’s X2 and Gamma correlation coefficients are shown on the right. C. Vascular tumor invasion (black squares) is associated with the indicated variables. D. Serum AFP levels are higher in HAPLN1 HIGH patients. High HAPLN1 protein correlates with high AFP serum levels. High PROM1/CD133 protein is detected in HAPLN1 HIGH HCCs. High HAPLN1 mRNA correlates with high cytoplasmic β-catenin.
DISCUSSION
We show here that Wnt-specific signals foster fibrogenic, myofibroblast-like populations with stem cell features and impair hepatocyte and biliary lineage commitment of liver progenitors. The pathway crosstalks elicited by Wnt-specific signals form a tight network in HCCs. Among the genes switched on within this network, HAPLN1 is associated with bad outcome and with the expression of markers of stemness and of tumor aggressiveness (Figure 9).
We showed that Wnt3a upregulates TGFB1, TGFB2 and BMP1 expression. In turn, TGFB activates Wnt signaling and plays a key role in mesenchymal commitment [4, 27]. Thus, Wnt/TGFB pathways may set up a positive feedback loop leading to tumor aggressiveness. In addition, R-spondin binding of LGR5 receptors enhances Wnt signaling and leads to long-term expansion of bipotent hepatic progenitor cells [13]. In this context, excess of TGFB/BMP signaling is inhibited by Noggin, thus avoiding growth arrest and epithelial-mesenchymal transition [13, 28]. Our data suggest that Wnt3a triggers finely-tuned signaling pathway crosstalks, driving progenitor cell reprogramming to replicative, invasive and fibrogenic myofibroblast-like cells. Probably because Wnt3a upregulates Noggin (Figure 3A and 3F), as well as MYC and BIRC5 (Figure 1C), TGFB/BMP signaling is counterbalanced, thus avoiding growth arrest and achieving high proliferation rates throughout a two-week assay (Figure 1F). Despite their fibrogenic activity, these myofibroblast-like progenitors kept high levels of the stem cells markers CD44 and LGR5. Thus, the rates of proliferation and the expression of stem cell markers suggest that these cells are not terminally differentiated. This phenotype is compatible with the dynamics of cancer progression.
Wnt/β-catenin lies at the crossroads of a growing list of signaling pathways, including Hedgehog and Notch [5, 29]. High LGR5 expression was achieved through Wnt3a induction of Indian Hedgehog. As both Wnt [13] and Hedgehog [30] pathways upregulate LGR5 expression, Wnt/Hedgehog cross-talks probably amplified the LGR5+ stem cell population. Along these lines, Notch signaling leads to biliary lineage commitment [28], but Wnt3a reduced Notch2 expression (Figure 1C) and upregulated the Notch inhibitor LFNG (Figure 3A), thus impairing biliary lineage commitment. The Wnt-triggered signaling network also included insulin growth factor pathway members, namely IGFL1; 2 and 3, IGF2BP3 and FHL2. IGF2BP3 is an oncofetal IGF-mRNA-binding protein de novo expressed and promoting aggressiveness in HCC [31]. FHL2 enhances β-catenin-dependent transcription of IGFBP5 [32]. IGFBP5, in turn, increases the bioavailability of IGF1 and IGF2 [33] and induces fibrogenesis [34].
Activation of Wnt signaling was reported in hepatic stem cell-like HCCs expressing the epithelial cell adhesion molecule EPCAM [35]. We found that the cancer epithelial cell markers EPCAM and cytokeratin 19 (KRT19) were highly correlated in 79 HCCs (R=0.87, Supplementary Table 7). However, neither EPCAM nor KRT19 did integrate the coexpression network of mesenchymal-committed hepatic progenitors. Consistently, the earliest mesoderm-committed progenitor cells derived from human embryonic stem cells and mesenchymal-competent HepaRG progenitors are EPCAM¯ [18, 26]. As EPCAM+ HCC cells are epithelial [35], and our work is based on mesenchyme-competent EPCAM¯ hepatic progenitors [16, 18], we hypothesize that Wnt signals may direct transit-amplifying progenitor cells toward a mesenchymal phenotype. Importantly, through activation of different gene networks, both works show that the final output is enhanced tumor aggressiveness.
Wnt signals led cells to ectopically express mesenchymal progenitor genes like HAPLN1, which regulates cell growth in developing cartilage [19] and heart valves [36]. Similarly, IGFBP5 and PI15 are involved in branching morphogenesis of epithelial organs [37]. In addition, HAPLN1 was expressed de novo upon mesenchymal commitment of embryonic stem cells and in two models of liver EMT. Conversely, HAPLN1 knockdown in hepatic progenitor cells downregulated LGR5, α-SMA, collagen IV and Snail. In HCCs, in consistency with in vitro data, high HAPLN1 levels were associated with basement membrane remodeling, myofibroblast activation and expression of the stem/progenitor markers CD44, LGR5 and CD133/PROM1; the Notch pathway inhibitor LFNG, the Wnt target gene LEF1 and the IGF pathway agonist IGFBP5 (Figure 8). Supporting our data, a bioinformatics study predicted HAPLN1 expression to lie downstream upregulated Wnt/β-catenin signaling in colorectal cancer [38] and proteomics analysis of tumor stroma reported HAPLN1 in highly metastatic melanomas [25]. Also, HAPLN1 has been associated with bad outcome in pleural mesothelioma [39].
In conclusion, Wnt-specific signals led hepatic progenitors to specialize into invasive, proliferating myofibroblast-like fibrogenic cells with stem cell features through a subtle equilibrium of signaling pathway crosstalks. Through the ectopic induction of mesenchymal progenitor genes in liver cancer cells, Wnt signals confer a high degree of biodiversity and, ultimately, selective advantages for cancer progression.
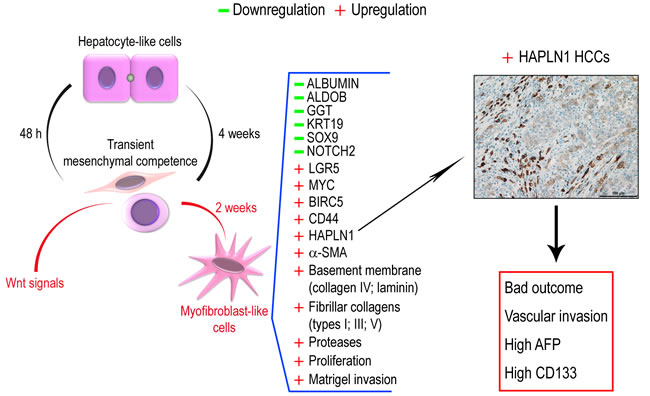
Figure 9: Upon low density plating, hepatocyte-like cells acquire transient mesenchymal competence within 48 h and dedifferentiate to bipotent progenitors that normally re-differentiate into hepatocyte-like cells. A cell microenvironment enriched in Wnt signals reprograms liver progenitor cells into invasive fibrogenic myofibroblast-like cells within two weeks. These cells de novo express the cartilage gene HAPLN1 among other gene modulations. In HCCs, HAPLN1 is expressed de novo in a subset of patients presenting with HCCs that feature bad outcome, vascular invasion, high AFP serum levels and CD133+ tumor cells.
Materials AND METHODS
Patients, clinical data management and tissue banking
Seventy HCC patients undergoing liver surgery with curative intention at Rennes University Hospital between January 1992 and December 2007 were included. Fifty-one were treated by partial hepatectomy and 19 by orthotopic liver transplantation (OLT). The microscopic features of tumors diagnosed as HCC were reviewed and annotated by a senior pathologist (BT). Combined hepatocellular-cholangiocarcinoma or fibrolamellar HCCs were not included. Demographic, clinical and biological data were retrieved from hospital charts by two experienced liver surgeons (LS & DB) and are described in Supplementary Table 8A. After resection, patients were followed up every 3 months for the first 2 years, and every 6 months thereafter. Follow-up, which was available for 65 patients, included clinical examination, serum AFP (alpha-fetoprotein), ultrasonography and computed tomography. Mean and median follow-up times were 60 and 53 months, respectively (95% CI= 50-70 months; range= 3-150 months). The end of follow-up was set between June and September 2012 or at the time of death. Only patients undergoing partial hepatectomy, with a follow-up ≥ 3 months and with complete tumor resection were included in the survival analyses. Early deaths, unrelated to the HCC, and patients without microscopically tumor-free resection margins, as well as patients treated by OLT, were excluded from survival analyses. Survival analysis flowchart is shown in Supplementary Table 8B. Eighty-two frozen tumors from 70 patients were analyzed. As controls, we included 66 matching non-tumor livers, 5 histologically normal livers from patients undergoing resection of liver metastases of extrahepatic primary cancers and 11 focal nodular hyperplasias. Non-tumor and normal liver samples were obtained at >2 cm from the tumors. Frozen human tissue samples were obtained after processing by the Biological Resource Center (BRC) at Rennes University Hospital (BB-0033-00056). Briefly, fresh tissues were frozen at -80°C in N2-cooled isopentane and stored at -80°C under quality-controlled conditions. Formalin-fixed, paraffin-embedded tissue blocks were obtained from the Anatomic Pathology laboratory. The study protocol complied with French laws and regulations and fulfilled the requirements of the local institutional ethics committee. Sample collection was reported to the Ministry of Education and Research (No. DC-2008-338).
Cell culture
Huh-7 [8] and 3P/3SP [23] cells were cultured [10] and Wnt3a-enriched conditioned medium was collected as described [9, 10]. HepaRG cells were expanded as liver progenitors, differentiated to hepatocytes and retrodifferentiated to liver progenitors [18]. To induce EMT in HepaRG cells, progenitor HepaRG cells were detached by trypsinization, seeded at low density (2x104 cells/cm2), detached three days later and seeded at the same density in complete HepaRG medium containing 50% Wnt3a-conditioned or 50% control L cell medium. In other experiments, purified mouse recombinant Wnt3a (R&D Systems, 7nM) and/or mouse FZD8_CRD (30 nM, R&D Systems) were added to serum-free HepaRG medium containing 0.1% BSA for the indicated periods of time according to the experiment. Independent culture experiments were performed in triplicate.
Tissue-microarray (TMA) and immunohistochemistry scoring of human tissues
For selection of tissue blocs for TMA construction, the whole set of archival HE-stained sections from formalin-fixed, paraffin-embedded tissue blocks from each one of the 67 available HCC patients and from five histologically normal livers were reviewed (Nikon 80i microscope) to ensure that the selected paraffin bloc was the mirror lesion of the frozen sample. A region of interest (ROI, 0.5 to 1 cm in diameter) was labeled with a permanent marker to precisely localize the punch zone. TMA construction was done with a MiniCore3 tissue arrayer (Alphelys, Plaisir, France). ROIs were punched in triplicate. Punch size was set at 1000 µm in diameter. HCC samples were randomly assigned to either one of two 132-punch receiver paraffin blocks. Randomization was performed with a “rand” function in Microsoft Excel. The five histologically normal liver controls were included in both array blocs #1 and #2 to control for signal and background intensities. Five-µm microtome sections were processed for immunohistochemistry with a Discovery XT from Ventana Medical Systems (Roche) slide staining system. See Supporting Table 2 for antibodies used. Stained slides were converted into high-resolution digital data with a NanoZoomer digital slide scanner (Hamamatsu, Massy, France). Digital slides were viewed using NDP.view software (Hamamatsu) in a Dell S2415H 24-Inch Screen LED-Lit Monitor. The signal was independently read by two observers (RD and OM) who were blinded to any information. Discrepancies were resolved by consensus reading. A 4-point scale (0-1-2-3-4) denoting increasing signal intensity was used. The relative importance of focal versus general expression of the proteins under study was weighted using the following formulas, where GS stands for general score and FS stands for focal score:
If general signal predominated, score = 2xGS + FS + 1; (where +1 was used to weight the relative importance of general signal with respect to focal signal).
If focal signal predominated, score = 2xFS + GS.
When applied, these formulas yielded scores varying from 3 to 13.
Cell growth assays
Cells were seeded at low density (5×103 cells per well in 96-well plates). HepaRG culture medium was supplemented with 5-bromo-2’-deoxyuridine (BrdU, GE Healthcare Lifesciences) for 1h30 and cells were fixed with 4% PFA. BrdU was detected with mouse anti-BrdU antibody (1:200, ab8152; Abcam) followed by anti-mouse IgG-DyLight 488 (1:500, 072-03-18-18, Eurobio, Les Ulis, France). Image acquisition and quantitation of BrdU-incorporating cells were performed with a Cellomics ArrayScan VTI HCS Reader (Thermo Scientific).
Cell invasion
Cell invasion was assessed in Matrigel-coated Boyden chambers (Transwell inserts, 8.0 µm pore size, Corning). After five days of culture following a routine passage, cells were detached, washed with PBS and suspended in serum-free medium supplemented with 0.1 % BSA. Then, 1x105 cells were seeded in the upper chamber of the Transwell insert. The lower chamber was filled with 800 µl of serum-free medium with or without recombinant mouse Wnt3a (R&D Systems). After 24 hr of incubation at 37°C, cells were fixed with 4% PFA and stained with 2.3% crystal violet. Non-invading cells were gently removed from the upper surface of the insert and images from five fields per membrane were randomly acquired (Axio observer, Carl Zeiss microscopy). Quantitation of invading cells was done with ImageJ (NIH).
β-catenin exon 3 sequencing and search for β-catenin exon 3 deletions
For mutation analysis, exon 3 and the flanking intronic sequences of the CTNNB1 gene were amplified by PCR using Kappa HiFi DNA polymerase (Kapa Biosystems, Wilmington, MA). Briefly, 50 ng of genomic DNA were used as template with βCATEX2F (5’-GAA-AAT-CCA-GCG-TGG-ACA-ATG-3’) and βCATEX4R (5’-TCG-AGT-CAT-TGC-ATA-CTG-TCC-3’) as oligonucleotide primers. The reaction included a 95°C denaturation step for 3 min followed by 35 cycles consisting of: 98°C denaturation for 15 sec, 60°C annealing for 15 sec, and elongation 72°C for 30 sec. Both strands of the amplified products were directly sequenced on an automated sequencer (ABI 3700; Perkin-Elmer, Boston, MA). Negative results on exon 3 mutations may overlook large exon 3 deletions in CTNNB1. Indeed, as somatic deletions are heterozygous, the wild-type allele may mask the deletion upon Sanger sequencing [40]. To avoid this pitfall, cases showing no exon 3 mutation were screened for β-catenin deletion by PCR on reverse-transcribed tumor cDNA. For PCR amplification of the exon 3 of CTNNB1, oligonucleotide primers (F1: 5’- GCG-TGG-ACA-ATG-GCT-ACT-CAA-G-3’; R1: 5’- TAT-TAA-CCA-CCA-CCT-GGT-CCT-C-3’; and R2: 5’- TTC-AGC-ACT-CTG-CTT-GTG-GTC-3’) were used. The primer pairs F1-R1 and F1-R2 yielded 517 bp (169 aa) and 1043 bp (344 aa) amplicons, respectively. As positive control, HepG2-derived cDNA was used, as these cells harbor a large CTNNB1 deletion (amino-acids 25-140) [40]. PCR was performed using the GoTaq Green Master Mix (Promega) and amplicons migrated in 1.5% Nusieve agarose (LifeTechnologies) gels.
DNA microarrays
In vitro transcription and labeling of 150 ng total RNA was carried out using an Agilent Low-Input QuickAmp Labeling kit (Agilent Technologies, Santa Clara, CA, USA) in the presence of Cy3-CTP following the manufacturer’s protocol. After purification using an RNeasy mini kit (Qiagen, Hilden, Germany), cRNA yield and specific activity were determined using a NanoDrop spectrophotometer, as previously described [10]. Equal amount of Cy3-labeled cRNA was subjected to fragmentation followed by 18-hr hybridization onto Agilent human 4x44K v2 pangenomic microarrays. Washing and scanning were performed according to the manufacturer’s instructions (Agilent Technologies). Gene expression data were further processed using Feature Extraction (version 10.7) and GeneSpring (version 11.0) softwares (Agilent Technologies). Filtration of array data resulted in the selection of nonflag-positive and significant gene features. Inter-array normalization was performed by using the 75th percentile signal value. Transcriptomic data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE68633.
Functional genomics analyses
Hierarchical clustering was performed with the hclust function in R software, applying the Ward method to correlation-based distances. Gene annotations based on gene ontology and enrichment for biological functions were done with the FuncAssociate 2.1 program [21]. Gene ontology functions were summarized by removing redundant terms using REVIGO [41]. Ingenuity pathway analysis was used to identify top biological functions, upstream regulators and transcription regulators (Ingenuity Systems, Redwood City, CA, USA). Gene set enrichment analysis (GSEA) was performed with the web-based tool developed at the Broad Institute [42].
Statistical analyses
After removing experimental batch effect with the SVA package [43], gene networks were generated by Weighted Correlation Network Analysis (WGCNA, WGCNA package) [44] using expression data. Gene networks were visually integrated with Cytoscape, with a correlation coefficient threshold ≥0.30 [45]. To analyze patient data, the Recommendations for Tumor Marker Prognostic Studies (REMARK) were applied [46]. Briefly, to screen our transcriptomic dataset for biologically relevant genes warranting further studies, we focused on those meeting two criteria: (i) response to both β-catenin targeting siRNA and FZD8_CRD in independent experiments and (ii) involvement in key signaling pathways. Eight genes meeting these criteria were analyzed by real-time PCR and TMA-based immunohistochemistry in human HCCs. Complying with the REMARK guidelines [46], the inclusion criteria for survival analysis are shown by a flowchart in Supplementary Table 8B. Survival analyses were performed using the Log Rank test and Kaplan-Meier curves and the Cox’s proportional hazards method for univariate or multivariate analyses, as indicated. To this end, real-time PCR results were calculated by the ΔΔCt method and calibrated with respect to the median of a standard range curve run in every experiment. Real-time PCR results were further normalized by the following equation: [(LNi-meanLNie→n)/SDLNi→n], where LN, natural logarithm; i→n, from the first to the last value. To obtain binary values for Kaplan-Meier curves, normalized real-time PCR results were binarized using the median as the cut-off value. TMA-based immunohistochemistry scores (0-1-2-3-4) were binarized by a ≥3 cut-off score for focal expression of specific proteins.
Correlation analyses between gene expression levels were performed with the Spearman rank order test (continuous variables, non-parametric distribution). Association analyses between binary or ordinal variables (expression scores, clinical, biological or anatomic pathology data) were performed with Pearson’s Chi2 or Fisher exact test. Group comparisons were done with non-parametric ANOVA and the Kruskal Wallis post hoc tests, unless otherwise indicated. Heatmaps showing associations between binary patient data were constructed with Excel (Microsoft Office 2010). Statistical calculations were performed with the R software (version 3.1.1) and the packages survival, FactoMineR, gplots, colorspace and marray or with Statistica 10 (Statsoft, Maisons-Alfort, France).
See Supplementary Materials and Methods for: RNA interference, nucleic acid extraction from human tissues and cells, real-time PCR and immunocytochemistry. See Supplementary Table 1 for real-time PCR primers and TaqMan probes and Supplementary Table 2 for antibodies.
Abbreviations
α-SMA, alpha smooth muscle actin; AFP, alpha-fetoprotein; APC, adenomatous polyposis coli; BRC, biological resource center; CTNNB1, DASL, cDNA-mediated Annealing, Selection, extension, and Ligation Assay ; EMT, epithelial-mesenchymal transition; FNH, focal nodular hyperplasia; FZD, frizzled; GGT1, gamma-glutamyltransferase 1; GSE, gene expression series; GSK3B, glycogen synthase kinase 3 beta; HAPLN1, hyaluronan and proteoglycan link protein 1; HCC, hepatocellular carcinoma; IGFBP5, insulin growth factor binding protein 5; LEF1, lymphoid enhancer-binding factor 1; LFNG, lunatic fringe homolog; MMP2, matrix metallopeptidase 2; NT, non-tumor; OLT, orthotopic liver transplantation; PI15, peptidase inhibitor 15; PROM1, prominin 1 (CD133); SFRPs, secreted frizzled-related proteins; siRNA, small interfering RNA; T, Tumor ; TGFB, transforming growth factor beta.
Acknowledgments
We thank the following core facilities: « Structure Fédérative de Recherche en Biologie et Santé de Rennes » UMS CNRS 3480/US INSERM 018 (Health Genomics, Jean Moser; High Precision Histopathology, Alain Fautrel); SynNanoVect electroporation facility (Pascal Loyer). We are indebted to Denise Glaise and Stéphanie Renaud for technical help, to Patricia Jouas and Michèle Le Guennec for secretarial support, to Edouard Curran for CTNNB1 deletion analysis, to Gaelle Angenard and Catherine Ribault for tissue banking. We thank Bert Vogelstein and Yingzi Yang for β-catenin cDNA vectors and Heidemarie Huber and Wolfgang Mikulits for the 3P and 3SP HCC cell lines. Financial support: Inserm, Université de Rennes 1, Région Bretagne, Agence Nationale de la Recherche, Institut National du Cancer, Fondation Recherche Médicale, Ligue Nationale Contre le Cancer (Comité des Côtes d’Armor), Feder, Contrat de Plan Etat Région 2007-2013, projet Cancéropôle. Conflicts of Interest: The authors declare no conflicts of interest.
Conflicts of interest
The authors declare no conflicts of interest.
Author contributions
Study design, OM, SM, RD, BC, CP; EMT reprograming, SM, MS, EL; Hepatocyte-like differentiation and retrodifferentiation, AC, HDPS, MS, SM; Real-time PCR, SM, MS, HDPS; Immunocytochemistry, MS, EL, RLG, OM; DNA microarray hybridization, slide scanning, normalization, clustering and expression ratios, CC; Functional genomics; RD, SM, OM; Statistical analyses, meta-analyses from mRNA expression datasets, RD, OM; Tissue microarray design, construction and immunohistochemistry, OM, PB, MD; Tissue microarray scoring, OM, RD; Frozen tissue bank management, nucleic acid extraction, quality controls, BT, MD, OM, SM; CTNNB1 exon 3 sequencing, CP, FC; Clinical, biological and follow-up data collection, LS, DB; Anatomic Pathology annotations, BT; Manuscript preparation, OM, SM, RD; Manuscript editing, all authors.
REFERENCES
1. Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, Watanabe G, Gabriel S, Friedman SL, Kumada H, Llovet JM and Golub TR. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer research. 2009; 69:7385-7392.
2. Lachenmayer A, Alsinet C, Savic R, Cabellos L, Toffanin S, Hoshida Y, Villanueva A, Minguez B, Newell P, Tsai HW, Barretina J, Thung S, Ward SC, Bruix J, Mazzaferro V, Schwartz M, et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clinical cancer research. 2012; 18:4997-5007.
3. Clevers H and Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012; 149:1192-1205.
4. Derynck R, Muthusamy BP and Saeteurn KY. Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Current opinion in cell biology. 2014; 31:56-66.
5. Steinway SN, Zanudo JG, Ding W, Rountree CB, Feith DJ, Loughran TP, Jr. and Albert R. Network modeling of TGFbeta signaling in hepatocellular carcinoma epithelial-to-mesenchymal transition reveals joint sonic hedgehog and Wnt pathway activation. Cancer research. 2014; 74:5963-5977.
6. Pez F, Lopez A, Kim M, Wands JR, Caron de Fromentel C and Merle P. Wnt signaling and hepatocarcinogenesis: molecular targets for the development of innovative anticancer drugs. Journal of hepatology. 2013; 59:1107-1117.
7. Surana R, Sikka S, Cai W, Shin EM, Warrier SR, Tan HJ, Arfuso F, Fox SA, Dharmarajan AM and Kumar AP. Secreted frizzled related proteins: Implications in cancers. Biochimica et biophysica acta. 2014; 1845(1):53-65.
8. Quelard D, Lavergne E, Hendaoui I, Elamaa H, Tiirola U, Heljasvaara R, Pihlajaniemi T, Clement B and Musso O. A cryptic frizzled module in cell surface collagen 18 inhibits Wnt/beta-catenin signaling. PLoS ONE. 2008; 3:e1878.
9. Hendaoui I, Lavergne E, Lee HS, Hong SH, Kim HZ, Parent C, Heuze-Vourc’h N, Clement B and Musso O. Inhibition of Wnt/beta-catenin signaling by a soluble collagen-derived frizzled domain interacting with Wnt3a and the receptors frizzled 1 and 8. PloS one. 2012; 7:e30601.
10. Lavergne E, Hendaoui I, Coulouarn C, Ribault C, Leseur J, Eliat PA, Mebarki S, Corlu A, Clement B and Musso O. Blocking Wnt signaling by SFRP-like molecules inhibits in vivo cell proliferation and tumor growth in cells carrying active beta-catenin. Oncogene. 2011; 30:423-433.
11. Hu J, Dong A, Fernandez-Ruiz V, Shan J, Kawa M, Martinez-Anso E, Prieto J and Qian C. Blockade of Wnt signaling inhibits angiogenesis and tumor growth in hepatocellular carcinoma. Cancer research. 2009; 69:6951-6959.
12. Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, Gupta S, Moore J, Wrobel MJ, Lerner J, Reich M, Chan JA, Glickman JN, Ikeda K, Hashimoto M, Watanabe G, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008; 359:1995-2004.
13. Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, Ellis E, van Wenum M, Fuchs SA, de Ligt J, van de Wetering M, Sasaki N, Boers SJ, Kemperman H, de Jonge J, Ijzermans JN, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015; 160:299-312.
14. Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, 3rd and Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003; 423:448-452.
15. Kumar S, Zigman M, Patel TR, Trageser B, Gross JC, Rahm K, Boutros M, Gradl D, Steinbeisser H, Holstein T, Stetefeld J and Ozbek S. Molecular dissection of Wnt3a-Frizzled8 interaction reveals essential and modulatory determinants of Wnt signaling activity. BMC Biol. 2014; 12:44.
16. Cerec V, Glaise D, Garnier D, Morosan S, Turlin B, Drenou B, Gripon P, Kremsdorf D, Guguen-Guillouzo C and Corlu A. Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology. 2007; 45:957-967.
17. Dianat N, Dubois-Pot-Schneider H, Steichen C, Desterke C, Leclerc P, Raveux A, Combettes L, Weber A, Corlu A and Dubart-Kupperschmitt A. Generation of functional cholangiocyte-like cells from human pluripotent stem cells and HepaRG cells. Hepatology. 2014; 60:700-714.
18. Dubois-Pot-Schneider H, Fekir K, Coulouarn C, Glaise D, Aninat C, Jarnouen K, Le Guevel R, Kubo T, Ishida S, Morel F and Corlu A. Inflammatory cytokines promote the retrodifferentiation of tumor-derived hepatocyte-like cells to progenitor cells. Hepatology. 2014; 60:2077-2090.
19. Watanabe H and Yamada Y. Mice lacking link protein develop dwarfism and craniofacial abnormalities. Nature genetics. 1999; 21:225-229.
20. Aspberg A. The different roles of aggrecan interaction domains. J Histochem Cytochem. 2012; 60:987-996.
21. Berriz GF, Beaver JE, Cenik C, Tasan M and Roth FP. Next generation software for functional trend analysis. Bioinformatics. 2009; 25:3043-3044.
22. Pehlivanova VN, Tsoneva IH and Tzoneva RD. Multiple effects of electroporation on the adhesive behaviour of breast cancer cells and fibroblasts. Cancer Cell Int. 2012; 12:9.
23. van Zijl F, Mall S, Machat G, Pirker C, Zeillinger R, Weinhaeusel A, Bilban M, Berger W and Mikulits W. A human model of epithelial to mesenchymal transition to monitor drug efficacy in hepatocellular carcinoma progression. Molecular cancer therapeutics. 2011; 10:850-860.
24. Mabbott NA, Baillie JK, Brown H, Freeman TC and Hume DA. An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genomics. 2013; 14:632.
25. Naba A, Clauser KR, Hoersch S, Liu H, Carr SA and Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012; 11:M111 014647.
26. Evseenko D, Zhu Y, Schenke-Layland K, Kuo J, Latour B, Ge S, Scholes J, Dravid G, Li X, MacLellan WR and Crooks GM. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:13742-13747.
27. Liu Y, Zhou R, Yuan X, Han N, Zhou S, Xu H, Guo M, Yu S, Zhang C, Yin T and Wu K. DACH1 is a novel predictive and prognostic biomarker in hepatocellular carcinoma as a negative regulator of Wnt/beta-catenin signaling. Oncotarget. 2015; 6:8621-8634. doi: 10.18632/oncotarget.3281.
28. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, Sato T, Hamer K, Sasaki N, Finegold MJ, Haft A, Vries RG, Grompe M and Clevers H. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013; 494:247-250.
29. Wang R, Sun Q, Wang P, Liu M, Xiong S, Luo J, Huang H, Du Q, Geller DA and Cheng B. Notch and Wnt/beta-catenin signaling pathway play important roles in activating liver cancer stem cells. Oncotarget. 2016; 7:5754-5768. doi:10.18632/oncotarget.6805.
30. Tanese K, Fukuma M, Yamada T, Mori T, Yoshikawa T, Watanabe W, Ishiko A, Amagai M, Nishikawa T and Sakamoto M. G-protein-coupled receptor GPR49 is up-regulated in basal cell carcinoma and promotes cell proliferation and tumor formation. Am J Pathol. 2008; 173:835-843.
31. Nguyen LH, Robinton DA, Seligson MT, Wu L, Li L, Rakheja D, Comerford SA, Ramezani S, Sun X, Parikh MS, Yang EH, Powers JT, Shinoda G, Shah SP, Hammer RE, Daley GQ, et al. Lin28b is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer cell. 2014; 26:248-261.
32. Renger A, Zafiriou MP, Noack C, Pavlova E, Becker A, Sharkova K, Bergmann MW, El-Armouche A, Zimmermann WH and Zelarayan LC. The four and a half LIM-domain 2 controls early cardiac cell commitment and expansion via regulating beta-catenin-dependent transcription. Stem Cells. 2013; 31:928-940.
33. Malaguarnera R and Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (Lausanne). 2014; 5:10.
34. Yasuoka H, Yamaguchi Y and Feghali-Bostwick CA. The membrane-associated adaptor protein DOK5 is upregulated in systemic sclerosis and associated with IGFBP-5-induced fibrosis. PloS one. 2014; 9:e87754.
35. Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, Jia H, Ye Q, Qin LX, Wauthier E, Reid LM, Minato H, Honda M, Kaneko S, Tang ZY and Wang XW. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009; 136:1012-1024.
36. DeLaughter DM, Christodoulou DC, Robinson JY, Seidman CE, Baldwin HS, Seidman JG and Barnett JV. Spatial transcriptional profile of the chick and mouse endocardial cushions identify novel regulators of endocardial EMT in vitro. J Mol Cell Cardiol. 2013; 59:196-204.
37. Gibbs GM, Roelants K and O’Bryan MK. The CAP superfamily: cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 proteins--roles in reproduction, cancer, and immune defense. Endocr Rev. 2008; 29:865-897.
38. Bebek G, Patel V and Chance MR. PETALS: Proteomic Evaluation and Topological Analysis of a mutated Locus’ Signaling. BMC Bioinformatics. 2010; 11:596.
39. Ivanova AV, Goparaju CM, Ivanov SV, Nonaka D, Cruz C, Beck A, Lonardo F, Wali A and Pass HI. Protumorigenic role of HAPLN1 and its IgV domain in malignant pleural mesothelioma. Clinical cancer research. 2009; 15:2602-2611.
40. de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A and Perret C. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998; 95:8847-8851.
41. Supek F, Bosnjak M, Skunca N and Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PloS one. 2011; 6:e21800.
42. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:15545-15550.
43. Leek JT, Johnson WE, Parker HS, Jaffe AE and Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012; 28:882-883.
44. Langfelder P and Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9:559.
45. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498-2504.
46. McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M and Clark GM. Reporting recommendations for tumor marker prognostic studies (REMARK). Journal of the National Cancer Institute. 2005; 97:1180-1184.