
INTRODUCTION
The misfolding and aggregation of specific proteins is a common hallmark of many neurodegenerative disorders, including highly prevalent illnesses such as Alzheimer’s disease and Parkinson’s disease, as well as prion diseases [1, 2]. One of the fundamental events related to TSE pathogenesis is the refolding of a host-encoded glycoprotein, the prion protein (PrPc), into a protease-insensitive isoform (PrPsc) that aggregates into deposits. The conversion into PrPsc is driven by the transition of the PrPc’s large N-terminal region from a random coil to a β-sheet structure, which dominates the α-helix content (43% vs. 30%)[3, 4]. These profound changes in PrPc physicochemical properties bring out a structural transition. While PrPc is soluble in non-denaturing detergents, PrPsc is not. PrPc is smoothly digested by proteases, whereas PrPsc is partially resistant [5]. Although the major role PrPsc plays in the origin and transmission of TSEs is well established, how the disease-specific prion protein exerts its harmful effects on neurons is unknown [6].
A synthetic peptide corresponding to amino acid residues 106-126 of human PrP, which forms fibrils in vitro, is toxic to cultured hippocampal neurons. PrP (106-126) possesses many of the pathogenic and physiologic properties of PrPsc, including the ability to induce apoptosis in hippocampal neurons and induce astrocyte proliferation [7]. Prion diseases are associated with misregulation of autophagy as shown by the formation of giant autophagic vacuoles in experimental scrapie in hamsters [8].
Autophagy is a conserved trafficking pathway that is highly correlated by environmental conditions [9]. Autophagy, a common morphological feature in dying cells, has more recently been thought to keep cells alive under stressful conditions[10]. There are three defined types of autophagy: macro-autophagy, micro-autophagy, and chaperone-mediated autophagy (CMA), all of which promote proteolytic degradation of intracellular components in the lysosome[11]. We focused on macro-autophagy in the present study. Upon autophagy induction, the phagophore expands and encloses a portion of cytoplasm resulting in the formation of a double-membraned structure called the autophagosome, which fuses with a lysosome for degradation[12]. Microtubule-associated protein light chain 3 (LC3) is localized, aggregating on the autophagosome, and is considered a marker of autophagy. LC3B undergoes lipidation and is recruited to the phagophore, where it correlates membrane elongation and closure [13]. LC3B then transforms from LC3B-I to LC3B-II during autophagosome formation[14]. Also involved in autophagy is P62, a multifunctional signaling molecule associated with a variety of cellular pathways. P62 is one of the best-known autophagic substrates and is extensively employed as an indicator of autophagic degradation [15]. SQSTM1/p62 can deliver ubiquitinylated cargo to the proteasome, though they are mainly degraded by autophagy [15, 16]. SQSTM1/p62 levels are generally inversely related to autophagic degradation, since the loss of Atg genes or factors required for autophagosome fusion with lysosomes all result in a marked increase in SQSTM1/p62-positive aggregates [17, 18]. By eliminating damaged intracellular organelles and aggregates, autophagy promotes cell surface antigen presentation and cellular senescence, protects against genome instability and prevents necrosis [11, 19]. Thus, autophagy has an essential role in preventing diseases such as neurodegeneration, cancer, cardiomyopathy, diabetes, liver disease, autoimmune diseases and infections [11, 20-22].
Hypoxia inducible factor-1 is a heterodimeric transcription factor that plays a pivotal role in regulating cellular oxygen homeostasis. It is composed of an oxygen-regulated HIF-1α subunit and a constitutively expressed HIF-1β subunit. Under hypoxic conditions, HIF-1α hydroxylation is inhibited, allowing its translocation into the nucleus where it binds to HIF-1β to form an active complex, HIF-1. HIF-1 then initiates the transcription of an array of target genes that are vital for cellular adaption to hypoxia [23]. Recently, many non-hypoxic stimuli, such as cytokines, free radicals, growth factors, and hormones, have been shown to activate HIF-1α under normoxic conditions. Based on these data, HIF-1α appears to have a neuroprotective effect in the ischemic brain [24].
Hinokitiol is a tropolone-related compound found in various natural sources such as the heartwood of several cupressaceous plants. Hinokitiol has been widely used as an antimicrobial agent in hair tonics, toothpastes, cosmetics, and food [25]. Jayakumar et al. suggested that hinokitiol treatment provides neuroprotection, improved recovery of infarcted tissue, and improved neurological outcomes in embolic stroke-induced ischemic rats [26]. In addition, some studies suggest that hinokitiol activates the HIF pathway [27, 28] and autophagy [29, 30]. Based on these studies, we investigated whether hinokitiol induces HIF-1α stabilization and autophagy and whether these pathways have apoptotic or protective effects. We report that hinokitiol enhances autophagy and protects against prion protein-induced neurotoxicity via HIF-1α stabilization.
RESULTS
Hinokitiol-attenuated, prion protein-induced cytotoxicity in neuronal cells
We investigated the influence of hinokitiol on PrP (106-126)-induced neurotoxicity in primary neuron cells using an annexin V assay. The primary neurons were exposed to hinokitiol with or without PrP (106-126). The viability of PrP (106-126)-treated cells was decreased approximately 50% compared to controls, and hinokitiol-treated cells had enhanced viability. Cell viability in hinokitiol-treated only cells was comparable to untreated controls. Importantly, hinokitiol treatment inhibited PrP (106-126)-induced neurotoxicity in primary neurons (Figure 1A, 1B). LDH release levels indicate that hinokitiol inhibited PrP (106-126)-induced apoptosis in a dose-dependent manner (Figure 1C), consistent with previous results. As seen in Figure 1D, hinokitiol attenuated PrP (106-126)-induced apoptosis, which was evident based on the amount of DNA strand breakage.
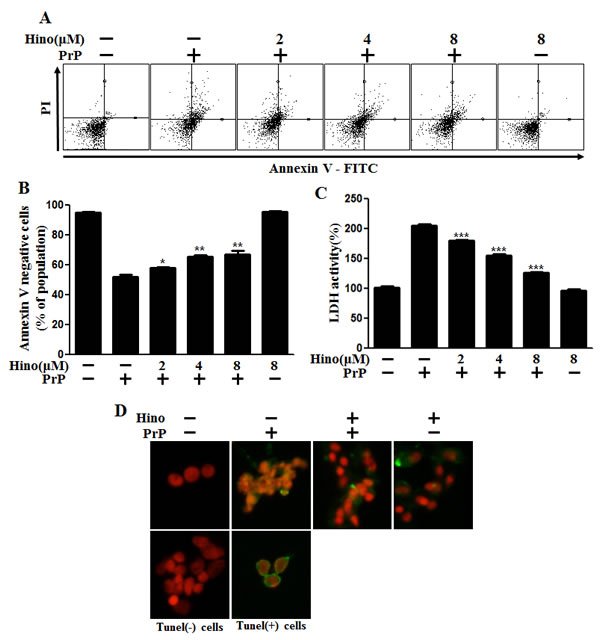
Figure 1: Hinokitiol attenuates PrP (106-126)-induced cytotoxicity in neuronal cells. A. The primary neuronal cells were pretreated with hinokitiol (6 h) in a dose-dependent manner and then exposed to 100 μM PrP (106-126) for 12 h. Cell viability was measured by the annexin V assay. Cells were treated with FITC-annexin V, which binds to phosphatidylserine on the plasma membrane during apoptosis. B. Bar graph indicating the averages of the annexin V-negative cells. C. A lactate dehydrogenase (LDH) assay was used to quantify LDH released into the medium. D. Representative immunofluorescence images of TUNEL-positive (green) cells 12 h after exposure to 100 μM of PrP (106-126) in the absence or the presence of hinokitiol (6 h). The cells were counterstained with PI (red) to show all cell nuclei. *p < 0.05, **p < 0.01, *** p < 0.001: Significant differences between the control and treatment groups. Hino, hinokitiol; PrP, PrP (106-126).
Hinokitiol-induced autophagy in neuronal cells
We explored autophagy as a survival strategy for prion-induced neurotoxicity. First, we examined whether hinokitiol increased the autophagy marker, LC3B. LC3 protein is localized and aggregates on autophagosomes and is, therefore, considered a marker of autophagy. LC3 transforms from LC3-I to LC3-II during autophagosome formation [14]. We observed that levels of the late autophagosome marker LC3-II increased in the hinokitiol-treated group in a dose-dependent manner compared to the control and PrP (106-126)-treated groups via Western blot analysis in mouse primary neurons (Figure 2A, 2B). To visualize the activation of autophagy through the formation of autophagosomes in neurons, the Premo Autophagy Sensor (LC3B-FP) BacMam 2.0 system was employed. LC3B-FP and LC3B (G120A)-FP viral vectors (MOI = 30) were transduced into SK-N-SH cells, enabling the expression of fluorescent LC3B protein, and consequently, allowing us to monitor autophagosome dynamics using inverted fluorescent microscopy. Negative controls were established using mutant chimera LC3B (G120A)-FP. According to the results reported in Figure 2C and 2D, BacMam LC3B (G120A)-FP transduced cells showed a marked cytosolic and diffuse expression pattern. SK-N-SH cells treated with hinokitiol presented with an increased punctate fluorescent distribution pattern, suggesting LC3B-FP protein accumulation in the autophagosomes. We analyzed this reduction in LC3-II and green fluorescent puncta, which is caused by lysosomal autophagosome degradation. To detect further autophagic flux, transmission electron microscopy was performed. As shown in Figure 2E, double-membraned autophagosomes containing entrapped cytoplasm or entire organelles were induced by hinokitiol treatment. These results suggest that hinokitiol activates autophagy in human and mice neuronal cells.
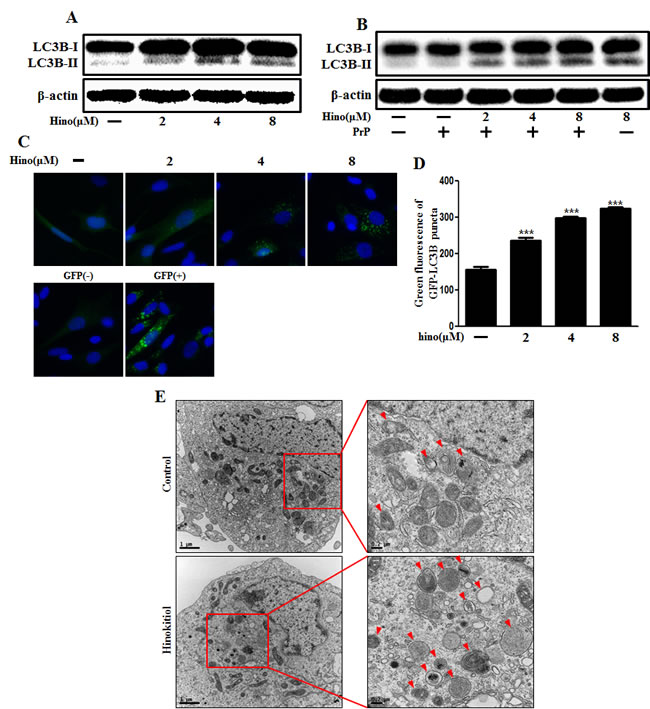
Figure 2: Hinokitiol induces autophagy in neuronal cells. The primary neurons were treated with 2, 4, and 8 μM of hinokitiol for 6 h A. and were then exposed to 100 μM PrP (106-126) for 6 h B.. The treated cells were assessed for LC3B production by Western blot analysis. SK-N-SH cells were mixed with a titration (30MOI) of BacMam GFP-LC3B virus over 18 h and were then treated with hinokitiol in a dose-dependent manner for 6 h C., D.. Negative control reagent and positive control reagent (CQ) at the same time. E. SK-N-SH cells were incubated with 8 μM of hinokitiol for 6 h and analyzed by TEM. Arrowheads indicate autophagosomes. *** p < 0.001; significant differences when compared with control and each treatment group. Hino, hinokitiol; PrP, Prion peptide (106-126); GFP (+), Positive control; GFP (-), Negative control.
According to our observations, SQSTM1/p62 protein was increased by hinokitiol treatment in neurons (Figure 3A, 3B). SQSTM1/p62 gene expression level was also increased with hinokitiol (Figure 3C, 3D). To determine the activity of the autophagic system during prion pathogenesis, alterations in the SQSTM1/p62 protein were assessed. The SQSTM1/p62 protein is a link between LC3 and ubiquitinated substrates and is incorporated into, and then degraded by, autolysosomes [31]. Usually, SQSTM1/p62 is degraded by autolysosomes when autophagy flux is activated. However, some studies suggest that p62 has a protective role and is required as a survival factor [32, 33]. In this case, we suggest that p62 is not involved in autophagy. By employing numerous experimental methods, we are confident that we have demonstrated autophagy.
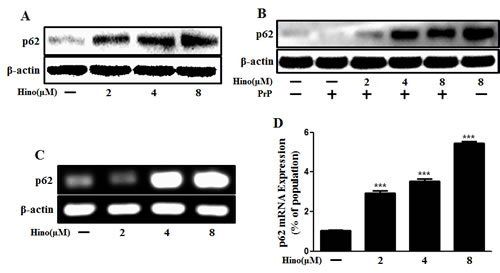
Figure 3: Hinokitiol increases p62/SQSTM1 mRNA and protein levels. Primary neuronal cells were treated with hinokitiol in a dose-dependent manner for 6 h A. and were then exposed to 100 μM of PrP (106-126) for 6 h B.. The treated cells were assessed for p62/SQSTM1 expression by Western blot analysis. C. RT-PCR for the p62/SQSTM1 gene was analyzed from primary neuron cells. D. Real-time PCR for the p62/SQSTM1 gene was analyzed from primary neurons. *** p < 0.001; significant differences when compared with control and each treatment group. Hino, hinokitiol; PrP, Prion peptide (106-126).
Hinokitiol inhibits PrP (106-126)-induced neuronal apoptosis by inducing autophagy
We recognize that the specific role of autophagy flux remains controversial. Therefore, we set out to determine whether autophagy flux has a protective function. We confirmed the effects of 3MA and wortmannin as autophagy inhibitors. We examined whether using these autophagy inhibitors could reduce the hinokitiol-induced neuroprotective effects against PrP (106-126). As shown in Figure 4A and 4B, the neuroprotective effects of hinokitiol diminished following treatment with the autophagy inhibitors. To visualize autophagy activation through the formation of autophagosomes, the Premo Autophagy Sensor (LC3B-FP) BacMam 2.0 system and Western blot analysis were employed as described above (Figure 4C, 4D, and 4E). We observed that the increase in LC3-II and green fluorescent puncta caused by the creation of autophagosomes was reduced with exposure to autophagy inhibitors. As shown in Figure 4D and 4E, LC-II protein expression level was decreased by the autophagy inhibitors. To confirm these results, transmission electron microscopy was performed (Figure 4F).
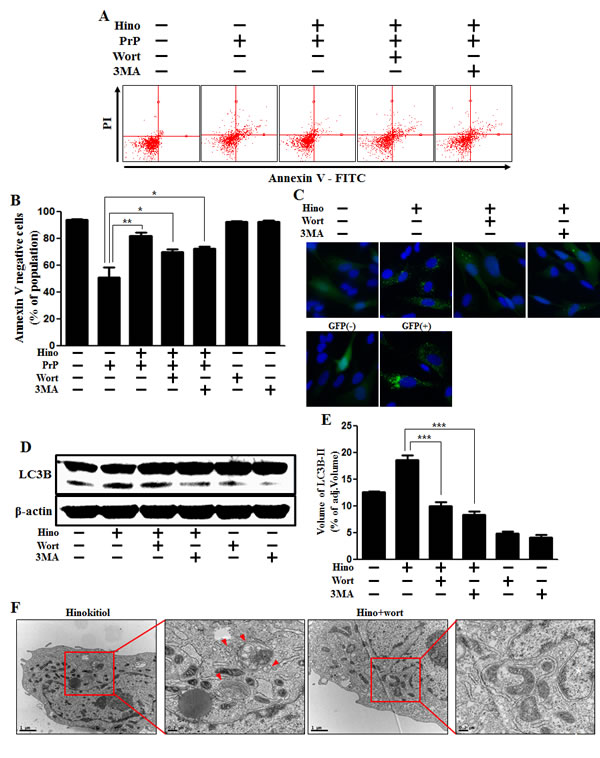
Figure 4: Hinokitiol-induced autophagy has a protective role in neuroblastoma cells. A. The SK-N-SH neuroblastoma cells were pretreated with 8 μM of hinokitiol in the presence of autophagy inhibitors (3MA or wortmannin) for 6 hr and were then exposed to 100 μM of PrP (106-126) for 12 h. Cell viability was measured by the Annexin V assay. B. Bar graph indicating the averages of annexin V-negative cells. C. SK-N-SH cells were mixed with a titration (30MOI) of BacMam GFP-LC3B virus for 18 h and then treated with hinokitiol and autophagy inhibitors for 6h, Negative control reagent and positive control reagent (CQ) at the same time. D. Western blot for LC3B production was analyzed from SK-N-SH cells. Beta-actin was used as a loading control. E. Bar graph indicating the averages of LC3B-II expression levels. F. SK-N-SH cells were incubated with hinokitiol at 8 μM with wortmannin for 6 h and were then analyzed by TEM. Arrowheads indicate autophagosomes. *p < 0.05, **p < 0.01, *** p < 0.001; significant differences when compared with the control and each treatment group. Hino, hinokitiol; PrP, Prion peptide (106-126); wort, wortmannin; 3MA, 3-Methyladenine; GFP (+), Positive control; GFP (-), Negative control.
Additionally, knockdown of ATG5 using ATG5 small interfering RNA (ATG5 siRNA) decreased hinokitiol-induced autophagy (Figure 5A and 5B), as well as reduced the neuroprotective effects induced by hinokitiol in neuronal cells (Figure 5C and 5D).
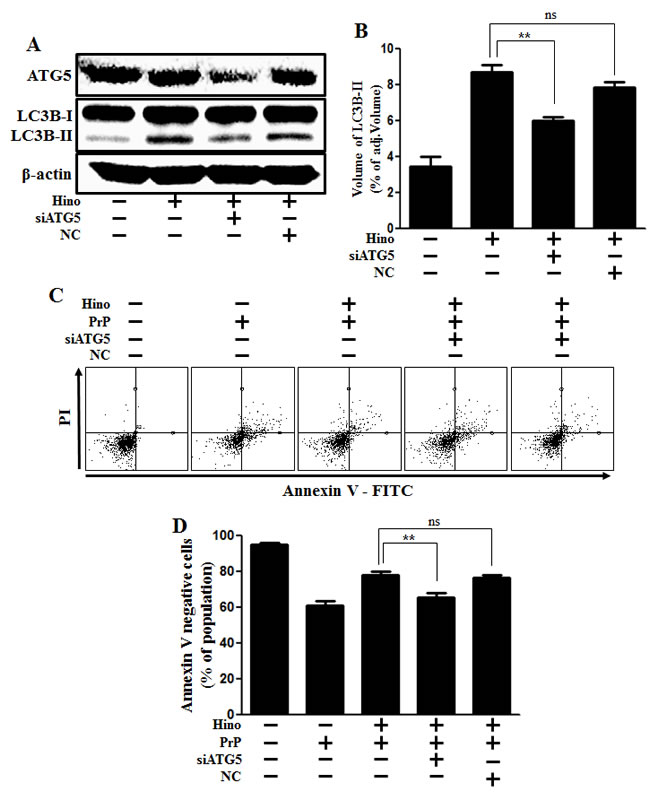
Figure 5: ATG5 knockdown decreases neuroblastoma cell viability. A. ATG5 small interfering RNA (siATG5)- or negative control siRNA (NC)-transfected SK-N-SH cells were incubated with 8 μM of hinokitiol for 6 h. Western blot for ATG5 and LC3B production was analyzed in SK-N-SH cells. Beta-actin was used as a loading control. B. Bar graph indicating the averages of LC3B-II expression levels. C. SK-N-SH cells were incubated with PrP (106-126) for 12 h in the presence of hinokitiol. Cell viability was measured by Annexin V assay. D. Bar graph indicating the averages of annexin V-negative cells. **p < 0.01; significant differences when compared with control and each treatment group. Hino, hinokitiol; PrP, Prion peptide (106-126); NC, Negative control.
Autophagy induced by hinokitiol inhibits the PrP (106-126)-induced apoptotic pathway via HIF-1α stabilization
Previously, it was suggested that hypoxia protects neuronal cells against PrP (106-126)-induced neurotoxicity and that this prevention is associated with hypoxia-mediated HIF-1α signaling [34]. Some reports suggest that hinokitiol activates the HIF pathway as described in the introduction. To assess expression of HIF-1α, primary neuronal cells were treated with hinokitiol, and both HIF-1α gene expression and protein levels increased in a dose-dependent manner (Figure 6A, 6B, and 6C). We confirmed this HIF-1α protein upregulation using immunocytochemistry (Figure 6D).
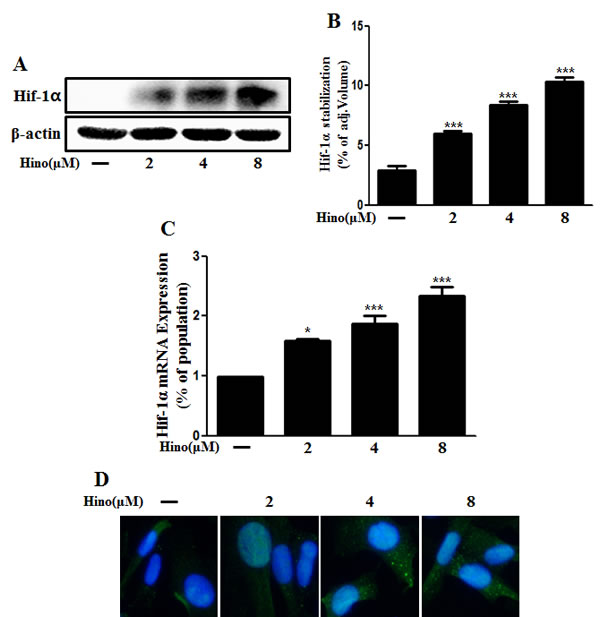
Figure 6: Hinokitiol induces Hif-1α stabilization. A. The primary neurons were treated with 2, 4, and 8 μM of hinokitiol for 6 h. The treated cells were assessed for Hif-1α by Western blot analysis. B. Bar graph indicating the averages of Hif-1α stabilization levels. C. Real-time PCR for the Hif-1α gene was analyzed from primary neuron cells. D. Immunocytochemistry for Hif-1α protein was performed on SK-N-SH cells. *p < 0.05, *** p < 0.001; significant differences when compared with control and each treatment group. Hino, hinokitiol; Hif-1α, Hypoxia-inducible factor-1 alpha; p-Akt, phosphorylation of Akt; PrPc, prion protein.
Furthermore, in order to confirm that hinokitiol protects against prion-induced apoptosis by HIF-1α stabilization, HIF-1α siRNA was used to knockdown HIF-1α gene expression. We found that knockdown of HIF-1α expression did block autophagy, as confirmed via Western blot (Figure 7A and 7B) and GFP-LC3 punta analysis (Figure 7C). In addition, we showed that knockdown of HIF-1α expression inhibited hinokitiol’s neuroprotective effects (Figure 7D and 7E). We conclude that hinokitiol prevents prion-induced neurotoxicity by inducing autophagy and activating HIF-1α.
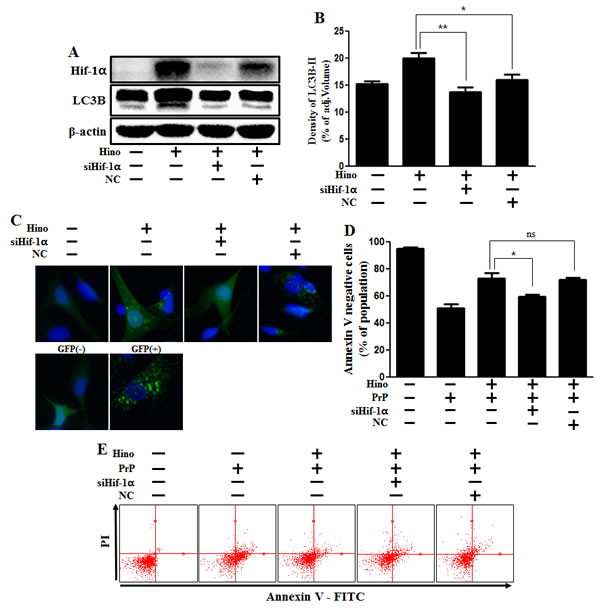
Figure 7: Hinokitiol-induced autophagy protects cytotoxicity via Hif-1α stabilization. A. Hif-1α small interfering RNA (siHif-1α)-transfected or negative control siRNA (NC)-transfected SK-N-SH cells were incubated with 8 μM of hinokitiol for 6 h. Western blots for Hif-1α, LC3B proteins were analyzed from SK-N-SH cells. B. Bar graph indicating the averages of LC3B-II levels. C. SK-N-SH cells were mixed with a titration (30MOI) of BacMam GFP-LC3B virus over 18 h and were then treated with hinokitiol for 6 h, Negative control reagent and positive control reagent (CQ) at the same time. D. Bar graph of cell viability indicating the averages of annexin V-negative cells. E. Cell viability was measured by Annexin V assay from siRNA-treated cells. *p < 0.05; significant differences when compared with control and each treatment group. Hino, hinokitiol; Prion peptide (106-126); Hif-1α, Hypoxia-inducible factor-1 alpha; p-Akt, phosphorylation of Akt; NC, Negative control.
DISCUSSION
This study demonstrates the possibility of attenuating prion protein-induced neurotoxicity with hinokitiol. It appears that hinokitiol attenuates neurotoxicity via autophagy, which is activated by HIF-1α stabilization. However, hinokitiol is not sufficient to prove a biological effect in neurons. We hope this study establishes a basis for further exploration of hinokitiol and its effects.
Autophagy is a dynamic lysosome-mediated process that involves the sequestration and delivery of cytoplasmic material to the lysosome where it is degraded and recycled [35, 36]. Several studies have proposed that autophagy is a double-edged sword, with both beneficial and harmful potential in cancer[37] and neurodegeneration[38]. Autophagy has several possible pathways depending on the circumstances, and the biochemical basis for its diverse functions is not well understood [39, 40]. Our results indicate that LC3-II was degraded by prion peptide treatment. Mizushima et al. suggested that LC3-II, which increases transiently upon induction of autophagy, is reduced after longer periods of autophagy activation [41]. For this reason, we suggest that degradation of LC3 indicates induction of autophagic flux.
Studies employing knockout, transgenic, and knock-in mice have shown that p62 plays a critical role in a number of cellular functions, including bone remodeling, obesity, and cancer [33, 42-44]. Recent findings link p62 activity to the extrinsic apoptosis pathway, and Mathew et al. showed that the modulation of p62 by autophagy is a key factor in tumorigenesis [45]. Previously, it was thought that p62 always has to be degraded when autophagy flux was induced. However, many reports suggest that p62 has various functions and correlates many signaling pathways. For example, upregulation of p62 may signify an inhibition of autophagic flux [46, 47]. In contrast, some studies report that the absence of p62 results in enhanced ROS production, a critical step for the induction of apoptosis [33]. Additionally, p62 depletion increases cell injury in neonatal rat ventricular myocytes (NRVMs) under basal conditions and during overexpression of misfolded proteins [48]. Haqa et al. suggested that mutant huntingtin can also form aggregates in the absence of p62, so they believe that the protective role of p62 may be to recruit autophagosomal components to the polyubiquitinylated protein aggregates rather than to facilitate the formation of these aggregates [32]. For these reasons, we believe that the increase in p62 in our case has a protective role in neuronal cells and does not influence autophagy. In addition, we checked increase of mRNA expression of p62. On the basis of our results and many studies, we suggest that it didn’t appear p62 degradation in spite of autolysosome degradation in autophagy flux because protein of p62 was more synthesized than degradation.
Based on our results, we propose that hinokitiol may have a critical role as a therapeutic target for prion disease. We demonstrate that hinokitiol induces autophagy via the HIF-1α pathway, which acts as the main neuroprotective mechanism against prion peptide-induced neurotoxicity. The prion peptide 106-126 sequence is a useful model for in vitro study of prion-induced cell death, in addition to in vivo retinal neuron models treated with intravitreous injection of PrP fragments [49, 50]. In the future, we will further study the neuroprotective effects of hinokitiol, autophagy and the HIF-1α pathway in mouse models to examine hinokitiol’s potential therapeutic role in prion disease.
MATERIALS AND METHODS
Cell culture
The primary neurons were isolated from embryonic day 18 mice. Briefly, tissues were collected in Hanks buffered saline solution without Mg2+ or Ca2+ (HBSS: Invitrogen-GIBCO, Grand Island, NY, USA), and digested in 0.25% trypsin with DNAse I (2000 units/mg) (Invitrogen, Carlsbad, CA, USA) for 20 min at 37°C. Cells were then mechanically dissociated and diluted in HBSS containing Mg2+ and Ca2+. Isolated neuron cells were diluted in DMEM containing 25 mM glucose (abbreviated in text as glucose medium; Thermo Scientific) supplemented with 10% FBS and were plated in flasks pre-coated with 50 μg/ml poly D-lysine. The human neuroblastoma cell line SK-N-SH was obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were cultured in minimum essential medium (MEM, Hyclone Laboratories, Logan, UT, USA) containing 10% fetal bovine serum (Invitrogen-GIBCO, Grand Island, NY, USA) and gentamycin (0.1 mg/mL) in a humidified incubator maintained at 37°C and 5% CO2.
PrP (106-126) treatment
Synthetic PrP (106-126) peptides (sequence: Lys-Thr-Asn-Met-Lys-His-Met-Ala-Gly-Ala-Ala-Ala-Ala-Gly-Ala-Val-Val-Gly-Gly-Leu-Gly) were synthesized by Peptron (Seoul, Korea). The peptides were dissolved in sterile dimethyl sulfoxide at a stock concentration of 10 mM and stored at -20°C.
Annexin V assay
Apoptosis in detached cells was assessed using an annexin V assay kit (Santa Cruz Biotechnology, Santa Cruz, CA, USA) according to the manufacturer’s protocol. Annexin V levels were determined by measuring fluorescence at 488 nm of excitation and 525/30 emission using a Guava easyCyte HT System (Millipore, Bedford, MA, USA).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
TUNEL analysis was performed to measure the degree of cellular apoptosis using an in situ ApoBrdU DNA fragmentation assay kit (BioVision, Mountain View, CA, USA) following the manufacturer’s instructions. Cells were counterstained with propidium iodide (PI) to show cell nuclei.
Lactate dehydrogenase assay
Cytotoxicity was assessed in the supernatants using a lactate dehydrogenase (LDH) cytotoxicity detection kit (Takara Bio, Inc., Tokyo, Japan) according to the manufacturer’s protocol. LDH activity was determined by measuring absorbance at 490 nm using a microplate reader (Spectra Max M2, Molecular Devices, Sunnyvale, CA, USA).
BacMam transduction
Wild-type or mutant GFP-tagged LC3B was expressed in cells by adding the appropriate concentrations of the appropriate virus from the Premo Autophagy Sensor LC3B-GFP kit (BacMam 2.0) (Life Technologies P36235) to the growth medium as indicated in the figure legends.
Immunocytochemistry
Immunocytochemical analyses were performed on neuroblastoma cells with anti-p62 (P0067, Sigma Aldrich) antibodies. Cells were cultured on glass slides (Nalge Nunc International, Naperville, IL). Cells were washed in sterilized TBST for 10 min, then blocked for 15 min with 5% FBS in TBST, and then incubated overnight at 4°C with the primary antibodies diluted with 5% FBS in TBST. Alexa Fluor 488-labeled donkey anti-rabbit IgG antibody diluted to 1:1000 (Molecular Probes, A21206) was used to visualize channel expression using fluorescence microscopy.
RNA interference
SK-N-SH cells were transfected with ATG5 small interfering RNA (siRNA: oligoID HSS114104: Invitrogen, Carlsbad, CA, USA) and HIF-1α siRNA (oligoID HSS104775: Invitrogen) using Lipofectamine 2000 according to the manufacturer’s instructions. After a 48-hr culture, knockdown efficiency was measured at the protein level by immunoblot analysis. Nonspecific siRNA (oligoID 12935-300: Invitrogen) was used as the negative control.
Western blot analysis
Primary neuronal cells and SK-N-SH cells were placed in lysis buffer [25 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.4, 100 mM NaCl, 1 mM EDTA (ethylene diamine tetra acetic acid), 5 mM MgCl2, 0.1 mM DTT (dithiothreitol), and a protease inhibitor mixture]. Whole cell proteins were electrophoresed on a 10-15% sodium dodecyl sulfate polyacrylamide gel and transferred to a nitrocellulose membrane. Immunoreactivity was detected through sequential incubation with primary antibodies, horseradish peroxidase-conjugated secondary antibodies, and an enhanced chemiluminescence reagent, i.e., West save gold detection kit (AbFrontier Inc.). The primary antibodies used for immunoblotting were anti-LC3B (#4108, Cell Signaling Technology), anti-P62 (#5114, Cell Signaling Technology), anti-ATG5 (#2630, Cell Signaling Technology) and anti-β-actin (A5441, Sigma Aldrich). Images were examined using a Fusion FX7 imaging system (Vilber Lourmat, Torcy Z.I., Sud, France). Signal band densitometry was analyzed using the Bio-1D software (Vilber Lourmat, Marne La Vallee, France).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total ribonucleic acid (RNA) was extracted from primary neuronal cells using the easy-spin™ Total RNA Extraction Kit (Intron Biotechnology, Seoul, Korea). The cDNA synthesis was carried out following the instructions in the TaKaRa PrimeScript TM 1st strand cDNA synthesis kit (TaKaRa Bio, Tokyo, Japan). For qRT-PCR, 1 μl of gene primers with SYBR Green (Bio-Rad Laboratories, Hercules, CA, USA) in 20 μl of reaction volume was applied. The primers were: p62/SQSTM1 (forward: 5′CTCCCCAGACTACGACTTGTGT3′, reverse: 5′TCAACTTCAATGCCCAGAGG3′), Hif-1α (forward: 5′CGCAAGTCCTCAAAGCACAGTTAC3′, reverse: 5′TGGTAGTGGTGGCATTAGCAGTAG3′), and β-actin (as an internal control) (forward: 5′GCAAGCAGGAGTATGACGAG3′, reverse: 5′CAAATAAAGCCATGCCAATC3′). All reactions with iTaq SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA) were performed on the CFX96 real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA).
Transmission electron microscopy analysis
TEM samples were analyzed by Transmission Electron Microscope (JEM-2010, JEOL) installed in the Center for University-Wide Research Facilities (CURF) at Chonbuk National University. After fixing SK-N-SH cell samples in 2.5% glutaraldehyde (TED PELLA, USA) in PBS (pH: 7.2), specimens were post-fixed in 1% osmium tetroxide (Heraeus, South Africa), dehydrated in graded ethanol and propylene oxide (Acros Organics, USA), and then embedded in epoxy resin (Embed812, NMA: Nadic methyl anhydride, DDSA: Dodenyl Succinic Anhidride, DMP-30, USA), as used previously. Serial ultrathin sections were cut on an LKB-III ultratome (LEICA, Germany). Ultrathin sections were stained with uranyl acetate (TED PELLA, USA) and lead citrate (TED PELLA, USA) and were examined using a Hitachi H7600 electron microscope (Hitachi, Japan) at an accelerating voltage of 100 kV.
Statistical analysis
Unpaired t-tests or Welch’s corrections were used to compare differences between the two groups. One-way ANOVAs followed by the Dunnett’s test were used for multiple comparisons. All statistical analyses were performed with GraphPad Prism software. Results were considered significant at * p < 0.05, ** p < 0.01 or *** p < 0.001.
ACKNOWLEDGMENTS
This study was supported by the National Research Foundation of the Korea Grant funded by the Korean Government (2013R1A1A2063931).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
References
1. Costanzo M and Zurzolo C. The cell biology of prion-like spread of protein aggregates: mechanisms and implication in neurodegeneration. The Biochemical journal. 2013; 452:1-17.
2. Aguzzi A and Calella AM. Prions: protein aggregation and infectious diseases. Physiological reviews. 2009; 89:1105-1152.
3. Caughey B, Raymond GJ, Ernst D and Race RE. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. Journal of virology. 1991; 65:6597-6603.
4. Ma J and Lindquist S. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science. 2002; 298:1785-1788.
5. Oesch B, Westaway D, Walchli M, McKinley MP, Kent SB, Aebersold R, Barry RA, Tempst P, Teplow DB, Hood LE and et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell. 1985; 40:735-746.
6. Aguzzi A. Staining, straining and restraining prions. Nature neuroscience. 2008; 11:1239-1240.
7. Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O and Tagliavini F. Neurotoxicity of a prion protein fragment. Nature. 1993; 362:543-546.
8. Boellaard JW, Kao M, Schlote W and Diringer H. Neuronal autophagy in experimental scrapie. Acta neuropathologica. 1991; 82:225-228.
9. Wang CW and Klionsky DJ. The molecular mechanism of autophagy. Molecular medicine (Cambridge, Mass). 2003; 9:65-76.
10. Levine B and Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008; 132:27-42.
11. Glick D, Barth S and Macleod KF. Autophagy: cellular and molecular mechanisms. The Journal of pathology. 2010; 221:3-12.
12. Mizushima N, Yoshimori T and Levine B. Methods in mammalian autophagy research. Cell. 2010; 140:313-326.
13. Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nature reviews Molecular cell biology. 2007; 8:931-937.
14. Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S and Klionsky DJ. In search of an “autophagomometer”. Autophagy. 2009; 5:585-589.
15. Sahani MH, Itakura E and Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014; 10:431-441.
16. Myeku N and Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. The Journal of biological chemistry. 2011; 286:22426-22440.
17. Bartlett BJ, Isakson P, Lewerenz J, Sanchez H, Kotzebue RW, Cumming RC, Harris GL, Nezis IP, Schubert DR, Simonsen A and Finley KD. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy. 2011; 7:572-583.
18. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012; 8:445-544.
19. Narita M. Quality and quantity control of proteins in senescence. Aging (Albany NY). 2010; 2:311-314. doi.10.18632/aging.100145.
20. Yue F, Li W, Zou J, Chen Q, Xu G, Huang H, Xu Z, Zhang S, Gallinari P, Wang F, McKeehan WL and Liu L. Blocking the association of HDAC4 with MAP1S accelerates autophagy clearance of mutant Huntingtin. Aging (Albany NY). 2015; 7:839-853. doi.10.18632/aging.100818.
21. Chapin HC, Okada M, Merz AJ and Miller DL. Tissue-specific autophagy responses to aging and stress in C. elegans. Aging (Albany NY). 2015; 7:419-434. doi. 10.18632/aging.100765.
22. Martini-Stoica H, Xu Y, Ballabio A and Zheng H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends in neurosciences. 2016.
23. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW and Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001; 292:468-472.
24. Sheldon RA, Osredkar D, Lee CL, Jiang X, Mu D and Ferriero DM. HIF-1 alpha-deficient mice have increased brain injury after neonatal hypoxia-ischemia. Developmental neuroscience. 2009; 31:452-458.
25. Saeki Y, Ito Y, Shibata M, Sato Y, Okuda K and Takazoe I. Antimicrobial action of natural substances on oral bacteria. The Bulletin of Tokyo Dental College. 1989; 30:129-135.
26. Jayakumar T, Hsu WH, Yen TL, Luo JY, Kuo YC, Fong TH and Sheu JR. Hinokitiol, a natural tropolone derivative, offers neuroprotection from thromboembolic stroke in vivo. Evidence-based complementary and alternative medicine : eCAM. 2013; 2013:840487.
27. Kim MK, Park HJ, Kim YD, Ryu MH, Takata T, Bae SK and Bae MK. Hinokitiol increases the angiogenic potential of dental pulp cells through ERK and p38MAPK activation and hypoxia-inducible factor-1alpha (HIF-1alpha) upregulation. Archives of oral biology. 2014; 59:102-110.
28. Lee MJ, Kim JW and Yang EG. Hinokitiol activates the hypoxia-inducible factor (HIF) pathway through inhibition of HIF hydroxylases. Biochemical and biophysical research communications. 2010; 396:370-375.
29. Li LH, Wu P, Lee JY, Li PR, Hsieh WY, Ho CC, Ho CL, Chen WJ, Wang CC, Yen MY, Yang SM and Chen HW. Hinokitiol induces DNA damage and autophagy followed by cell cycle arrest and senescence in gefitinib-resistant lung adenocarcinoma cells. PLoS One. 2014; 9:e104203.
30. Wang WK, Lin ST, Chang WW, Liu LW, Li TY, Kuo CY, Hsieh JL and Lee CH. Hinokitiol induces autophagy in murine breast and colorectal cancer cells. Environmental toxicology. 2014.
31. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H and Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. The Journal of cell biology. 2005; 171:603-614.
32. Haga S, Ozawa T, Yamada Y, Morita N, Nagashima I, Inoue H, Inaba Y, Noda N, Abe R, Umezawa K and Ozaki M. p62/SQSTM1 plays a protective role in oxidative injury of steatotic liver in a mouse hepatectomy model. Antioxidants & redox signaling. 2014; 21:2515-2530.
33. Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT and Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer cell. 2008; 13:343-354.
34. Jeong JK, Seo JS, Moon MH, Lee YJ, Seol JW and Park SY. Hypoxia-inducible factor-1 alpha regulates prion protein expression to protect against neuron cell damage. Neurobiology of aging. 2012; 33:1006.e1001-1010.
35. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD and Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005; 122:927-939.
36. Yang Z and Klionsky DJ. An overview of the molecular mechanism of autophagy. Current topics in microbiology and immunology. 2009; 335:1-32.
37. White E and DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clinical cancer research. 2009; 15:5308-5316.
38. Wei K, Wang P and Miao CY. A double-edged sword with therapeutic potential: an updated role of autophagy in ischemic cerebral injury. CNS neuroscience & therapeutics. 2012; 18:879-886.
39. Czyzyk-Krzeska MF, Meller J and Plas DR. Not all autophagy is equal. Autophagy. 2012; 8:1155-1156.
40. Cuervo AM. Autophagy: many paths to the same end. Molecular and cellular biochemistry. 2004; 263:55-72.
41. Mizushima N and Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007; 3:542-545.
42. Duran A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP, Moscat J and Diaz-Meco MT. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Developmental cell. 2004; 6:303-309.
43. Moscat J, Diaz-Meco MT, Albert A and Campuzano S. Cell signaling and function organized by PB1 domain interactions. Molecular cell. 2006; 23:631-640.
44. Rodriguez A, Duran A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM, Serrano M, Auwerx J, Diaz-Meco MT and Moscat J. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell metabolism. 2006; 3:211-222.
45. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V and White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009; 137:1062-1075.
46. Johansen T and Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011; 7:279-296.
47. Nezis IP and Stenmark H. p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxidants & redox signaling. 2012; 17:786-793.
48. Zheng Q, Su H, Ranek MJ and Wang X. Autophagy and p62 in cardiac proteinopathy. Circulation research. 2011; 109:296-308.
49. Ettaiche M, Pichot R, Vincent JP and Chabry J. In vivo cytotoxicity of the prion protein fragment 106-126. The Journal of biological chemistry. 2000; 275:36487-36490.
50. Singh N, Gu Y, Bose S, Kalepu S, Mishra RS and Verghese S. Prion peptide 106-126 as a model for prion replication and neurotoxicity. Frontiers in bioscience. 2002; 7:a60-71.