
Introduction
KRAS/K-RAS is the most frequently mutated transforming oncogene in tumors of the pancreas, and colorectum [1], COSMIC:http://www.sanger.u.k. Indeed, KRAS mutations occur in 22% of all tumors analyzed (the highest among RAS isoforms), while HRAS and NRAS mutations are less frequent (3% and 8%, respectively) [2]. KRAS was originally identified in Kirsten sarcoma virus (Ki-SV) DNA [3, 4]. It was named kis, although the viral Kras (v-Kras) was first named Kirsten ras [5]; its product was identified as a 21 kDa protein (p21) with guanine nucleotide-binding activity [6, 7] in Ki-SV-transformed cells. The protein shared antigenicity with the viral p21 Hras oncogene (v-Hras), a product of the Harvey sarcoma virus (Ha-SV) [6].
Harvey and Kirsten sarcoma viruses were isolated as sarcoma-inducing retroviruses during the passaging of murine leukemia virus (MLV) in rats [8, 9]. The transforming genes were thought to be derived from the rat genome, while a temperature-sensitive (ts) Ki-SV was reported for transformation [10]. The viral RNA sequences were shown to share homology with rat leukemia virus (RaLV) RNA [11, 12]. Subsequently, a rat genome retrovirus-like RNA sequence (VL30) was found to be incorporated into RaLV particles [13, 14]. However, there was no evidence of either gene amplification or the presence of additional sequences in VL30 from rat tumor DNA [14]. Thus, viral ras oncogenes could not be identified until these viral genomic DNAs [3, 15] were cloned and sequenced in 1981-1982 [4, 16, 17]. During the same years, the mouse ortholog (bas) of v-Hras was identified in BALB-murine sarcoma virus (MSV) (BALB-MSV), which had been isolated from a BALB/c mouse hemangio-sarcoma [18, 19].
Meanwhile, studies of human transforming genes were initiated using an entirely different method: DNA transfection. In 1972, transforming activity was reported in cellular DNA fragments transferred into other cells. The DNA had been extracted from hamster cells transformed by the ts mutant of Rous sarcoma virus (RSV) [20]. This method was successfully applied in Weinberg’s laboratory [21], followed by Cooper’s, Wigler’s, and Barbacid’s laboratories, for mouse and human cancer cell DNA fragments that transformed “normal” mouse NIH3T3 [22-25]
In 1982-3, orthologs of viral ras oncogenes with point mutations were identified in transforming DNA fragments from human cancer cells both for HRAS [26-31] and KRAS [32-34]. This identification of RAS genes as oncogenes marked the beginning of molecular oncology in human cancer research. The Ras oncogene research was reviewed by Malumbres and Barbacid [35], and retroviral oncogenes were reviewed by Vogt [36]. Other oncogenes first identified in retroviruses and later as drivers in human cancer include myc, raf, erbB1 (EGFR: Epidermal growth factor receptor), AKT, and sis (PDGF: platelet derived growth factor, subunit B). Subsequently, these genes were found to be involved in the growth signaling cascade [35].
This review describes (i) the historical background and experimental basis of the “oncogene” concept, (ii) the details of the discovery of the transforming viral and human KRas oncogenes along with HRas/Bas, and (iii) how the word “oncogene” was integrated into human cancer research literature as one of the most important keywords, according to PubMed database records (https://www.ncbi.nlm.nih.gov/). Recent advancements in the field and studies of the clinical relevance of KRAS as an oncogenic driver in human cancer pathogenesis, and as a therapeutic target, are reviewed.
Timeline view: the “oncogene theory, ” the discovery of viral and human Ras oncogenes, and the clinical relevance of KRAS mutations
Figure 1 shows yearly tabulated numbers of publications extracted from the PubMed database from 1969 to the present, using the keywords “human carcinogenesis” plus one of the following: “oncogene, ” “carcinogen, ” “tumor virus, ” “ras, ” “src, ” or “kras/k-ras/ki-ras.” In 1969, the word “oncogene” was introduced by Huebner and Todaro [37] to explain the mechanism underlying carcinogenesis. The figure shows how the word was integrated into human cancer research.
The first retroviral oncogene (src) sequence was found to be transduced from the chicken genome in 1976. The sequence was also present in genomes of other vertebrates, including humans [38], indicating that the src retroviral oncogene had a cellular origin. However, Figure 1 shows that “oncogene” or “src” was rarely used as a keyword in human carcinogenesis research literature prior to 1981. Further, the number of publications with “oncogene” began to rise when alterations in cellular oncogenes related to retroviruses were found in human cancer. Transforming Ras oncogenes were discovered in the genomes of both Harvey and Kirsten sarcoma viruses and human cancer cells in 1982-1983. Publications using the keyword “ras” were the most common among those using “oncogene.” The discovery of (i) the enhanced expression of the human cellular MYC gene (c-MYC) following its translocation to the immunoglobulin heavy chain enhancer locus in Burkitt’s lymphoma [39], and (ii) the immortalization of primary rat embryo cells by promoter-linked human c-MYC [40] also contributed to this rise in “oncogene” studies, as did studies of other retroviral oncogenes [36] and oncogenes in DNA tumor viruses.
At the end of 1983, a chemical carcinogen was found to induce a ras-activating mutation in rat mammary tumors [41]. In 1985, the number of publications with “oncogene” as a keyword exceeded those with “carcinogen.” Thus, the word “oncogene” was integrated into human cancer research as one of the most important keywords. During the following two decades, studies of Ras were focused mainly on its biological and biochemical characteristics in cancer and normal cells [35]. Additionally, the growth signaling cascade in which Ras is involved was established. This knowledge was later translated into cancer therapies, such as blocking growth factor receptor function [42]. In 2007, the European Medicines Evaluation Agency (EMEA) approved the use of a drug (panitumumab) for anti-EGFR monoclonal antibody therapy for metastatic colorectal cancer (mCRC) patients with wild-type, but not mutant, KRAS; KRAS mutations were reported to reduce the efficacy of this therapy, as reviewed by Normanno et al. [43]. These findings stimulated research on KRAS/K-RAS as shown in Figure 1.
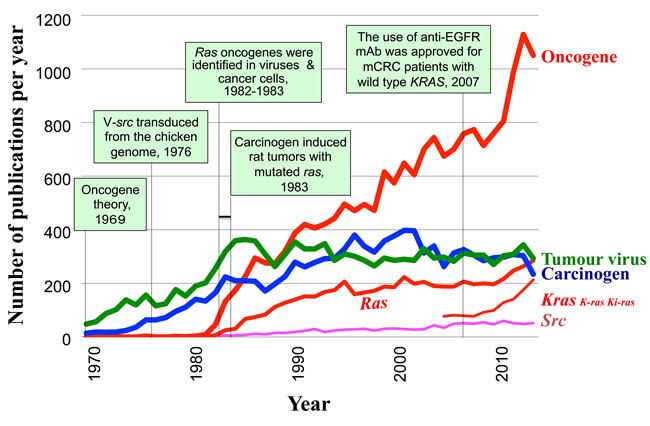
Figure 1: Time line showing the influence of “ras” discovery in human cancer research and the key events related to oncogene history (boxed). Yearly tabulated numbers of publications between 1969 and 2014 with the keywords “human + carcinogenesis” and one of the following: “oncogene, ” “tumor virus, ” “carcinogen, ” “ras, ” “Kras/K-ras/ki-ras, ” or “src.” The numbers for Kras/K-ras/ki-ras were counted separately for Kras, K-ras, or ki-ras, but overlapping publications were only counted once. Numbers were tabulated from 2005, when the Human Genome Nomenclature Committee updated the KRAS2 (c-ki-ras2) gene symbol to KRAS. Numbers were counted based on the PubMed database (NCBI, NIH) in July 2015.
“Oncogene theory”
The historical finding as the backbone of the theory
The “oncogene theory” was essentially based on the two main mechanisms of carcinogenesis: chemical and radiation carcinogenesis, and viral carcinogenesis. These mechanisms were discovered in the 1910s, when RSV and coal tar (which contained an “oncogene” and “carcinogens, ” respectively) were shown to induce cancers in experimental animals.
The first keystone discovery for viral carcinogenesis was made in 1911 by Peyton Rous (Rockefeller University). He succeeded in inducing a sarcoma by inoculating RSV in the form of a filtrate of sarcoma tissue extracts into a healthy chicken [44]. Later, Rubin established a method for quantifying transforming activities in vitro called a focus assay. The assay was based on the number of transformed cell colonies (focus forming units, FFU) that grew on a flat cell layer of chick fibroblasts [45]. The mouse mammary tumor virus and murine leukemia virus (MLV) retroviruses were also isolated in 1936 and in 1951, respectively. However, an appropriate method to assay the transforming activities of these viruses was not available in vitro. The first human cancer virus, Epstein-Barr virus (EBV), was discovered by Epstein, Henle, Achong, and Barr in 1965 [46]. EBV, one of the DNA tumor viruses, induces Burkitt’s lymphoma, a cancer of B cells. EBV is involved in lymphocyte immortalization caused by telomerase dysfunction, which is associated with MYC activation [47]. Human adenovirus type 12 (Ad 12) was found to induce tumors not in humans but in hamsters, which the virus could infect non-productively. These studies contributed to the elucidation of the general mechanisms underlying cellular transformation. For a review of the history of tumor virology, as reviewed by Javier and Butel [48].
The second keystone discovery for chemical carcinogenesis was made by Yamagiwa and Ichikawa of the University of Tokyo [49]. They succeeded in inducing carcinoma on the ears of rabbits by repeatedly rubbing coal tar, a mixture of many chemical substances, on them. This finding was followed by demonstrations that single compounds in this mixture, such as 3, 4-benzopyrene and α-aminoazotoluene, induced tumors in experimental animals [50, 51]. Chemical carcinogens exhibited genotoxic activities, leading to DNA lesions. Loeb and Harris reviewed advances in chemical carcinogenesis [52].
Extensive overlap was noticed between carcinogens and mutagens [53]. Although these studies did not clearly indicate the existence of a gene(s) that might be directly involved in carcinogenesis, studies from Weinberg’s laboratory did [21]. DNA extracted from mouse cancer cells, which had been transformed with chemical carcinogens, were found to induce “foci” upon transfection into “normal” NIH3T3 mouse cells. This observation suggested the presence of a single DNA fragment with transforming activities. This finding was consistent with the idea that a gene(s) was mutated by the carcinogen, thereby conferring transforming activities on a single fragment of the mouse cell DNA.
Experimental basis for the theory
As described above, Huebner and Todaro (National Cancer Institute, NIH) proposed the “oncogene theory” in 1969, in a paper titled “Oncogenes of RNA tumor viruses as determinants of cancer” (RNA tumor viruses were later named retroviruses) [37]. It had been proposed that carcinogens could solely explain the development of human cancers, as well as the increased incidence of cancer associated with aging [54].
“Oncogene theory” was based on cancer (mostly leukemia) incidence in the following different inbred mouse strains: AKR (high incidence), BALB/c (intermediate incidence), and C57BL (low incidence). Without exposure to other known cancer risk factors, the incidence was strain-specific and depended on age. Furthermore, incidence levels paralleled the expression levels of endogenous retrovirus and the viral antigen. In addition, type C viruses, similar to RSV particles, were found in human and other vertebrate cells. The presence of retroviral information (endogenous virus) in vertebrate genomes was hypothesized based on these findings. The abstract of this study states: “It is postulated that the viral information (the virogene), including that portion responsible for transforming a normal cell into a tumor cell (the oncogene), is transmitted from animal to progeny animal and from cell to progeny cell in a covert form. Carcinogens, irradiation, and the normal aging process all favor the partial or complete activation of these genes.”
By postulating the presence of oncogenes in the cellular genome, this theory offered an explanation of the mechanisms of carcinogenesis induced by carcinogens and by endogenous viral oncogene genetic sequences, along with aging. The theory was also supported by knowledge of the RSV genome, as investigations of both the “virogene” and the “oncogene” had been reported by that time. Although RSV is an exogenous virus, RSV genetic information could exist in host genome as a provirus [55] that remained after viral infection.
Two criticisms of the “oncogene theory” surfaced later: (i) the relationship between the production of endogenous viruses and cancer incidence was clearly shown only in mouse models, and (ii) the oncogene (src) and “virogenes” were found on different chromosomes in chickens [56]. Therefore, the term “oncogene” was rarely used to refer to a transforming gene, while “src” was used as a synonym for “transforming gene (oncogene).” However, in 1981, the Retrovirus Study Group of the International Committee on Taxonomy of Viruses named at least 13 distinct “src” genes as individual oncogenes, to avoid confusion [5].
Road to viral Kras/K-ras oncogene identification
Transforming virus-specific sequences of rat, mouse and avian sarcoma viruses studied in the 1970s
One year after the “oncogene theory” was proposed, reverse transcriptase was discovered in retroviral particles [57, 58]. This discovery not only supported the provirus hypothesis, but also made it possible to study the origins of transforming (oncogene) sequences in sarcoma viruses to test the validity of the “oncogene theory”. The replication mechanism of leukemia viruses, which had not yet been clarified, was also studied. Thus, cDNA prepared from viral genomic RNA was used to identify viral oncogene sequences in sarcoma viruses, as well as sequences needed for viral replication (gag, pol, and env).
Sarcoma-virus specific sequences were first prepared from viral particles produced by mouse 58-2T cells, developed by Masakazu Hatanaka, which produced a large excess of Ki-SV over helper MLV [59, 60]. As described earlier, Ki-SV production might have resulted when MLV transduced a sarcomagenic rat sequence (see Figure 2A). Ki-SV specific 58-2TS cDNA was prepared from 58-2T viral cDNA by removing the hybridizable MLV sequence with hydroxyapatite [59]. 58-2TS cDNA detected RNA species that were the same size as 30S RNA (this RNA species was later named VL30) in normal rat cells and RaLV-producing cells [13], consistent with the idea that the Ki-SV was formed by transducing the VL30 sequence. However, most of the 58-2TS cDNA sequences were later found to be those of rat VL30, while less than one-fifth were estimated to be the v-Kras oncogene sequence [17] (Figure 2A). It was thus difficult to identify the cellular Kras sequence in normal rat cells at that time. However, this strategy of preparing sarcoma-virus specific cDNA was also used to identify the RSV and Mo-SV oncogene sequences as described below.
Two years later, Stehelin, Varmus, Bishop, and Vogt [38] reported the isolation of RSV-specific cDNA (cDNAsrc). As shown in Figure 2B, the RSV genome contained the transforming sarcoma virus-specific sequence (src) in addition to gag, pol, and env, which make RSV replication-competent. cDNA fragments complementary to gag, pol, and env sequences were removed from RSV cDNA preparation. The resulting cDNAsrc formed a duplex with chicken genomic DNA at the highest melting temperature (Tm). The duplex with other vertebrate DNAs, including human DNA, had a lower Tm relative to evolutionary distance to the chicken, indicating that the src sequence originated in the chicken genome and was conserved in vertebrates, including humans. However, it was difficult to find biochemical differences in src genes between normal and cancer cells at that time. Moreover, the src sequence in human cancer cells was not known to have transforming activities.
Although transforming human src stop-codon mutations were reported in colon cancer more than 10 years later, additional analyses failed to confirm these and other activating transforming mutations, as reviewed by Russello and Shore [61]. As mentioned earlier, the 1976 finding thus did not lead to an immediate increase in publications with “src” or “oncogene” as a keyword in human carcinogenesis research (see Figure 1). However, accumulating evidence showed that cellular SRC kinase expression levels and/or activity were often enhanced in human cancer. It has been reported that src family kinases phosphorylated tyrosine residues of the Bcr-Abl protein, which conferred oncogenic activities [62].
It should be mentioned that the src protein was the first tyrosine kinase to be discovered, as reviewed by Hunter [63]. This led to the discovery of the src family of receptor tyrosine kinases (RTK), as reviewed by Parsons and Parsons in 2004 [64]. The SH (src homology) domain was later found to be crucial for binding to other cellular proteins involved in signaling, as reviewed by Pawson and Schlessinger [65].
Also in 1976, Frankel, Heubauer, and Fischinger reported the isolation of Mo-SV-specific cDNA (then denoted cDNAsarc) [66] (see Figure 2A). At that time, sarc was used to denote the cellular counterpart of viral src (oncogene). The Mo-SV-specific sequence, later named mos, was found to have originated from the mouse genome and to be present in the human genome as well [67]. However, the incidence of mos mutations was low in human cancer cells, while enhanced expression was observed [68]. It was reported that mos kinase activated ERK in the Ras-Raf pathway [69]. In addition, the roles of c-mos in meiotic maturation were shown, as reviewed by Vande Woude [70].
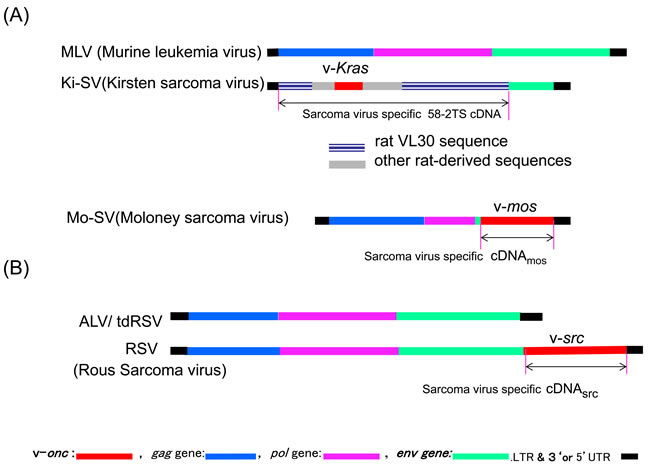
Figure 2: Comparisons of Ki-SV, Mo-SV, RSV, and the corresponding leukemia viral genomes. A. Genomic structures of Murine leukemia virus (top structure), Kirsten sarcoma virus [17, 72], and Moloney sarcoma virus [141]. B. ALV/tdRSV (avian leukemia virus/transformation defective RSV, the upper structure) and RSV (bottom structure). Leukemia viruses/tdRSV mutant genomes are shown for comparison. Each sarcoma virus figure shows regions covered by sarcoma virus-specific cDNA by an arrow.
Molecular cloning of genomic DNA of sarcoma viruses carrying v-Kras or v-Hras
Molecular cloning of the Ki-SV DNA was conducted in Tsuchida’s laboratory at the Wistar Institute in Philadelphia when NIH guidelines for recombinant DNA experiments on retrovirus genomes were relaxed from the P4 to the P2 level (Federal Register Dec 22, 1978 part VI: DHEW NIH, recombinant DNA research, revised guideline). Unintegrated circular genomic DNA of Ki-SV was cloned into a plasmid vector (clone 4) [3, 71]. The linear form of the Ki-SV DNA with restriction sites, aligned with the 5’ to 3’ orientation of viral RNA [60], and both clone 4 and clone 4(E) inserted, as shown in Figure 3A. As a result of the transforming activities of restriction-enzyme-cleaved clone 4(E), at least one XbaI site and one PvuII site were involved in transformation [3]. This result is not consistent with the findings of Chien et al. [72], who reported that v-Kras had been estimated to be in the 5’ terminal 0.7 Kb region of viral RNA, since the corresponding region of the viral DNA contained neither a XbaI nor a PvuII site [3] (Figure 3A).
Molecular cloning of the Ha-SV DNA carrying v-Hras was conducted in the P4 Mobile Containment Laboratory at the NIH in Bethesda before the NIH guidelines were relaxed. The results were reported by Scolnick and his colleagues [15]. Unintegrated circular genomic Ha-SV DNA was inserted into a lambda phage vector (clone H-1) (Figure 3B). In 1980, the transforming gene of the Ha-SV genome was located in a region between 0.4 kb and 1.5 kb from the 5’ end of the viral RNA genome [73].
A molecular clone of BALB-MSV DNA was also isolated (clone p9) [74]. BALB-MSV DNA had the transforming gene bas, the mouse ortholog of v-Hras [19], as described above.
Comparisons of Ki-SV and Ha-SV and their sub-clones to detect human KRAS or HRAS sequences
To localize the viral ras oncogenic sequences, the cloned Ki-SV and Ha-SV DNA segments (pKBE-2 and pHB-11, respectively), shown as thick blue lines in Figure 3A and 3B, respectively, were compared by heteroduplex analysis, since the ras proteins and genomes of both viruses shared common antigenicity and VL30 sequences, respectively [6, 72]. However, there was a discrepancy between the finding of Chien et al. [72], and that of Tsuchida and Uesugi [3], as described earlier. The analysis showed homology of a short 0.35-kb region, 1.3-1.65 Kb from the 5’ end of Ki-SV RNA, but not the 5’ terminal 0.7 Kb region. The upper XbaI recognition sequence of Ki-SV DNA was placed adjacent to 3’ side of this common region [17] (see Figure 3A). Further, this common region was later found to be highly homologous to the G domain (165 N-terminal amino acids) of v-Kras and v-Hras, as described below. Common tryptic peptides of both Kirsten and Harvey p21 proteins were suggested to be encoded by this region [17]. Consequently, two DNA fragments that encompassed the common region were subcloned from K-SV clone 4: the 1.0 kb HiHi-3 and the 0.38 kb HiHi-380 [17].
Furthermore, HiHi-380 of v-Kras and BS-9 [75] of v-Hras (both of these clones lack sequences corresponding to genomic exon 4 sequences) were able to detect respective EcoRI fragments of different sizes in vertebrate genomes; thus, it was possible to distinguish human DNA fragments from mouse DNA fragments in Southern hybridization [17]. HiHi-3 and KBE-2 clones for v-Kras and BS-9/pHB-1 for v-Hras/v-bas were used to identify human KRAS or HRAS exon-containing fragments of NIH3T3 cells transfected with human cancer DNAs (Figure 4). pHB-1 was reported to have sequences to detect all 4 HRAS exons of human cancer cells [74, 76].
Identification of the oncogenes (v-Kras/kis and v-Hras/has) in the Kirsten and Harvey sarcoma viral genomes
Scolnick and his colleagues identified the 1.0 kb nucleotide segment of the Ha-SV DNA encompassing the 0.35 Kb region that duplexed with the Ki-SV DNA. The v-Hras/has ORF in this segment was found to encode a 30 kDa protein starting 156 nucleotides upstream of the ATG codon of the internal p21 ORF [16] (Figure 3B). Later, the two upstream initiation codons were shown to be unnecessary for transforming activities [77]. The viral Kras/kis gene was localized by analysis of the transforming activities of deletion mutants of clone 4(E). Based on this, the v-Kras ORF was localized to the region shown in Figure 3A [4]. This ORF encoded 189 amino acids. Both v-Hras and v-Kras sequences were presented in the same issue of Science. Portions of the p21 ORF sequences of both v-Kras and v-Hras were verified in analyses of the tryptic peptides [17, 78].
v-Kras and v-Hras amino acid sequences of p21 ORFs revealed 90% homology in the N-terminal G domain, while the carboxyl terminal 166 to 189 region (highly variable region: HVR) [79] only shared 27% homology (Figure 3C). Therefore, the 0.35 kb homologous region detected by heteroduplex analysis between the two viral DNAs was confirmed to be within the G-domain sequences. When HVR amino acid sequences of the human exon 4 (E4) of HRAS [80] and exon 4A (E4A) of KRAS [33] were compared to the respective viral proteins, they were 100% and 96% similar, respectively, (Figure 3C), confirming that KRas and HRas were highly phylogenetically conserved independent genes. They were located on different human chromosomes [81, 82].
In 1983, Wigler and his colleagues isolated a new transforming NRAS oncogene in a neuroblastoma cell line, and it shared extensive homology with both v-Kras/KRAS and v-Hras/bas/HRAS [83]. They also identified a common intron-exon structure with 4 exons, except for KRAS, which had two 4th exons, 4A and 4B and highly homologous sequences in the G domain. However, the C-terminal HVR sequences were unique for each RAS protein (Figure 3C). The highly homologous G-domain encoded both GTPase [84] and the sequence necessary for binding common downstream effector proteins, as reviewed by McCormick and Wittinghofer [85].
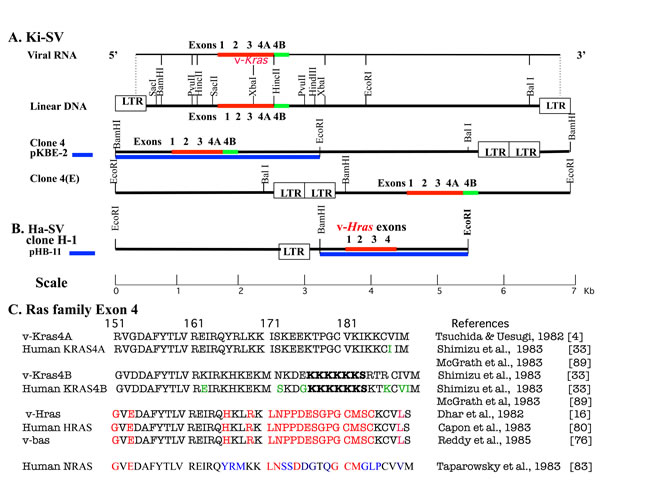
Figure 3: Physical maps and comparison of viral and cellular amino acid sequences of Ras isoforms. A. Physical map of Ki-SV and its restriction sites [3, 4, 17, 33, 60]. The positions corresponding to KRAS exons 1, 2, 3, and 4A, and 4B are shown in red and green, respectively, for (1) viral RNA, (2) the linear form DNA with LTRs at both ends, aligned with viral RNA, (3) clone 4 DNA (circular DNA was linearized at BamHI and inserted into plasmid vector) with subclone KBE-2 underneath as the blue thick line, and (4) clone 4(E) DNA (circular DNA was linearized at EcoRI and inserted into a vector). Restriction sites are shown for viral DNA. B. Physical map of Ha-SV DNA Clone H-1 [15, 16, 17, 75, 80]. Circular DNA with LTR was linearized at EcoRI, and the positions corresponding to HRAS exons 1, 2, 3, and 4 (in red) and restriction sites are shown. The sub-clone pHB-11 for heteroduplex analysis is shown with the blue thick line, underneath clone H-1. The scale is in Kb for both Ki-SV DNA and Ha-SV DNA. C. Comparisons of viral and cellular exon 4 amino acid sequences of Ras isoforms and KRAS exon 4B. Amino acid sequences of v-KRAS, v-HRAS and v-BAS, and those corresponding human sequences of exon 4 and NRAS are presented. Viral KRAS exon 4B is the sequence of the corresponding rat sequence inferred from the corresponding viral sequence [33]. Amino acid residues that are different from that of viral KRAS are shown in red or blue, and differences between the human cellular and the viral sequences are shown in green. In viral and cellular KRAS 4B, polylysine residues and serine 181 phosphorylated with PKC are shown with bold letters.
Road to the first human RAS oncogenes
HRAS/BAS oncogene
The following timeline describes the discovery of the ortholog of a retroviral Hras/bas oncogene as the first transforming oncogene in human cancer cells.
In 1979, two important discoveries eventually led to the identification of the first human oncogene: (a) the identification of transforming activities in a DNA fragment of carcinogen-transformed mouse cell [21], and (b) molecular cloning of Ha-SV genomic DNA [15]. In 1981-1982, transforming DNA fragments from human cancer cell lines were reported [22-25]. They were molecularly cloned using a human-specific Alu sequence as a probe. In 1980 and 1981, viral probes (BS-9 and pHB-1) were constructed from Ha-SV and BALB-MSV DNA, respectively [75, 74].
In 1982, the viral Hras/bas probes identified a ~7 kb BamHI fragment in transforming human DNA [26-28]. The V-Hras ORF sequence for p21 was reported [16].
A difference was found in a single base of the 12th codon in the 1st exon that conferred transforming activities on the gene. This difference resulted in the substitution of glycine for valine. This was reported by the Weinberg [29], Barbacid [30], and Wigler [31] groups.
It should be noted that the first report of a HRAS/BAS codon 12 mutation from Weinberg’s group was carried out as collaboration with Scolnick’s group [29]. The v-Hras BS-9 probe was used in the laboratories of Weinberg, Cooper, Wigler, and Levinson, while viral pHB-1 was used to identify the transforming BAS/HRAS fragment in the laboratories of Barbacid and Aaronson (Figure 4).
KRAS/K-RAS oncogene
Structure and activation mechanism of KRAS oncogene were elucidated in the following timeline.
In 1981 the structure and functions of Ki-SV genomic DNA clones were reported [3], though cloning of Ki-SV DNA corresponding to clone 4 (Figure 3A) was presented at the Cold Spring Harbor meeting in May, 1980 [71].
In 1981, the approximate position of v-Kras was estimated based on a comparison with Ha-SV genomic DNA. Based on this research, a subclone (HiHi-3) was constructed as a v-Kras probe [17] used to detect human KRAS DNA in NIH3T3 transfectants of human cancer cell DNAs (see Figure 4).
In 1982, Cooper’s group [26] detected a transforming human DNA fragment homologous to v-Kras in a lung cancer cell line, LX-1. Similarly, Barbacid, Aaronson, and colleagues detected the Kras transforming fragment in additional human lung, colon, gall bladder, pancreas, and rhabdomyosarcoma cell lines [86]. The nucleotide sequence of the viral Kras/kis ORF was reported [4].
In 1983, the Wigler [32] and Weinberg and Lowy [87] groups found a transforming human KRAS DNA fragment in Calu-1 and SW480 cancer cells using the v-Kras probe and/or the cellular KRAS probe (a c-ki-ras2 [88] fragment originally isolated with a v-Kras probe), respectively. Wigler’s group cloned a transforming ALU-containing DNA fragment, in which the v-Kras KBE-2 probe mapped the exon-containing EcoRI fragments. An activating point mutation was found in KRAS codon 12 in Calu-1 cells [33]. Based on the findings that Calu-1 and SW480 contained a transforming KRAS DNA fragment, Levinson’s group isolated KRAS cDNA clones from both cell lines with a v-Kras probe and identified codon 12 mutations with amino acid substitutions by comparing the KRAS cDNA sequences in normal and cancer cells [34].
Additionally, Levinson’s laboratory constructed a restriction map of the KRAS gene in normal human cells [89]. Wigler’s and Levinson’s groups found that the human Kras gene contained exons 1+2+3+4A+4B. The two 4th exons, A and B, resulted in the synthesis of two mRNA species by alternative splicing [33, 89]. Viral pKBE-2 also contained sequences corresponding to exons 1+2+3+4A+4B [33], but the ORF corresponding to exons 1+2+3+4A was possibly translated [4].
Activating mutations in KRAS, the transforming fragment of which was 5 times larger, were identified 10 months after the HRAS oncogene. However, once the v-Kras ORF sequence was published [4], researchers could easily elucidate its structure and point mutations in cancer cells. Viral Kras and the corresponding human protein KRAS4A also had 189 amino acid residues with differences at only 7 positions, one of which was a codon 12 mutation in both genes [4, 33, 89, 90].
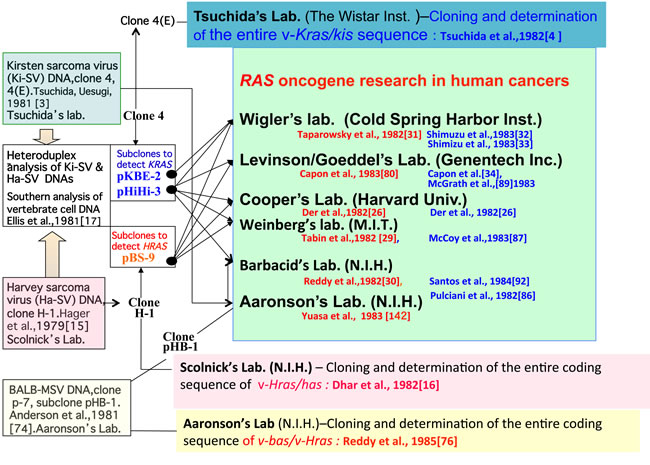
Figure 4: Identification of the viral ras oncogenes and distribution of sub-clones. Cloning of viral genomes and identification of their oncogenes were performed in the laboratories of Tsuchida (v-Kras), Scolnick, (v-Hras), and Aaronson (v-Bas/v-Hras). Subclones containing viral oncogenes (pHiHi-3, and/or pKBE-2 of v-Kras, pBS-9 of v-Hras) were used in the laboratories of Wigler, Cooper, Weinberg, and Levinson/Goeddel. The laboratories of Aaronson and Barbacid used their BALB-MSV clone pHB-1 for HRAS/BAS detection and clone 4(E) and HiHi-3 for KRAS detection. Publications of RAS genes from these eight laboratories are listed as references (indicated in red for HRAS/BAS and in blue for KRAS.
Ras genes in normal cells, cancer cells, and retroviruses
Analyses of human cancer cell RAS genes showed that they differed from those in normal cells in single nucleotides of codons, such as 12, 13, 59, 61, and 63; hot spot mutations in exons 1 and 2 [91] were associated with transforming activities. Consequently, transforming cellular genes were also called oncogenes based on the same criteria used to identify viral oncogenes, while RAS in normal cells was called a “proto-oncogene.” Thus, the word “oncogene” in cancer cells described a gene that could “transform a normal cell into a tumor cell, ” as first defined by Huebner and Todaro [37]. However, proto-oncogenes and oncogenes were later shown to play unique signaling roles in both normal and cancer cells, respectively, (see below) [35]. In addition, a KRAS mutation detected in a lung carcinoma was not present in normal tissues from the same patient, indicating an association between activation of oncogenes and the development of certain human cancers [92]. Additionally, the identification of human transforming genes that corresponded to viral Hras and Kras suggested that human RAS oncogenes could induce cancer just as Ki-SV and Ha-SV did in rodents.
In addition to point mutational activation, three other events are involved in the activation of a proto-oncogene causing enhanced expression of the proto-oncogene product, as reviewed by Vogt [37]: (1) gene amplifications, (2) promoter/enhancer insertion in the vicinity of the proto-oncogene, and (3) chromosomal translocation. Among these, RAS gene amplification has been reported in various tumors [93], but events (2) and (3) have not been reported. Chromosomal translocation also results in the formation of an activated oncogene product as a fusion protein, such as BCR-ABL [94] and RET-PTC [95]. Moreover, leukemia viruses induce transduction either through oncogenes/proto-oncogenes or by generating a gag-fusion gene, thereby changing into transforming viruses. This process was thought to be important for the generation of RSV, other sarcoma viruses, and acute leukemia viruses [37]. The oncogene (p29 gag-ras ) for Rasheed rat sarcoma virus (RaSV) is reportedly a fusion gene of RaLV gag with rat cellular Hras [ 96].
In Ki-SV and Ha-SV, two transduction steps might have occurred: (i) cellular ras was inserted into the endogenous rat VL30 retrovirus-like sequence, and (ii) VL30 RNA with ras was subsequently transduced into the MLV genome [13, 17, 74 97].
Merging of “carcinogenesis” research with oncogene research
As shown in Figure 1, Barbacid and colleagues found, in December 1983, an activated Hras oncogene in a mammary carcinoma that was induced in rats with N-nitroso-N-methylurea (NMU), a chemical carcinogen [41]. This finding revealed the following molecular mechanism underlying in vivo chemical carcinogenesis: (i) the carcinogen causes mutagenic damage of a proto-oncogene DNA [9], (ii) the proto-oncogene is fixed as an oncogene in the cell genome, and (iii) tumor formation originates from a cell bearing the activated oncogene. Thus, this finding led to the merging of carcinogen research with oncogene research, thereby consolidating human carcinogenesis research and oncogene research at the molecular level (see Figure 1). Consequently, the series of findings on ras oncogenes, together with other retroviral oncogenes, greatly influenced human cancer research, and these findings opened up the new research field of molecular oncology.
Clinical relevance of KRAS mutations
KRAS is the most important biomarker in anti-EGFR therapy for mCRC
In normal cells, upon growth factor (EGF) stimulation and the activation of its receptor (EGFR), the inactive RAS protein (GDP-bound RAS) changes transiently to the active form (GTP-bound RAS) (Figure 5). However, KRAS, which is frequently mutated in mCRC, sustains the activated RAS-GTP form. This causes constitutive activation of downstream effectors, mainly RAF in the RAF-MEK-ERK pathway, PIK3CA (p110α) in the PI3K-AKT-mTOR pathway, and RALGDS in the RALGDS pathway, as reviewed previously [35]. The activated RAS-GTP form takes on an “open” conformation in the Switch I and Switch II regions (amino acids 28-63), allowing the effector domain of residues 32 to 40 within Switch I to interact with the Ras binding domain (RBD) of RAF, while PIK3CA (p110α), and RALGDS bind at both the Switch I and Switch II regions [85]. The mechanisms involved in signaling to downstream effectors and membrane anchoring of Ras isoforms were reviewed by Castellano and Santos [99].
Inhibition of overexpressed EGFR has been used in cancer therapies [42]. For mCRC, it was reported that patients with amplified EGFR copies respond well to anti-EGFR treatment [100]. However, starting in 2006, anti-EGFR therapy with a monoclonal anti-body (mAb) or tyrosine kinase inhibitor (TKI) was reported to be less effective in mCRC patients with KRAS mutations at codon 12/13 [101, 102] because the mutated KRAS oncogene is unaffected by EGFR signaling (Figure 5). Additional studies have shown that patients with NRAS, BRAF, and PIK3CA oncogene mutations (shown by circles with thick lines) are also resistant to anti-EGFR therapy [103]. The proportions of patients with each of these activated oncogenes among anti-EGFR-resistant patients and patients with large intestine cancer (COSMIC) were similar - i.e., highest for KRAS, followed by PIK3CA, BRAF, and NRAS [103]. Thus, activated KRAS mutations were the first to be reported in anti-EGFR-resistant mCRC patients. Some patients had both PIK3CA and KRAS mutations [103]. This finding was consistent with the presence of another direct signaling pathway from EGFR to PI3K [104]. PTEN mutations also contribute to anti-EGFR treatment resistance [105], since, like PIK3CA mutations, they enhance AKT phosphorylation [106].
All activated oncogenes detected through anti-EGFR therapy was found to be a transformation-driver in vitro using cultured cells [35, 107-109]. Further, with the exception of NRAS, they were also the first to be identified in retroviruses [36]. These mutated oncogenes were thus acquired/selected during anti-EGFR therapy, and work individually as “oncogenic drivers for tumor initiation and progression and therefore conferred primary/acquired resistance to cells during anti-EGFR therapy. Figure 5 shows that these activated oncogenes transfer malignant signaling to their downstream effectors to promote tumor growth and act as tumor drivers independently of upstream signaling. By contrast, their wild-type forms are regulated by upstream proteins.
Growth factor receptors, including MET, HER2, and HER2/HER3 heterodimer, activate both the RAS pathway and a direct PI3K pathway. Amplification/activation of these receptors or enhanced expression of their ligands, HGF and TGF alpha, also conferred resistance to anti-EGFR therapy [110-113]. Acquired EGFR mutation, which abrogates the binding of the mAb cetuximab has been reported [114]. The mechanisms underlying resistance to anti-EGFR therapies have been reviewed for mCRC [115-117].
As KRAS mutations predict the efficacy of anti-EGFR therapy, KRAS mutation testing is mandatory, and anti-EGFR therapy is only prescribed for mCRC patients with wild-type KRAS (see Figure 1). Consequently, molecular epidemiological data of Kras mutations in mCRC patients has accumulated. KRASG12D and KRASG12C were the most common KRAS genotypes in mCRC and lung cancer patients, respectively [118]. Each mutant genotype has been correlated to clinical outcomes [119-121].
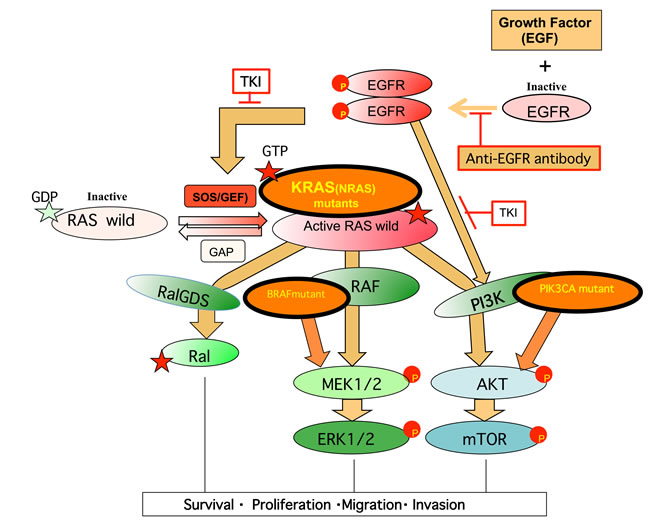
Figure 5: Growth signaling and mutated signal proteins that confer resistance to anti-EGFR-targeted therapy in mCRC. Growth factor stimulation in the normal cell generally proceeds as follows: (i) activation of the receptor by phosphorylation and dimerization, (ii) activation of RAS and PI3K, and then (iii) activation of the RAS effector pathways RAF, PI3K, and RALGDS through binding to RBD of each protein. In mCRC, EGFR copy number was often increased. EGFR-targeted therapy prevents steps (i) and (ii) by using anti-EGFR monoclonal antibodies (mAb) or a tyrosine kinase inhibitor (TKI). An oncogenic driver mutation of the EGFR effector (KRAS, NRAS, or PIK3CA) or the RAS effector (PIK3CA or BRAF) makes cancer patients refractory to EGFR-targeted therapy, while patients with wild-type KRAS/ NRAS/PIK3CA/BRAF are mostly sensitive to this therapy. Activated tumor driver proteins are shown as frames with thick lines. Refer to the text for details.
Activated KRAS as a therapeutic target
As KRAS was found to be the most frequently mutated oncogene in human adenocarcinomas, the development of drugs targeting activated KRAS/KRAS has been pursued. However, initial attempts to block Ras family proteins by lipid modification of HVR were unsuccessful, as described by Ledford [122]. Targeting KRAS /KRAS sequences in genomic DNA, mRNAs, and proteins, in addition to cancer cells with mutated KRAS, has been explored as described below.
1) Targeting oncogenic codon 12 sequences of chromosomal KRAS DNA.
This was accomplished by alkylating adenine N3 at the target sequence. The modification induced DNA strand cleavage and thereby suppressed the growth of KRAS-mutated human colon cancer cells [123]. This approach was suitable for inactivating either G12D or G12V mutated genes, which are frequent in mCRC, [118].
2) Silencing of KRAS mRNAs using siRNA [124, 125] or miRNAs (such as Let-7 [126] or miR143 [127]), which indirectly suppress levels of mutated KRAS proteins in cancer cells. A new administration method was developed to maintain siRNA stability and to introduce siRNA slowly into KRAS G12D-mutated pancreatic carcinoma tissue [125].
3) KRASG12C protein was modified by guanine nucleotide (GN) analogs so that the analogs formed a covalent bond to the cysteine residue at codon 12 in the GTP/GDP pocket [128]. The modified KRAS protein is unable to bind to downstream effector proteins, and oncogenic signaling is thereby attenuated, as reviewed by Wang et al. [129]. Most KRASG12C mutations are due to G to T nucleotide transversion in lung cancer [2].
4) Therapeutic immunization (GI-4000 vaccine) with whole cell lysates of recombinant yeast that expresses a mutated KRAS protein has been reported [130, 131].
5) Eradication of KRAS-activated cancer cells in vivo by T-cell response. Anti-CTLA-4, anti-PD-1, or anti-PD-L1 antibodies generated T-cell responses against cancer-specific neo-antigens. This method was effective in patients suffering from malignant melanoma with mutated NRAS or BRAF [132]. The possible use of these types of antibodies was reported for KRAS- mutated lung cancer patients, since higher PD-1 or PD-L1 levels were observed in these patients [133, 134].
Two splice variants of KRAS mRNAs differing mainly in HVR are produced from the KRAS oncogene by alternative splicing: KRAS4A and KRAS4B. Relative amounts of the mRNA species were approximately equal in most samples from 17 fresh human colorectal tumors, although the KRAS4B species is more abundantly expressed in cell lines [135].
Activated KRAS4B had been reported to stimulate cell migration more strongly than KRAS4A, while the opposite was true for focus formation and anchorage-independent growth. Cell motility facilitates angiogenesis, invasion, and ultimately metastasis. It had been suggested that both splice variants might act together to confer the fully malignant phenotype induced by activated KRAS [136]. KRas4A shares lipid modifications of HVR as well as a G-domain sequence with NRAS and Hras, while the sequence and lipid modification of KRas4B HVR are distinct from the above 3 RAS proteins (see Figure 3C). Therapy inhibiting both KRAS splice variants or their downstream effectors, such as the combination of MEK and PI3K inhibitors may prove an effective strategy for the treatment of KRAS-mutated cancers [ 137-139]. Therapeutic strategies against KRAS were recently reviewed by McCormick [140].
In conclusion, the discovery of Kirsten Ras oncogene sequences in both viral genomes and transforming DNA fragments in cancer cells lead to the discovery of crucial roles for KRAS in human carcinogenesis, as well as the high frequency of KRAS mutations in human cancer. Furthermore, recent results regarding anti-EGFR therapeutic efficacy in patients bearing wild-type or mutant KRAS suggested that it is the most important cancer driver in vivo. Improved knowledge of the complex mechanisms involved in KRAS activation and signaling is necessary in order to develop new therapeutic strategies directed against mutated KRAS.
ACKNOWLEDGMENTS
Sincere thanks to Masakazu Hatanaka for providing the opportunity to work on Kirsten ras, to Kei Fujinaga and Masa-aki Ikeda for critically reading the manuscript, and to Giancarlo Vecchio for his continuous interests in Kras work. Works for the cloning of the Ki-SV genomic DNA and the identification of viral Kras oncogene were supported by NIH grant CA-22701 to N.T. as the P.I.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
References
1. Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L, Lee JC, Nicoletti R, Hatton C et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007; 39:347-351.
2. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in Cancer. Cancer Res. 2012; 72:2457-2467.
3. Tsuchida N, Uesugi S. Structure and functions of the Kirsten murine sarcoma virus genome: Molecular cloning of biologically active Kirsten murine sarcoma virus. J Virol. 1981; 38:720-727.
4. Tsuchida N, Ryder T, Ohtsubo E. Nucleotide sequence of the oncogene encoding the p21 transforming protein of Kirsten murine sarcoma virus. Science. 1982; 217:937- 939.
5. Coffin JM, Varmus HE, Bishop JM, Essex M., Hardy WD.Jr., Martin GS, Rosenberg NE, Scolnick EM, Weinberg RA, Vogt PK. Proposal for naming host cell-derived inserts in retrovirus genomes. J Virol. 1981; 40:953-957.
6. Shih TY, Weeks MO, Young HA, Scolnick EM. Identification of a sarcoma virus-coded phosphoprotein in non-producer cells transformed by Kirsten murine sarcoma virus. Virology. 1979; 257:64-79.
7. Scolnick EM, Papageorge AG, Shih TY. Guanine nucleotide-binding activity as an assay for src protein of rat-derived murine sarcoma viruses. Proc Natl Acad Sci USA. 1979; 76:5355-5359.
8. Harvey JJ. An unidentified virus which causes the rapid production of tumours in mice. Nature. 1964; 204:1104-1105.
9. Kirsten WH, Mayer LA. Morphologic responses to a murine erythroblastosis virus. J Natl Cancer Inst. 1967; 39:311-335.
10. Scolnick EM, Stephenson JR, Aaronson SA. Isolation of temperature-sensitive mutants of murine sarcoma virus. J Virol. 1972; 10:653-657.
11. Scolnick EM, Rands E, Williams D, Parks WP. Studies on the nucleic acid sequences of Kirsten sarcoma virus: a model for formation of a mammalian RNA-containing sarcoma virus. J Virol. 1973 ;12:458-463.
12. Scolnick EM, Parks WP. Harvey sarcoma virus: a second murine type C sarcoma virus with rat genetic information. J Virol. 1974;13:1211-1219.
13. Tsuchida N, Gilden RV, Hatanaka M. Sarcoma-virus-related RNA sequences in normal rat cells. Proc Natl Acad Sci U S A. 1974; 71:4503-4507.
14. Andersen GR, Robbins KC. Rat sequences of the Kirsten and Harvey murine sarcoma virus genomes: nature, origin, and expression in rat tumor RNA. J Virol. 1976 ;17:335-351.
15. Hager GL, Chang EH, Chan HW, Garon CF, Israel MA, Martin MA, Scolnick EM, Lowy DR. Molecular cloning of the Harvey sarcoma virus closed circular DNA intermediates: initial structural and biological characterization. J Virology. 1979; 31: 795-809.
16. Dhar R, Ellis RW, Shih TY, Oroszlan S, Shapiro B, Maisel J, Lowy D, Scolnick E. Nucleotide sequence of the p21 Harvey sarcoma virus. Science. 1982; 217: 934-937.
17. Ellis RW, DeFeo D, Shih TY, Gonda MA, Young HA, Tsuchida N, Lowy DR, Scolnick EM. The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nature 1981; 292:506-510.
18. Aaronson SA, Barbacid M. Origin and biological properties of a new BALB/c mouse sarcoma virus. J. Virol. 1978 ;27:366-373.
19. Andersen PR, Devare SG, Tronick SR, Ellis RW. Aaronson SA and Scolnick EM. Generation of BALB-MuSV and Ha-MuSV by type C virus transduction of homologous transforming genes from different species. Cell 1981; 26:129-134.
20. Hill M, Hillova J. Recovery of the temperature-sensitive mutant of Rous sarcoma virus from chicken cells exposed to DNA extracted from hamster cells transformed by the mutant. Virology. 1972 ;49:309-313.
21. Shih C, Shilo B-Z, Goldfarb MP, Dannenberg A, Weinberg RA. Passage of phenotypes of chemically transformed cells via transfection of DNA and chromatin. Proc Natl Acad Sci USA. 1979; 76:5714-5718.
22. Krontiris T and Cooper G. Transforming activity of human tumor DNAs. Proc.Natl. Acad.Sci.USA. 1981; 78: 1181-1184.
23. Shih C, Padhy LC, Murray M, Weinberg RA. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature. 1981; 290 :261-264.
24. Perucho M, Goldfarb M, Shimizu K, Lama C, Fogh J, Wigler M. Human-tumor-derived cell lines contain common and different transforming genes. Cell. 1981;27:467-476.
25. Pulciani S, Santos E, Lauver AV, Long LK, Barbacid M. Transforming genes in human tumors. J Cell Biochem. 1982:20:51-61.
26. Der CJ, Krontiris TG, Cooper GM. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci USA. 1982; 79:3637-3540.
27. Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ Bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. 1982; 297:474-478.
28. Santos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M. T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes. Nature. 1982; 298:343-347.
29. Tabin CJ, Bradley SM, Bargmann CI, Weinberg RA, Papageorge AG, Scolnick EM, Dhar R, Lowy DR, Chang EH. Mechanism of activation of a human oncogene. Nature. 1982; 300:143-149.
30. Reddy EP, Reynolds RK, Santos E, Barbacid M. A point mutation is responsible for the activation of transforming properties by the T24 human bladder carcinoma oncogene. Nature. 1982; 300:149-152.
31. Taparowsky E, Squard Y, Fasano O, Goldfarb M, Wigler M. Activation of the T24 bladder carcinoma transforming gene is linked to a single amino acid change. Nature. 1982; 300:762-765
32. Shimizu K, Goldfarb M, Suard Y, Perucho M, Li Y, Kamata T, Feramisco J, Stavnezer E, Fogh J, Wigler MH. Three human transforming genes are related to the viral ras oncogenes. Proc Natl Acad Sci USA. 1983; 80:2112-2116.
33. Shimizu K, Birnbaum D, Ruley MA, Fasano O, Suard Y, Edlund L, Taparowsky E, Goldfarb M, Wigler M. Structure of the Ki-ras gene of the human lung carcinoma cell line Calu-1. Nature. 1983; 304:497-500.
34. Capon DJ, Seeburg PH, McGrath JP, Hayflick JS, Edman U, Levinson AD, Goeddel DV. Activation of Ki-ras2 gene in human colon and lung carcinomas by two different point mutations. Nature. 1983; 304: 507-513.
35. Malumbres M, Barbacid M. Ras oncogene: the first 30 years. Nature Rev Cancer. 2003; 3:459-465.
36. Vogt PK. Retroviral oncogenes: a historical primer. Nature rev Cancer. 2012; 12:639- 648.
37. Huebner RJ, Todaro GJ. Oncogenes of RNA tumor viruses as determinants of cancer. Proc Natl Acad Sci USA. 1969; 64:1087-1094.
38. Stehlin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976; 260:170-173.
39. Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P. Translocation of the c-myc gene of the imminogloblin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci USA. 1982; 79:7837-7841.
40. Land H, Parada LF. Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983; 304:596-602.
41. Sukumar S, Notario V, Maein-Zanca D, Barbacid M. Induction of mammary carcinoma by nitroso-methylurea involves malignant activation of H-ras-1 locus by single point mutation initiation. Nature. 1983; 306:358-361.
42. Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy.Oncogene. 2000;19:6550-6565.
43. Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nature Rev Clinical Oncology. 2009; 6:519-527.
44. Rous P. A sarcoma of fowl transmissible by an agent separable from the tumor cells. J Exp Med. 2011; 108:397-411.
45. Rubin H. The early history of tumor virology: Rous, RIF, and RAV. Proc Natl Acad. Sci USA. 2011; 108:14389-14398.
46. Epstein MA, Henle G, Achong BG, Barr YM. Morphological and biological studies on a virus in cultured lymphoblasts from Burkitt’s lymphoma. J Exp Med. 1965; 121:761-770.
47. Kamranvar SA, Gruhne BA, Szeles A, Masucci MG. Epstein-Barr virus promotes genomic instability in Burkitt’s lymphoma. Oncogene. 2007; 26:5115-5123.
48. Javier RT, Butel JS. The history of tumor virology. Cancer Res. 2008; 68:7693-7706.
49. Yamagiwa K, Ichikawa K. Uber die kunstliche erzeugung von papillom. V Jap Pathol Ger. 1915; 5:142-158.
50. Kennaway EL. Further experiments on cancer-producing substances. Biochem J. 1930; 24:497-504.
51. Sasaki T, Yoshida T. Experimentelle Erzeugung des Leberkarzinoma durch Fuetterung mit o-amidoazotoluol. Virchow Arch Path Anat. 1935; 295:175-200.
52. Loeb LA, Harris CC. Advances in chemical carcinogenesis: a historical review and prospective. Cancer Res. 2008; 68:6863-6872.
53. Ames BN, Lee FD, Durston WE. An improved bacterial test system for the detection and classification of mutagens and carcinogens. Proc Natl Acad Sci U S A. 1973; 70:782-786.
54. Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J of Cancer. 1954; 8:1-12.
55. Temin HM. Nature of the proviruss of Rous sarcoma virus. J Natl Cancer lnst Monograph. 1964; 17:557-570.
56. Padgett E, Stublefield PE. Varmus HE. Chicken macrochromosomes contain an endogenous provirus and microchromosomes contain sequences related to the transforming gene of ASV. Cell. 1977; 10:649-657.
57. Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature. 1970; 226:1209-1211.
58. Temin HM. Nature of the provirus of Rous sarcoma virus. Nat Cancer Inst Monograph. 1964; 17:557-570.
59. Tsuchida N, Shih MS, Gilden RV, Hatanaka M. Sarcoma and helper-specific RNA tumor virus subunits in transformed nonproducer mouse cells activated to produce virus by treatment with bromodeoxyuridine. J Virol. 1974; 14:1262-1267.
60. Tsuchida N, Kominami R, Hatanaka M, Uesugi S. Identification of unintegrated forms of Kirsten murine sarcoma DNA and restriction endonuclease cleavage map of linear DNA. J Virol. 1981; 38:797-803.
61. Russello SV, Shore SK. Src in human carcinogenesis. Frontiers in Bioscience. 2004; 9:139-144.
62. Meyn III MA, Wilson MB, Abdi FA, Fahey N, Schiavone AP, Wu J, Hochrein JM, Engen JR, Smithgall TE. Src family kinases phosphorylate the Bcr-Abl SH3-SH2 region and modulate Bcr-Abl transforming activity. J Biol Chem. 2006; 281: 30907-30916.
63. Hunter T. Discovering the first tyrosine kinase. Proc Natl Acad Sci USA. 2015; 112:7877-7882.
64. Parsons SA, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004; 23:7906-7909.
65. Pawson T, Schlessinger J. SH2 and SH3 domains. Curr Biol. 1993; 3:434-442.
66. Frankel AE, Neubauer RL, Fischinger PJ. Fractionation of DNA nucleotide transcripts from Moloney sarcoma virus and isolation of sarcoma virus-specific complementary DNA. J Virol. 1976; 18:481- 490.
67. Frankel AE, Fischinger PJ. Nucleotide sequences in mouse DNA and RNA specific for Molony sarcoma virus. Proc Natl Acad Sci USA. 1976; 73:3705-3709.
68. Cooper CS. The role of non-ras transforming genes in chemical carcinogenesis. Environmental Health Perspectives. 1991; 93:33-40.
69. Webb CP, Van Aelst L, Wigler MH, Vande Woude GF. Signaling pathways in Ras-mediated tumorigenicity and metastasis. Proc Natl Acad Sci USA. 1998; 95:8773-8778.
70. Vande Woude GF. On the loss of Mos. Nature 1994; 370:20-21.
71. Tsuchida N, Uesugi S. Molecular cloning of Ki-MSV genome size DNA. At the Cold Spring Harbor Meeting on RNA tumor viruses. May 1980. Abstract page 35.
72. Chien U-H, Lai M, Shih TY, Verma IM, Scolnick EM, Roy-Burman P, Davidson N. Heteroduplex analysis of the sequence relationships between the genomes of Kirsten and Harvey sarcoma viruses, their respective parental murine leukemia viruses, and the rat endogenous 30S RNA. J Virol. 1979; 31:752-760
73. Wei CM, Lowy DR, Scolnick ED. Mapping of transforming region of the Harvey murine sarcoma virus genome by using insertion-deletion mutants constructed in vitro. Proc Natl Acad Sci USA. 1980; 77:4674-4678.
74. Andersen PR, Tronick SR and Aaronson SA. Structural organization and biological acitivity of molecular clones of the integrated genome of a BALB/c mouse sarcoma virus. J Virol. 1981; 40:431-439.
75. Ellis RW, DeFeo D, Maryak JM, Young HA, Shih TY, Chang EH, Lowy DR, Scolnick EM. Dual evolutionary origin for the rat genetic sequences of Harvey murine sarcoma virus. J Virol. 1980; 36:408-420.
76. Reddy EP, Lipman DP, Tronick S, Aaronson SA. Nucleotide sequence analysis of the BALB/c murine sarcoma virus transforming gene. J Virol. 1985; 53:984-987.
77. Willumsen BM, Ellis RW, Scolnick EM, Lowy DR. Further genetic localization of the transforming sequences of the p21 v-ras gene of Harvey murine sarcoma virus. J Virol. 1984; 49:601-603.
78. Shih TY, Stokes PE, Smythers GW, Dharl R, Oroszlan S. Characterization of the phosphorylation sites and the surrounding amino acid sequences of the p21 transforming proteins coded for by the Harvey and Kirsten strains of murine sarcoma viruses. J Biol Chem. 1982; 257:11767-11773.
79. Laude AJ, Prior IA. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J Cell Science. 2008; 121:421-427.
80. Capon DJ, Chen EY, Levinson AD, Seeburg PH, Goeddel DV. Complete nucleotide sequences of the T24 human bladder carcinoma oncogene and its normal homologue. Nature 1983; 302:33-37.
81. Jhanwar SC, Neel BG, Hayward WS, Chaganti RS. Localization of c-ras oncogene family on human germ-line chromosomes. Proc Natl Acad Sci U S A. 1983; 80: 4794- 4797.
82. Popescu NC, Amsbaugh SC, DiPaolo JA, Tronick SR, Aaronson SA, Swan DC. Chromosomal localization of three human ras genes by in situ molecular hybridization. Somat Cell Mol Genet. 1985; l11:149-155.
83. Taparowsky E, Shimizu K, Goldfarb M, Wigler M. Structure and activation of the human N-ras gene. Cell. 1983; 34:581-586.
84. Sweet RW, Yokoyama S, Kamata T, Feramisco JR, Rosenberg M, Gross M. The product of ras is a GTPase and the T24 oncogenic mutant is deficient in this activity. Nature. 1984 ;311:273-275.
85. McCormick F and Wittinghofer A. Interactions between Ras proteins and their effectors. Curr Opin Biotechnol. 1996;7:449-456.
86. Pulciani S, Santos, E, Lauver AV, Long LK, Aaronson SA, Barbacid M. Oncogenes in solid human tumours. Nature1982; 300:539-542.
87. McCoy MS, Toole JJ, Cunningham JM, Chang EH, Lowy DR, Weinberg RA. Characterization of a human colon/lung carcinoma oncogene. Nature. 1983; 302:79- 81.
88. Chang EH, Gonda MA, Ellis RW, Scolnick EM, Lowy DR. Human genome contains four genes homologous to transforming Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci USA. 1982; 79:4848-4852.
89. McGrath JP, Capon DJ, Smith DH, Ellson S, Chen EY, Seeburg PH, Goeddel DV, Levinson AD. Structure and organization of the human Ki-ras proto-oncogene and a related processed pseudogene. Nature. 1983; 304:501-506.
90. Iritani A, Katayama N, Tahira T, Hayashi K, Tsuchida N. Nucleotide sequence of exon I of the rat c-K-ras gene. Bull Tokyo Med Dent Univ. 1986; 33:35-40.
91. Bos JL. ras oncogenes in human cancer:a review Cancer Res 1989; 49:4682-4689.
92. Santos E, Martin-Zanca D, Reddy EP, Pierotti MA, Della Porta G, Barbacid M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science. 1984; 223:661-664.
93. Bos JL The ras gene family and human carcinogenesis. Mutat Res. 1988 ;195:255-271.
94. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007; 7:233-245.
95. Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, Vecchio G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas.Cell. 1990;60:557-563.
96. Gonda MA, Young HA, Elser JE, Rasheed S, Talmadge CB, Nagashima K, Li CC, Gilden RV. Molecular cloning, genomic analysis, and biological properties of rat leukemia virus and the onc sequences of Rasheed rat sarcoma virus. J Virol. 1982 ;44:520-529.
97. Scolnick EM, Goldberg RJ, Williams D. Characterization of rat genetic sequences of Kirsten sarcoma virus: distinct class of endogenous rat type C viral sequence. J Virol. 1976;18:559-566.
98. Zarbl H, Sukumar S, Arthur AV, Martin-Zanca D, Barbacid M. Direct mutagenesis of Ha-ras-1 oncogene by N-nitroso-N-methylurea during initiation of mammary carcinogenesis in rats. Nature. 1985; 315:382-385.
99. Castellano E, Santos E. Functional Specificity of Ras Isoforms: So similar but so different. Genes & Cancer. 2011; 2:216-231. doi: 10.1177/1947601911408081.
100. Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, Gambacorta M, Siena S, Bardelli A. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005; 6:279-286.
101. Lievre A, Bachet J-B, Le Corre D, Boige V. Landi B, Emile J-F, Cote J-F, Tomasic G, Penna C, Ducreux M, Rougier, P, Penault-Llorca, F, Laurent-Puig P. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006; 66:3992-3995.
102. Di Fiore, F. Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech JJ Sesboüé R, Tuech JJ, Queuniet AM, et al. . Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by cetuximab plus chemotherapy. Br J Cancer. 2007; 96:1166-1169.
103. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010; 11:753-762.
104. Onishi-Haraikawa Y, Funaki M, Gotoh N, Shibuya M, Inukai K, Katagiri H, Fukushima Y, Anai M, Ogihara T, Sakoda H, Ono H, Kikuchi, M, Oka Y, Asano T. Unique phosphorylation mechanism of Gab1 using PI3-kinase as an adaptor protein. Biochem Biophys Res Comm. 2001; 288:476-482.
105. Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer. 2007; 97:1139-1145.
106. Perrone F, Lampis A, Orsenigo M, Di Bartolomeo M, Gevorgyan A, Losa M, Frattini M, Riva C. Andreola S, Bajetta E, Bertario L, Leo E, Pierotti MA, et al. PIK3CA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol. 2009; 20:84-90.
107. Davies HI, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett, MJ, Bottomley W, Davis N, Dicks E, Ewing R, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417: 949-954.
108. Vogt PK, Kang S, Elsliger MA, Gymnopoulos M. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem Sci. 2007; 32:342-349.
109. Murugan AK, Munirajan AK, Tsuchida N. Genetic deregulation of the PIK3CA oncogene in oral cancer. Cancer Lett. 2013; 338:193-203.
110. Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, Apicella M, Migliardi G, Galimi F, et al. Amplifcation of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658-673.
111. Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M et al (2011) Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 3: 99ra86
112. Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, Morgillo F, Capasso A, Sforza V, Nappi A, De Palma R, D’Aiuto E, Berrino L, et al. Increased TGF- as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. 2013; 19: 6751-6765.
113. Liska D, Chen CT, Bachleitner-Hofmann T, Christensen JG, Weiser MR. HGF Rescues Colorectal Cancer Cells from EGFR Inhibition via MET Activation. Clin Cancer Res. 2011; 17: 472-482-
114. Montagut C1, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, Minoche A, Seshagiri S, Serrano S. et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 201;218:221-223.
115. Van Emburgh BO, Sartore-Bianchi A, Di Nicolantonio F, Siena S, Bardelli A. Acquired resistance to EGFR-targeted therapies in colorectal cancer. Molecular Oncology 2014; 8:1084-1094.
116 Leto SM, Trusolino L. Primary and acquired resistance to EGFR-targeted therapies in colorectal cancer: impact on future treatment strategies. J Mol Med. 2014; 92:709:722.
117. Dienstmann R, Salazar R, Tabernero J. Overcoming resistance to Anti-EGFR therapy in colorectal cancer. Am Soc Clin Oncol Educ Book; 2015:e149-156.
118. Vasan N, Boyer JL, Herbst RS. A RAS renaissance: emerging targeted therapies for Kras-mutated non-small cell lung cancer. Clin Cancer Res. 2014; 20:3921-3930.
119. Rachagani S, Senapati S, Chakraborty S, Ponnusamy MP, Kumar S, Smith LM, Jain M, Batra SK. Activated KrasG12D is associated with invasion and metastasis of pancreatic cancer cells through inhibition of E-cadherin. Br J Cancer. 1985; 53:1038- 1048.
120. Valsangkar NP, Ingkakul T, Correa-Gallego C, Mino-Kenudson M, Masia R, Lillemoe KD, Fernández-del Castillo C, Warshaw AL, Liss AS, Thayer SP. Survival in ampullary cancer: potential role of different KRAS mutations. Surgery. 2015 ;157:260-268.
121. Margonis GA, Kim Y, Spolverato G, Ejaz A, Gupta R, Cosgrove D, Anders R, Karagkounis G, Choti MA, Pawlik TM. Association between specific mutations in KRAS codon 12 and colorectal liver metastasis. JAMA Surg. 2015;150:722-729.
122. Ledford H. The RAS renaissance. Nature 2015, 278 -280.
123. Hiraoka K, Inoue T, Taylor RD, Watanabe T, Koshikawa N, Yoda H, Shinohara K, Takatori A, Sugimoto H, Maru Y, Denda T, Fujiwara K, Balmain A, et al. Inhibition of KRAS codon 12 mutants using a novel DNA-DNA-alkylating pyrrole-imidazole polyamide conjugate. Nat Commun. 2015; 6:6706.
124. Yuan TL, Fellmann C, Lee CS, Ritchie CD, Thapar V, Lee LC, Hsu DJ, Grace D, Carver JO, Zuber J, Luo J, McCormick F, Lowe SW. Development of siRNA payloads to target KRAS-mutant cancer. Cancer Discov. 2014; 4:1182-1197.
125. Golan T, Khvalevsky EZ, Hubert A, Gabai RM, Hen N, Segal A, Domb A, Harari G, David EB, Raskin S, Goldes Y, Goldin E, Eliakim R, et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015; 6: 24560-24570. doi: 10.18632/oncotarget.4183.
126. Jiang QH, Peng HX, Zhang Y, Tian P, Xi ZL, Chen H. rs712 polymorphism within let-7 microRNA-binding site might be involved in the initiation and progression of colorectal cancer in Chinese population. Onco Targets Ther. 2015; 22:3041-3045.
127. Gomes SE, Simões AE, Pereira DM, Castro RE, Rodrigues CM, Borralho PM. miR-143 or miR-145 overexpression increases cetuximab-mediated antibody-dependent cellular cytotoxicity in human colon cancer cells. Oncotarget. 2016; 7:9368-87. doi: 10.18632/oncotarget.7010.
128. Ostrem JM, Peters U, Sos ML, James A, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013; 503:548- 553.
129. Wang Y, Kaiser CE, Frett B, Li HU. Targeting mutant KRAS for anticancer therapeutics: A review of novel small molecule modulators. J Med Chem. 2013; 56:5219-5230.
130. Lu Y, Bellgrau D, Dwyer-Nield LD, Malkinson AM, Duke RC, Rodell TC, Franzusoff A. Mutation-selective tumor remission with ras-targeted, whole yeast-based immunotherapy. Cancer Res. 2004; 64:5084-5088.
131. Chaft JE, Litvak A, Arcila ME, Pate P, D’Angelo SP, Krug LM, Rusch V, Mattson A, Coeshott C, Park B, Apelian DM, Kris MG, Azzoli CG. Phase II study of the GI-4000 KRAS vaccine after curative therapy in patients with stage I-III lung adenocarcinoma harboring a KRAS G12C, G12D, or G12V mutation. Clin Lung Cancer. 2014; 15: 405- 410.
132. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371:2189-2199.
133. D’Incecco A, Andreozzi MV, Ludovini V, Rossi1 E, Capodann A, Landi A, Tibaldi C, Minuti GJ, Salvini J, Coppi E, Chella A, Fontanini G, Filice ME, et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Brit J Cancer 2015; 112: 95-102,
134. Davar D, Socinski MA, Dacic S, Burns TF. Near complete response after single dose of nivolumab in patient with advanced heavily pre-treated KRAS mutant pulmonary adenocarcinoma. Exp Hematol Oncol 2015: 4:34 DOI 10.1186/s40164-015-0029-7
135. Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, Gierut JJ, Cox AD, Haigis KM, Philips MR. K-Ras 4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 2015; 112:779-784.
136. Voice JK, Klemke RL, Ann Le A, Jackson JH. Four human ras homologs differ in their abilities to activate Raf-1, induce transformation, and stimulate cell motility J Biol. Chem. 1999; 274:17164-17170.
137. Turacli D, Ozkan AC, Ekmekci A. The comparison between dual inhibition of mTOR with MAPK and PI3K signaling pathways in KRAS mutant NSCLC cell lines. Tumour Biol. 2015; 36: 9339-9345.
138. Ischenko I, Petrenko O, Hayman MJ A MEK/PI3K/HDAC inhibitor combination therapy for KRAS mutant pancreatic cancer cells. Oncotarget. doi: 10.18632/oncotarget.4538.
139. Van Dort ME, Galbán S, Wang H, Sebolt-Leopold J, Whitehead C, Hong H, Rehemtulla A, Ross BD. Dual inhibition of allosteric mitogen-activated protein kinase (MEK) and phosphatidylinositol 3-kinase (PI3K) oncogenic targets with a bifunctional inhibitor. Bioorg Med Chem. 2015 ;23:1386-1394.
140. McCormick F. KRAS as a therapeutic target. Clin Cancer Res. 2015; 21:1797-1801.
141. Donohue DJ, Hunter T. Recombinational Junctions of varients of Moloney murine sarcoma virus: generation and divergence of a mammalian transforming gene. J Virol. 1983; 45:607-617.
142. Yuasa Y, Srivastava SK, Dunn CY, Rhim JS, Reddy EP, Aaronson SA. 1983. Acquisition of transforming properties by alternative point mutations within c-bas/has human proto-oncogene. Nature. 1983; 303:775-779.