
INTRODUCTION
Mitochondria are not only vital organelles for producing ATP and intermediates for eukaryotic cancer cells, but they are also major regulators of cell apoptosis and key sources of ROS generation, making them a promising target for cancer therapy [1–3]. Moreover, recent studies showed that targeting mitochondria may inhibit tumorigenesis and tumor growth [2]. Several compounds including clinically applied drugs that inhibit mitochondrial bioenergetic capacity, redox capacity and biosynthetic production have been used for cancer therapy [1, 2].
The mitochondrial respiratory chain comprises five enzyme complexes, and the inhibition of the mitochondrial respiratory chain has demonstrated great value in cancer models [4]. Mitochondrial complex II, known as succinate dehydrogenase (SDH), is the only complex that participates in the Krebs cycle as well as the electron transport chain. Mitochondrial complex II consists of four subunits: SDHA, SDHB, SDHC and SDHD. SDHA exhibits FAD (flavin adenine dinucleotide) and a succinate binding site, which catalyzes the oxidation of succinate to fumarate. The two electrons generated from this progress are transferred to the SDHB subunit. The SDHB contains [2Fe-2S], [4Fe-4S] and [3Fe-4S] iron-sulfur clusters, which deliver the electrons to SDHC and SDHD, resulting in the electrons reduction of UbQ (ubiquinone) to UbQH2 in the presence of heme. The electrons are then passed from complex II to complex III via UbQH2 and may combine with oxygen to induce superoxide generation and apoptosis in cancer cells when UbQ-binding sites were occupied by complex II inhibitors [5, 6].
It has been shown that more than 60% of the anti-cancer agents used today are derived from natural products. Plant-derived anti-cancer agents including vinca alkaloids, podophyllotoxin derivatives, taxanes and campothecin derivatives have been commonly used for the treatment of many types of cancers [7, 8]. Our previous data showed that a new derivative of danshensu (DSS) and tetramethylpyrazine (TMP), namely ADTM, displayed strong cardioprotective effects both in vitro and in vivo [9]. ADTM inhibited platelet aggregation and thrombus formation by targeting ERp57 both in vitro and in vivo [10]. ADTM also conferred relaxation effects on rat mesenteric arteries [11]. Further study showed that ADTM inhibited the growth of breast cancer cells. However, the ester bond of ADTM between TMP and DSS is not stable [12]. To improve the stability and activities of ADTM, a novel conjugate of DSS and TMP, with increased steric hindrance, DT-010, was synthesized. In the present study, the effects of DT-010 on cytotoxicity and cell proliferation of breast cancer cells will be evaluated. We will also investigate the underlying mechanism by examining the mitochondrial respiration, mitochondrial membrane potential, ATP levels, ROS levels and mitochondrial complex II activity of breast cancer cells after DT-010 treatment.
RESULTS
DT-010 inhibited the proliferation of breast cancer cells
Figure 1 showed the structures of DT-010, ADTM and the parental compounds DSS and TMP As shown in Figure 2A and 2B, DT-010 treatment for 24 h inhibited cell proliferation and increased cytotoxicity in MCF-7 and MDA-MB-231 cells in a dose-dependent manner. DT-010 at the indicated concentrations was much more effective than ADTM, DSS, TMP and DSS+TMP in decreasing cell numbers of MCF-7 and MDA-MB-231 cells (Figure 2C and 2D). Figure 2E illustrates that DT-010 treatment can also promote cells cycle arrest in both MCF-7 and MDA-MB-231 cells. There was an increase of cells in the G1 phase with a marked decrease in the S phase of cells after DT-010 treatment.
Figure 1: Chemical structures of DSS, TMP, and DT-010.
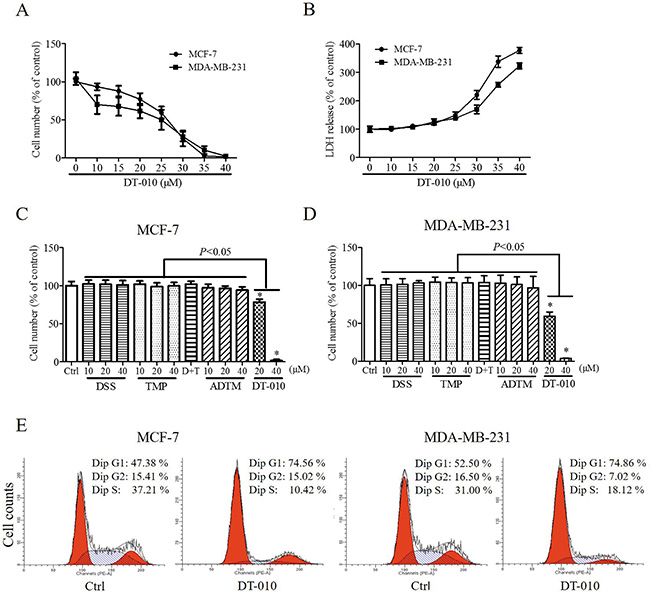
Figure 2: DT-010 inhibited the proliferation of breast cancer cells. Cell numbers (A) and cytotoxicity (B) of MCF-7 and MDA-MB-231 cells were determined after 24 h of DT-010 treatment. The cytotoxicity of cells were measured by lactate dehydrogenase assay. (C and D) Treatment with DT-010 but not ADTM, DSS, TMP or D+T (DSS+TMP) significantly decreased the numbers of MCF-7 and MDA-MB-231 cells. (E) DT-010 induced cell cycle arrest in MCF-7 and MDA-MB-231 cells. Breast cancer cells were stained with PI after 24 h of DT-010 (20 μM) treatment and the cell cycle was analyzed by flow cytometry. Error bars represent mean ± S.D. n = 3. *P < 0.05 vs. Ctrl.
DT-010 inhibited mitochondrial respiration
The effects of DT-010 on the metabolic state of cells were investigated by the Seahorse XF Extracellular Flux Analyzer. After 12 h of DT-010 treatment, the OCR in MCF-7 cells was monitored (Figure 3A). We found that DT-010 significantly inhibited the basal respiration of MCF-7 (Figure 3B). Moreover, DT-010 treatment decreased ATP turnover (Figure 3C) and maximal respiration (Figure 3D) of MCF-7, as compared with the control group. Further studies indicated that continuous treatment with DT-010 decreased the values of OCR after FCCP injection, which could be restored after DT-010 removal (i.e. DT-010 12 h recovery), suggesting that the inhibitory effects of DT-010 on mitochondrial respiration are reversible (Figure 3E).
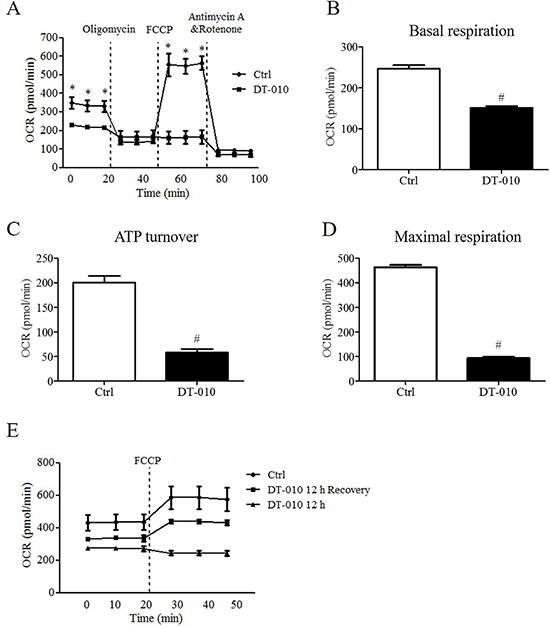
Figure 3: DT-010 inhibited mitochondrial respiration in breast cancer cells. Effects of DT-010 on OCR in MCF-7 cells were determined. MCF-7 cells were treated with DT-010 for 12 h, the OCR values in MCF-7 cells have been monitored with XF24 extracellular flux analyzer (A). A representative graph of OCR showing basal respiration (B), ATP turnover (C) and maximal respiration (D) of MCF-7 cells. (E) The inhibitory effect of DT-010 on mitochondrial respiration in MCF-7 cells was reversible. MCF-7 cells were continuously treated with DT-010 for 12 h (labeled with DT-010 12 h) or the cells were incubated in DT-010 for 12 h followed by 12 h of recovery after removal of DT-010 (labeled with DT-010 12 h recovery). The values of OCR were measured XF24 extracellular flux analyzer. Error bars represent mean ± S.D. n = 3. *P < 0.05 vs. DT-010 treated group. #P < 0.05 vs. Ctrl group.
DT-010 caused mitochondrial dysfunction
The effects of DT-010 on the mitochondrial function of breast cancer cells were determined. Figure 4A and 4B shows that the mitochondrial membrane potential of MCF-7 and MDA-MB-231 cells were decreased after DT-010 treatment. Similarly, treatment of DT-010 attenuated ATP generation in MCF-7 and MDA-MB-231 cells (Figure 4C and 4D). A previous study indicated that cancer cells are more sensitive to mitochondrial dysfunction after glucose deprivation [13]. We investigated whether glucose starvation enhanced DT-010-induced cell death in MCF-7 and MDA-MB-231 cells. Our results showed that DT-010 decreased cell numbers in both cells in glucose-containing medium, which were further enhanced by DT-010 treatment in glucose-free medium (Figure 4E and 4F). The results indicated that DT-010 induced mitochondrial dysfunction.
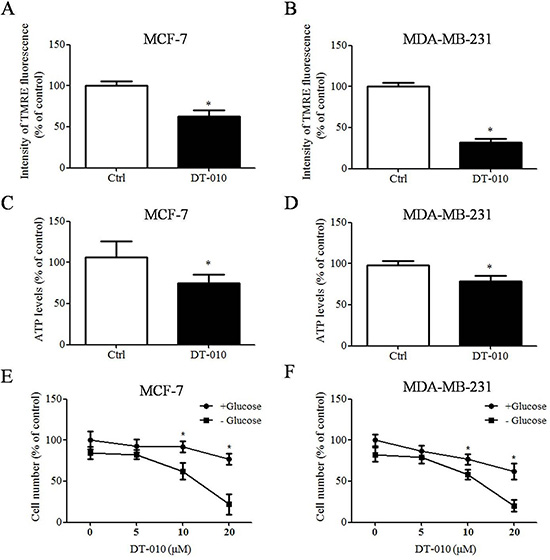
Figure 4: DT-010 induced mitochondrial dysfunction. (A and B) MCF-7 and MDA-MB-231 cells were treated with DT-010 for 24 h. Mitochondrial membrane potential was detected by TMRE staining and measured using flow cytometry (*P < 0.05 vs. Ctrl group). (C and D) DT-010 decreased ATP production in breast cancer cells. The levels of ATP in MCF-7 and MDA-MB-231 cells were measured after 12 h of DT-010 treatment (*P < 0.05 vs. Ctrl group). (E and F) MCF-7 and MDA-MB-231 cells were treated with DT-010 in the glucose-free or glucose-containing medium. Cell numbers were counted after 24 h of DT-010 treatment (*P < 0.05 vs. glucose-deprived group). Error bars represent mean ± S.D. n = 3.
DT-010 induced ROS generation in breast cancer cells
It has been shown that SDH is a major site for ROS generation and the inhibition of SDH results in ROS generation [14, 15, 16]. To investigate whether DT-010 may increase ROS levels, MCF-7 and MDA-MB-231 cells were treated with DT-010 for different time periods. As shown in Figure 5A and 5B, DT-010 boosted ROS generation in a time-dependent manner. On the other hand, DT-010 also caused a time-dependent rise in the levels of mitochondrial superoxide in MCF-7 and MDA-MB-231 cells as evidenced by increased MitoSox fluorescence (Figure 5C and 5D). Furthermore, DT-010 increased cytotoxicity in both MCF-7 and MDA-MB-231 cells which was significantly reversed by NAC, a ROS scavenger (Figure 5E and 5F). These findings imply that DT-010 induced cellular toxicity in breast cancer cells, at least partly via ROS generation.
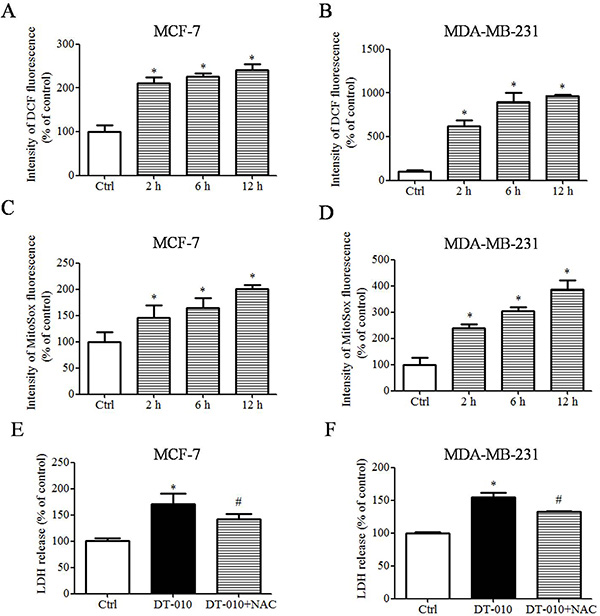
Figure 5: DT-010 induced ROS generation in breast cancer cells. (A and B) After treatment with DT-010 at the indicated time points, MCF-7 and MDA-MB-231 cells were stained with ROS indicator (CM-H2DCFDA) and ROS levels were measured by flow cytometry. (C and D) The levels of mitochondrial superoxide in MCF-7 and MDA-MB-231 cells were measured with MitoSOX red staining and detected by flow cytometry. (E and F) MCF-7 and MDA-MB-231 cells were treated with DT-010 in the presence or absence of 2 mM NAC. The cytotoxicity of cells was determined by LDH assay. Error bars represent mean ± S.D. n = 3. *P < 0.05 vs. Ctrl group. #P < 0.05 vs. DT-010 treated group.
DT-010 inhibited the activity of mitochondrial complex II
Since the OCR of breast cancer cells was inhibited by DT-010, we hypothesized that DT-010 may block mitochondrial respiration by targeting mitochondrial complexes of the electron transport chain. Figure 6A shows that OCR values were increased after injection of saponin, ADP, rotenone and complex II substrate, succinate, which were remarkably inhibited by DT-010 treatment. Western blot analysis showed that DT-010 did not significantly alter the expression of SDHA (succinate dehydrogenase complex, subunit A) protein, a subunit of complex II (Figure 6B). The inhibitory effects of DT-010 on complex II activity have also been confirmed by complex II activity assay. Figure 6C and 6D showed that treatment with DT-010 for 4 h decreased the complex II-mediated MTT reduction in a dose-dependent manner in MCF-7 and MDA-MB-231 cells. These data are consistent with the results showing that mitochondrial complex II activity of MCF-7 and MDA-MB-231 cells significantly decreased after treatment with DT-010, which was measured by Succinate Dehydrogenase Activity Assay kit (Figure 6E and 6F).
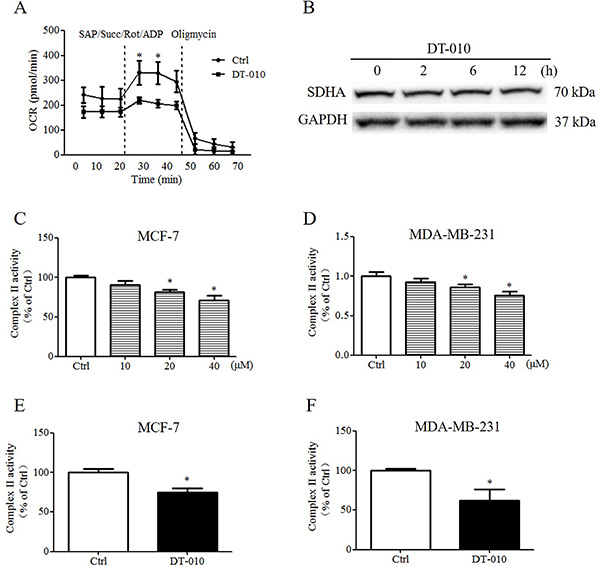
Figure 6: DT-010 inhibited the activity of mitochondrial complex II in breast cancer cells. (A) MCF-7 cells were treated with DT-010 for 12 h, and then saponin, ADP, rotenone and succinate were added to the cells, OCR was monitored by XF24 extracellular flux analyzer (*P < 0.05 vs. DT-010 treated group). (B) The expression of SDHA protein was determined by Western blotting. (C and D) MCF-7 and MDA-MB-231 cells were treated with DT-010 at the indicated concentration for 4 h, the cells were then incubated with MTT in the presence of 20 mM succinate for 2 h. The formazan crystals were then dissolved in DMSO and the absorbance was measured at 570 nm (*P < 0.05 vs. Ctrl group). Breast cancer cells were collected after treatment with DT-010 for 12 h, the inhibitory effects of DT-010 on the complex II activity of MCF-7 (E) and MDA-MB-231 (F) cells were confirmed by measuring the absorbance of 2, 6-dichlorophenol-indophenol (DCPIP) at 600 nm in the absence or presence of DT-010 according to Succinate Dehydrogenase Activity Assay kit (*P < 0.05 vs. Ctrl group). Error bars represent mean ± S.D. n = 3.
Antitumor effects of DT-010 on breast cancer cells in vivo
To study the antitumor effects of DT-010 in vivo, the compound was intraperitoneally injected into Balb/c mice bearing breast tumors for 13 days. DT-010 treatment resulted in a significant reduction in both the volume and weight of tumor cells (Figure 7).
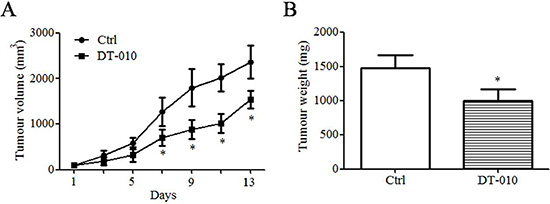
Figure 7: DT-010 suppressed breast tumor growth in vivo. (A) Antitumor effect of DT-010 on 4T1 breast cancer. Tumor sizes were measured every 2 days (n = 5 in Ctrl group; n = 6 in DT-010-treated group). (B) The tumors were excised and weighed after mice were sacrificed. Error bars represent mean ± S.D. n = 3. *P < 0.05 vs. Ctrl group.
DISCUSSION
Danshensu (DSS) is an active ingredient of the Chinese herb Salvia miltiorrhiza (Danshen), which not only shows cardioprotective effects including inhibition of platelet aggregation and myocardial protection against ischemia/reperfusion in vivo [17, 18], but also displays anti-tumor effects in melanoma [19]. On the other hand, tetramethylpyrazine (TMP), also known as ligustrazine, is a major active component of the Chinese medicinal herb Chuanxiong (Ligusticum Wallichii Franch). The anti-cancer properties of TMP have been observed in many types of cancer. It was shown that TMP induced ROS generation and activated mitochondrial-mediated apoptotic pathway in human gastric cancer [20]. TMP may inhibit proliferation of lymphocytic leukemia and gastric carcinoma cells [21, 22]. TMP also showed anti-fibrotic activities by promoting cell cycle arrest and inducing mitochondrial-dependent apoptosis in hepatic stellate cells [23]. In our previous study, ADTM, aconjugate of DSS and TMP, was synthesized, which is more potent than its parental compounds in protecting against t-BHP-induced H9c2 cells injury in vitro [9]. Furthermore, we have found that the stability and activities of ADTM may improve after increasing bulky groups between DSS and TMP [24]. Based on these findings, DT-010 was synthesized by substitution with two allyl groups at the linkage position between DSS and TMP. In the present study, we found that DT-010 at the indicated concentrations displayed stronger anti-proliferation effects in breast cancer cells as compared with ADTM, DSS, TMP alone or DSS and TMP combination (Figure 2C and 2D). DT-010 but not DSS or TMP induced LDH release in MCF-7 and MDA-MB-231 cells (Figure 2B and Supplementary Figure S1A and S1B). However, DT-010 treatment for 24 h did not induce apoptosis in MCF-7 cells and MDA-MB-231 cells (Supplementary Figure S2).
Controlling mitochondrial metabolism and function are emerging strategies for cancer therapy [2, 13]. Mitochondrial targeted compounds, Mito-chromanol (Mito-ChM) and Mito-chromanol acetate (Mito-ChMAc), have been reported to inhibit mitochondrial energy metabolism, leading to increased cytotoxicity and decreased cell proliferation in breast cancer cells [25]. A small molecule named VLX600 displays potent antitumor effects both in vitro and in vivo, decreasing mitochondrial respiration and inducing mitochondrial dysfunction by inhibition of mitochondrial complex I, II and IV [13]. Fenofibrate, a lipid-lowering drug, showed anticancer effects in vitro and in vivo through promoting mitochondrial respiration impairment and dysfunction via complex I inhibition [26].
In the present study, the effects of DT-010 on mitochondrial metabolism and function were investigated. To detect the subcellular distribution of DT-010 in cells, a biotinylated-DT-010 analogue, named BAA, was applied, which was synthesized in our previous study [10]. We found that BAA also displayed anti-tumor effects in MCF-7 cells at higher concentrations (Supplementary Figure S3A). The fluorescence of streptavidin-FITC-BAA has been shown to accumulate in the mitochondria of MCF-7 cells (Supplementary Figure S3B), which suggests that DT-010 can enter the mitochondria of cells. Our results showed that mitochondrial function, including basal respiration, ATP turnover and maximal respiration, was significantly inhibited after DT-010 treatment (Supplementary Figure 3A, 3B, 3C and 3D), associated with a decrease of mitochondrial membrane potential (Figure 4A and 4B) and ATP levels (Figure 4C and 4D) in breast cancer cells, indicating that DT-010 may inhibit mitochondrial metabolism and promote mitochondrial dysfunction in those cells. In contrast, ATP levels in breast cancer cells did not change after DSS or TMP treatment, which suggested that DSS or TMP alone at the indicated concentration did not affect the mitochondrial function of breast cancer cells (SupplementaryFigure S1C and S1D). Previous studies showed that cancer cells with mitochondrial dysfunction are more sensitive to glucose starvation [13]. This was consistent with our data showing that the toxicity of DT-010 in MCF-7 and MDA-MB-231 cells was increased in glucose-free medium (Figure 4E and 4F). In the present study, OCR values, an indicator of mitochondrial respiration, was also lower after DT-010 treatment in normal cell line H9c2 myoblasts (Supplementary Figure S4). Previous reports indicated that the basic respiration of MCF-7 cells was higher than that of normal kidney epithelial cells. Moreover, the basic respiration of cancer cells such as glioblastoma cell lines, LN-229 and U-87MG, was much higher than that of normal human astrocytes, which could be the reason that LN-229 and U-87MG were more sensitive to fenofibrate [26, 27]. Similarly, our results showed the basic and maximal respiration of H9c2 cells were much lower than those of MCF-7 cells. Differences in basic and maximal respiration levels of H9c2 and MCF-7 cells may contribute to their sensitivity to DT-010. On the other hand, It was shown that DT-010 treatment at the indicated concentrations did not induce ROS generation in H9c2 cells and HUVEC (Supplementary Figure S5B and S5C), which may also contribute to our findings that H9c2 cells and HUVEC are more tolerant to DT-010 compared to cancer cells (Supplementary Figure S5A). Also, in a xenograft mice model in vivo, DT-010 treatment resulted in a significant reduction in both the volume and weight of tumor cells (Figure 7). However, DT-010 treatment did not significantly decrease the body weight and survival rate of mice, as compared with control group (data not shown).
In this study, we further investigated the molecular targets involved in the mitochondrial respiration deficiency and mitochondrial dysfunction triggered in breast cancer cells by DT-010. Mitochondrial complex II appears to be a novel target for cancer therapy, and the inhibition of mitochondrial complex II may result in anti-cancer activities [6, 14, 16]. α-TOS, an analogue of vitamin E, targets a number of cancer cells including breast cancer, lung cancer, and colon cancer – as well as mesothelioma – and has less toxicity toward to normal cells. α-TOS may induce ROS mediated-apoptosis in cancer cells via targeting mitochondrial complex II, which may be inhibited after the mutation of complex II [14]. On a limited scale, a clinical study showed that the life expectancy of a mesothelioma patient was improved after receiving α-TOS [6]. MitoVES is another analogue of vitamin E, which mainly accumulates in the mitochondria of cells and is more potent than α-TOS in inducing ROS generation in vitro and decreasing tumor volume in vivo [6]. Similarly, our results showed that DT-010 inhibited succinate-induced mitochondrial respiration and the activity of mitochondrial complex II enzyme (Figure 6), suggesting that it may promote mitochondrial respiration deficiency and mitochondrial dysfunction in breast cancer cells via mitochondrial complex II inhibition. Treatment with DT-010 also triggered ROS and mitochondrial superoxide generation in a time-dependent manner in MCF-7 and MDA-MB-231 cells (Figure 5A–5D), while the cytotoxicity mediated by DT-010 was reduced after NAC co-treatment (Figure 5E and 5F).
In conclusion, our findings demonstrated that DT-010 displayed antitumor effects in breast cancer cells both in vivo and in vitro via ROS generation and mitochondrial dysfunction, as mediated by the inhibition of mitochondrial complex II. The study suggests that DT-010 represents a promising candidate for the development of anticancer agents.
MATERIALS AND METHODS
DT-010 was synthesized at Jinan University, China. DSS and TMP were purchased from Xi’an Honson Biotechnology (China) and Shanghai Banghai Chemical Company (China), respectively. The purity of DT-010 was > 98%. Fetal bovine serum (FBS), Dulbecco’s Modified Eagle’s Medium (DMEM) and penicillin/streptomycin were purchased from Gibco Life Technologies (USA). 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazoliumbromide (MTT) was purchased from Sigma-Aldrich (USA). A Cytotoxicity Detection Kit was purchased from Roche Applied Science (Germany). Tetramethylrhodamine, ethyl ester (TMRE), MitoSox Red Mitochondrial Superoxide Indicator and 5-(and-6-)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) were purchased from Life Technologies (USA). An XF Cell Mito Stress Test Kit was purchased from Seahorse Bioscience (USA).
Cell cultures
MCF-7, MDA-MB-231, 4T1 and H9c2 cells were derived from ATCC and cultured in DMEM (high glucose) medium supplemented with 10% FBS at 37°C, 5% CO2 in a humidified incubator. HUVECs (human umbilical endothelial cells) were purchased from Life Technologies and cultured in F-12K medium supplemented with 10% FBS and 0.1 mg/ml heparin. Cells were used when they reached 70%–80% confluence.
Measurement of cellular toxicity, cell numbers and ATP level
Cellular toxicity was determined by lactate dehydrogenase assay. MCF-7 and MDA-MB-231 cells were seeded into 96-well plates at a density of 6 × 103/well for 24 h and were then treated with different concentrations of DT-010 for 24 h. The LDH released into the culture medium was measured by Cytotoxicity Detection Kit according to the manufacturer’s protocol. The absorbance was detected at 490 nm using a SpectraMax M5 plate reader (Molecular Device, USA). Cell numbers were determined as described previously [28]. Briefly, cells were stained with DAPI after fixation and were visualized by In Cell Analyzer 2000 system. Cell numbers were calculated by the Developer Toolbox software. MCF-7 and MDA-MB-231 cells were treated with DT-010 (20 μM) for 12 h. ATP levels were measured by ATP assay from Promega (USA) and luminescence was detected using a SpectraMax M5 plate reader.
Determination of cell cycle by flow cytometry
MCF-7 and MDA-MB-231 cells were plated on 6-well plates at 1.5 × 105 cells/well. After 24 h of DT-010 treatment, the cells were fixed with 70% ethanol (in water) overnight at –20°C. After fixation, the cells were collected and stained with PI (50 μg/ml) in the presence of RNase A (200 μg/ml) for 15 minutes at room temperature. The samples were then analyzed using the PE channel of a BD FACSCanto Flow Cytometer (BD Biosciences, USA).
Measurement of ROS level and mitochondrial membrane potential
MCF-7 and MDA-MB-231 cell suspension was added to each well of 6-well plates (1.5 × 105 cells/well) and cultured in an incubator for 24 h. After treatment with DT-010 for the indicated time periods, cells were collected and loaded with CM-H2DCFDA (10 μM), MitoSox Red (5 μM) and TMRE (50 nM) for the measurement of intracellular ROS level, mitochondrial superoxide production and mitochondrial membrane potential, respectively. The levels of ROS, mitochondrial superoxide production and mitochondrial membrane potential were measured by the BD FACSCanto Flow Cytometer.
Assessment of metabolic parameters
Changes in the oxygen consumption rate (OCR) of cells was monitored by an Extracellular Flux Analyzer as previously described [26]. Cells were plated into Seahorse 24-well tissue culture microplates at a density of 1 × 104 cells/well for 24 or 36 h. After 12 h of DT-010 treatment, the cells were washed and cultured in Seahorse base medium containing 4.5 g/L glucose (pH = 7.4) and then incubated in non-CO2 incubator (37°C) for 45–60 minutes. The OCR values were monitored after the injection of metabolic reagents from the XF Cell Mito Stress Test Kit. The parameters of mitochondrial function were calculated as previously described [29]. Basal respiration (OCR values of baseline respiration-OCR values of antimycin A/rotenone post injection), ATP turnover (OCR values of baseline respiration-OCR values of oligomycin post injection) and maximal respiration (OCR values of FCCP post injection-OCR values of antimycin A/rotenone post injection). The complex II-meditated respiratory activity was measured as previous described [30]. Briefly, cells were incubated in MAS-BSA solution (70 mM sucrose, 220 mM Mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA and 4 mg/ml BSA) and co-injected with saponin, ADP, rotenone and succinate. The OCR values were monitored after the injection. The final concentrations of substrates were as follows: saponin 10 mM, ADP 1 mM, rotenone 1 μM and succinate 10 mM.
Mitochondrial complex II activity
The complex II activity of MCF-7 and MDA-MB-231 cells was measured by MTT assay and SDH activity assay kit. According to the MTT assay described previously [14], cells were seeded into 96-well plates at a density of 6 × 103/well for 24 h. The cells were exposed to 1 mg/ml MTT in DMEM medium containing 20 mM succinic acid for 2 h at 37°C. The formazan was then dissolved in 150 μL DMSO and the absorbance was measured at 570 nm. For Mitochondrial complex II activity assay, MCF-7 and MDA-MB-231 cells were cultured in 6-well plates for 24 h. The cells were collected and the activity was detected according to Succinate Dehydrogenase Activity Assay kit (BioVision). The absorbance was measured at 600 nm.
Western blotting
Whole cell protein samples were prepared and separated by SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) and transferred onto polyvinylidene fluoride membrane. The membrane then was incubated overnight at 4°C with the primary antibodies against GAPDH and SDHA (Cell Signaling). After incubation with secondary horseradish peroxidase-conjugated anti-rabbit antibodies, the blots were developed with the Enhanced ECL System (GE Healthcare).
Animal experiments
Female Balb/c mice were purchased from Guangdong Medical Laboratory Animal Center. Tumors were generated by subcutaneous injection of 1 × 105 of 4T1 cells with 100 μL of PBS. After tumors developed, mice were treated with 40 mg/kg DT-010 in PEG400/5% ethanol by intraperitoneal injection every 3 days. Control mice were injected with an equal quantity of vehicle. Tumor sizes were measured every 2 days and tumor volume was calculated by the formula ab2/2 (where a and b are the long and short axes of tumor, respectively). After treatment for 13 days, the mice were sacrificed and the tumors were weighed.
Statistical analysis
Data are presented as means ± standard deviation (SD). One-way ANOVA followed by Turkey’s multiple comparison tests were used to compare the differences between the groups. A P-value < 0.05 was considered as statistically significant.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the grants from Research Committee of University of Macau (Grant No. MYRG139(Y1-L4)-ICMS12-LMY, MYRG2015-00182-ICMS-QRCM, MYRG2015-00214-ICMS-QRCM and MRG022/DLJ/2015/FHS) and Science and Technology Development Fund of Macau SAR (Grant No. FDCT/025/2014/A1 and FDCT/134/2014/A3).
CONFLICTS OF INTEREST
None.
REFERENCES
1. Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010; 9:447–464.
2. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015; 11:9–15.
3. Byun HO, Kim HY, Lim JJ, Seo YH, Yoon G. Mitochondrial dysfunction by complex II inhibition delays overall cell cycle progression via reactive oxygen species production. J Cell Biochem. 2008; 104:1747–1759.
4. Rohlena J, Dong LF, Neuzil J. Targeting the mitochondrial electron transport chain complexes for the induction of apoptosis and cancer treatment. Curr Pharm Biotechnol. 2013; 14:377–389.
5. Schwall CT, Greenwood VL, Alder NN. The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim Biophys Acta. 2012; 1817:1588–1596.
6. Kluckova K, Bezawork-Geleta A, Rohlena J, Dong L, Neuzil J. Mitochondrial complex II, a novel target for anti-cancer agents. Biochim Biophys Acta. 2013; 1827:552–564.
7. Demain AL, Vaishnav P. Natural products for cancer chemotherapy. Microbiol Biotechnol. 2011; 4:687–699.
8. Cragg GM, Grothaus PG, Newman DJ. Impact of natural products on developing new anti-cancer agents. Chem Rev. 2009; 109:3012–3043.
9. Cui G, Shan L, Hung M, Lei S, Choi I, Zhang Z, Yu P, Hoi P, Wang Y, Lee SM. A novel Danshensu derivative confers cardioprotection via PI3K/Akt and Nrf2 pathways. Int J Cardiol. 2013; 168:1349–1359.
10. Cui G, Shan L, Guo L, Chu IK, Li G, Quan Q, Zhao Y, Chong CM, Zhang Z, Yu P, Hoi MP, Sun Y, Wang Y, et al. Novel anti-thrombotic agent for modulation of protein disulfide isomerase family member ERp57 for prophylactic therapy. Sci Rep. 2015; 5:10353.
11. Li RW, Yang C, Shan L, Zhang Z, Wang Y, Kwan YW, Lee SM, Hoi MP, Chan SW, Cheung AC, Cheung KH, Leung GP. Relaxation effect of a novel Danshensu/tetramethylpyrazine derivative on rat mesenteric arteries. Eur J Pharmacol. 2015; 761:153–160.
12. Li S, Shan L, Zhang Z, Li W, Liao K, Sheng X, Yu P, Wang Y. Pharmacokinetic, Metabolic Studies of ADTM: A Novel Danshensu Derivative Confers Cardioprotection by HPLC-UV and LC-MS/MS. J Chromatogr Sci. 2015; 53:872–878.
13. Zhang X, Fryknas M, Hernlund E, Fayad W, De Milito A, Olofsson MH, Gogvadze V, Dang L, Pahlman S, Schughart LA, Rickardson L, D’Arcy P, Gullbo J, et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat Commun. 2014; 5:3295.
14. Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK, Freeman R, Swettenham E, Valis K, Liu J, Zobalova R, Turanek J, Spitz DR, et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene. 2008; 27:4324–4335.
15. Dong LF, Jameson VJ, Tilly D, Cerny J, Mahdavian E, Marin-Hernandez A, Hernandez-Esquivel L, Rodriguez-Enriquez S, Stursa J, Witting PK, Stantic B, Rohlena J, Truksa J, et al. Mitochondrial targeting of vitamin E succinate enhances its pro-apoptotic and anti-cancer activity via mitochondrial complex II. J Biol Chem. 2011; 286:3717–3728.
16. Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012; 287:27255–27264.
17. Yin Y, Guan Y, Duan J, Wei G, Zhu Y, Quan W, Guo C, Zhou D, Wang Y, Xi M, Wen A. Cardioprotective effect of Danshensu against myocardial ischemia/reperfusion injury and inhibits apoptosis of H9c2 cardiomyocytes via Akt and ERK1/2 phosphorylation. Eur J Pharmacol. 2013; 699:219–226.
18. Yu C, Qi D, Lian W, Li QZ, Li HJ, Fan HY. Effects of danshensu on platelet aggregation and thrombosis: in vivo arteriovenous shunt and venous thrombosis models in rats. PloS one. 2014; 9:e110124.
19. Zhang LJ, Chen L, Lu Y, Wu JM, Xu B, Sun ZG, Zheng SZ, Wang AY. Danshensu has anti-tumor activity in B16F10 melanoma by inhibiting angiogenesis and tumor cell invasion. Eur J Pharmacol. 2010; 643:195–201.
20. Yi B, Liu D, He M, Li Q, Liu T, Shao J. Role of the ROS/AMPK signaling pathway in tetramethylpyrazine-induced apoptosis in gastric cancer cells. Oncol Lett. 2013; 6:583–589.
21. Wang XJ, Xu YH, Yang GC, Chen HX, Zhang P. Tetramethylpyrazine inhibits the proliferation of acute lymphocytic leukemia cell lines via decrease in GSK-3beta. Oncol Rep. 2015; 33:2368–2374.
22. Ji AJ, Liu SL, Ju WZ, Huang XE. Anti-proliferation effects and molecular mechanisms of action of tetramethypyrazine on human SGC-7901 gastric carcinoma cells. Asian Pac J Cancer Prev. 2014; 15:3581–3586.
23. Zhang F, Kong DS, Zhang ssZL, Lei N, Zhu XJ, Zhang XP, Chen L, Lu Y, Zheng SZ. Tetramethylpyrazine induces G0/G1 cell cycle arrest and stimulates mitochondrial-mediated and caspase-dependent apoptosis through modulating ERK/p53 signaling in hepatic stellate cells in vitro. Apoptosis. 2013; 18:135–149.
24. Sun YW, Tan ZC, Liang ZB, Wang L, Shan LC, Yu P, Lee SM, Wang YQ. Synthesis and relationship of stability and biological activity of new DSS and TMP conjugates. Medchemcomm. 2015; 6:586–591.
25. Cheng G, Zielonka J, McAllister DM, Mackinnon AC, Jr., Joseph J, Dwinell MB, Kalyanaraman B. Mitochondria-targeted vitamin E analogs inhibit breast cancer cell energy metabolism and promote cell death. BMC cancer. 2013; 13:285.
26. Wilk A, Wyczechowska D, Zapata A, Dean M, Mullinax J, Marrero L, Parsons C, Peruzzi F, Culicchia F, Ochoa A, Grabacka M, Reiss K. Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol Cell Biol. 2015; 35:182–198.
27. Nilsson H, Lindgren D, Mandahl Forsberg A, Mulder H, Axelson H, Johansson ME. Primary clear cell renal carcinoma cells display minimal mitochondrial respiratory capacity resulting in pronounced sensitivity to glycolytic inhibition by 3-Bromopyruvate. Cell Death Dis. 2015; 6:e1585.
28. Wang L, Zhang X, Chan JY, Shan L, Cui G, Cui Q, Wang Y, Li J, Chen H, Zhang Q, Yu P, Han Y, Lee SM. A novel danshensu derivative prevents cardiac dysfunction and improves the chemotherapeutic efficacy of doxorubicin in breast cancer cells. J Cell Biochem. 2015.
29. Lopert P, Day BJ, Patel M. Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. PloS one. 2012; 7:e50683.
30. Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc. 2014; 9:421–438.