
INTRODUCTION
Esophageal cancer exhibits a common malignancy with a poor prognosis, ranking the 10th most common cancers and taking a toll of ~400,000 deaths in 2012 worldwide [1], and esophageal squamous cell carcinoma (ESCC), accounting for ~90% esophageal cancer, is the predominant histological subtype throughout the world [2]. The hugest burden of ESCC occurs in the “Asian esophageal cancer belt”, which extends from Caspian littoral of Iran, east to China, and north to Russia [3]. Our previous epidemiological study has shown an extremely high incidence of ESCC (74.47/100,000) in Nan’ao Island of Shantou, an isolated Chaoshan littoral region of southern China, from 1995 to 2004 [4], highlighting an urgent need for deeper understanding of its pathogenesis. In most cases, the development of ESCC is associated with esophageal squamous dysplasia. As a consequence, esophageal squamous dysplasia is regarded as the precursor lesion and risk factor for ESCC. A 13-year prospective follow-up study has demonstrated that patients with mild, moderate, or severe dysplasia have a risk of ESCC that is increased by a factor of 2.9, 9.8, or 28.3, respectively [5]. Hence, it is of great importance to explore the underlying mechanisms for the development of esophageal dysplasia.
The association between chronic inflammation and tumorigenesis was first proposed by Rudolf Virchow over a century ago, when he noticed the presence of inflammatory cells in neoplastic tissues [6]. Since then, accumulating evidence from preclinical and clinical studies has shown that recurrent and persistent chronic inflammation may be a causative factor for oncogenesis [7]. It has previously been estimated that chronic infection and related inflammation contributed to approximately 20% to 25% of all cancer cases worldwide [8].
Chronic inflammation-induced reactive oxygen and nitrogen species (RONS) could cause damage to important cellular components (e.g., DNA, proteins and lipids), which consequently contributes to malignant cell transformation [9]. According to our previous study regarding chronic inflammation-related DNA damage response (DDR) in gastric cardia carcinogenesis [10], the level of DDR was increased with the progression of precancerous lesions in human gastric cardia tissues.
All the aforementioned evidence begs the question of how chronic inflammation influences tumor initiation. In the study presented herein, we therefore investigated the relationship between DNA damage status and degree of chronic inflammation as well as precursor lesions in human esophageal tissues. Our findings demonstrated that the level of DNA damage was positively correlated with degree of chronic inflammation and precursor lesions. Taken together, chronic inflammation-related DNA damage (genomic instability) may trigger the initiation of esophageal squamous dysplasia.
RESULTS
Chronic inflammation correlated with esophageal precancerous lesions
To clarify the role of chronic inflammation during the course of esophageal carcinogenesis, we examined a total of 109 endoscopic esophageal biopsies from non-tumor individuals (Table 1). We noticed that esophageal precancerous lesions (e.g., mild, moderate, and severe dysplasia) were frequently accompanied by varying degree of chronic inflammation. The above histological evidence aroused our attention on the association between chronic inflammation and esophageal tumorigenesis. As expected, severe chronic inflammation was commonly observed in severe dysplastic esophageal epithelia (Figure 1A–1D). Correlation analysis revealed that chronic inflammation was positively correlated with histological severity (rs = 0.37, P < 0.01) (Figure 1E). Interestingly, we found that inflammatory cells migrated into esophageal squamous epithelia, and the neighboring epithelial cells constantly exhibited dysplastic changes (Figure 1F). One possible explanation could be that chronic inflammation may serve as a critical role in malignant transformation of esophageal epithelial cells instead of an accidental phenomenon.
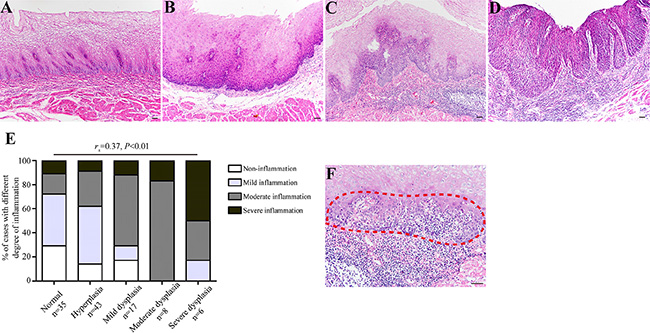
Figure 1: The degree of chronic inflammation correlates with esophageal histological severity. (A–D) Representative images showing normal epithelium without evident inflammation (Panel A), hyperplastic epithelium with mild inflammation (Panel B), mild dysplastic epithelium with severe inflammation (Panel C), and severe dysplastic epithelium with severe inflammation (Panel D), respectively. (E) Esophageal precursor lesions were associated with the degree of chronic inflammation. For statistical analysis, Spearman rank correlation analysis was used, with P values indicated in the figure. (F) The association between chronic inflammation and esophageal histological severity was evidenced by microscopic appearance of intraepithelial inflammation, intraepithelial infiltrating inflammatory cells and dysplastic esophageal epithelial cells are indicated with red dashed line. Scale bars correspond to 50 μm in A–D and F.
Table 1: Clinicopathological data of 109 endoscopic biopsy specimens
Variable |
Value |
|---|---|
Age, yrs, n = 109 |
54.2 ± 16.2 |
Sex, n = 109 |
|
Male (%) |
71 (65) |
Female (%) |
38 (35) |
Histological condition, n = 109 |
|
Normal (%) |
35 (32) |
Hyperplasia (%) |
43 (39) |
Mild dysplasia (%) |
17 (16) |
Moderate dysplasia (%) |
8 (7) |
Severe dysplasia (%) |
6 (6) |
Degree of inflammation, n = 109 |
|
Normal (%) |
19 (17) |
Mild (%) |
38 (34) |
Moderate (%) |
36 (34) |
Severe (%) |
16 (15) |
Oxidative DNA damage correlated with chronic inflammation and histological severity
Although inflammation is a critical function of the innate immune system, chronic inflammation is often accompanied by the excessive formation of reactive oxygen species (ROS) that may potentially inflict damage on nucleic acids, proteins and lipids in neighboring healthy epithelial and stromal cells [11]. As a result, this process can lead to mutations in tumor-related genes, which increases cancer risk. We, therefore, speculate that ROS-induced oxidative DNA damage may explain the close relationship between chronic inflammation and esophageal precancerous lesions. We then stained all the 109 cases of endoscopic esophageal biopsies using anti-8-OHdG antibody for detection of oxidative DNA damage. Intriguingly, the immunostaining intensity of 8-OHdG was prominently stronger in tissues with higher degree of chronic inflammation, whereas only a small fraction of non-inflammation tissues showed positive immunostaining (Figure 2A–2E). Correlation analysis of the immunohistochemical results demonstrated that the immunostaining intensity of 8-OHdG was positively correlated with the degree of chronic inflammation (rs = 0.21, P = 0.03) (Figure 2F), indicating that higher level of oxidative DNA damage occurred during chronic inflammatory process, particularly in the setting of severe chronic inflammation.
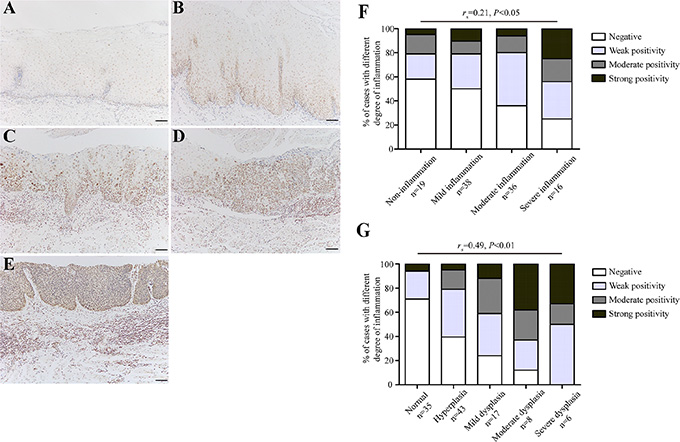
Figure 2: The level of oxidative DNA damage correlates with the degree of chronic inflammation as well as esophageal histological severity. (A–E) Representative images showing 8-OHdG immunostaining in tissues with non-inflammtion (normal tissue) (Panel A), mild inflammation (hyperplasia) (Panel B), moderate inflammation (mild dysplasia) (Panel C), moderate inflammation (severe dysplasia) (Panel D), and severe inflammation (severe dysplasia) (Panel E), respectively. (F–G) Correlation analysis for the level of oxidative DNA damage and the degree of chronic inflammation (F), as well as esophageal histological severity (G). For statistical analysis, Spearman rank correlation analysis was used, with P values indicated in the figure. Scale bars correspond to 50 μm in A–E.
Based on aforementioned results, we have reasons to hypothesize that oxidative DNA damage ought to be evident in esophageal preneoplastic lesions in case of its involvement in tumor initiation. To clarify this, we evaluated the oxidative DNA damage status in esophageal epithelial tissues grouped by different pathological changes. Being consistent with our assumption, the level of oxidative DNA damage increased progressively in the sequential stages from histologically normal esophageal epithelia to dysplastic esophageal epithelia (Figure 2A–2E). In addition, correlation analysis demonstrated a significantly positive association between oxidative DNA damage and esophageal precancerous lesions (rs = 0.49, P < 0.01) (Figure 2G). These results suggest that chronic inflammation-related oxidative DNA damage probably triggers the initiation of esophageal carcinogenesis.
The level of DSBs increased with esophageal histological severity
We have confirmed that the accumulation of oxidative DNA damage in inflamed or dysplastic esophageal tissues. Additionally, we have previously reported that chronic inflammation-related DNA damage response is significantly increased in dysplastic tissues of human gastric cardia compared with that in normal gastric cardia tissues [10]. The phosphorylation of serine 139 in the C-terminal tail of the histone H2A variant, γH2AX, is a rapid and sensitive cellular response to the presence of DSBs [12], and its aggregation, in a way, is a predictor of genomic instability [13]. Hence, we attempted to evaluate the status of DSBs in esophageal tissues with different histological severity during esophageal carcinogenic process (Table 2). Importantly, immunostaining of γH2AX was evident in dysplastic epithelial cells, especially in severe dysplastic epithelial cells, whereas the immunoreactivity was almost absent in histologically normal tissues (Figure 3A–3D). Immunohistochemical analysis also confirmed this phenomenon with statistical significance (P < 0.001) (Figure 3E). Similarly, immunohistochemical analysis of γH2AX in tissues with different inflammation status demonstrated that the positive rates of γH2AX were significantly higher in samples affected by severe inflammation than that in tissues with milder inflammation (P < 0.01) (Figure 3F).
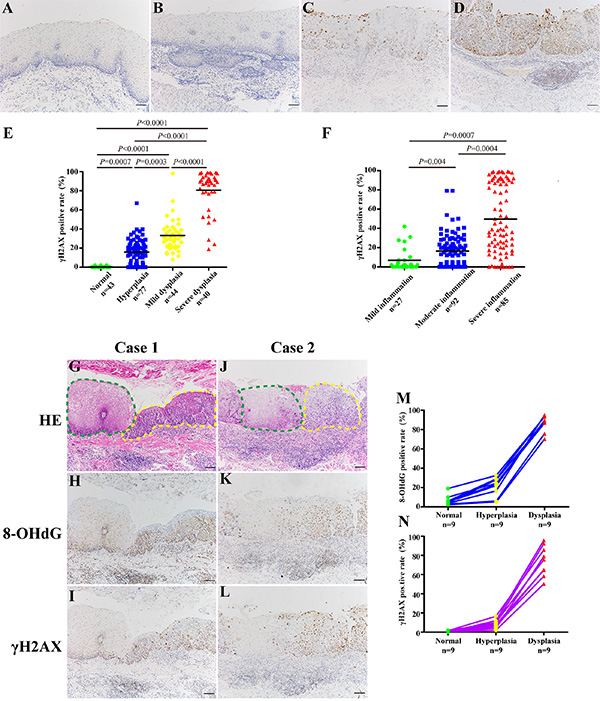
Figure 3: The level of DSBs correlates with esophageal histological severity. (A–D) Representative images of γH2AX immunostaining in normal epithelium (Panel A), hyperplastic epithelium (Panel B), mild dysplastic epithelium (Panel C), and severe dysplastic epithelium (Panel D), respectively. (E–F) Quantification of γH2AX positive cells in tissue samples with different histological severity (E) and different degree of chronic inflammation (F). For statistical analysis, one-way ANOVA was used, with P values indicated in the figure. Tamhane’s T2 tests were selected to identify specific differences between each two groups. For multiple comparisons, the P values of or less than 0.008 (E) or 0.016 (F) were considered statistically significant adjusted by Bonferroni correction. Black bars indicate mean values. (G–L) Representative images showing immunostaining of 8-OHdG and γH2AX in samples with transitional pathological changes from the same ESCC patient (Case 1 and Case 2). Green dashed line indicates normal epithelium; yellow dashed line indicates dysplastic epithelium. (M, N) Quantification of 8-OHdG (M) and γH2AX (N) positive cells in samples with transitional pathological changes from the same patient. Scale bars correspond to 50 μm in A–D, G–L.
Table 2: Clinicopathological data of 204 tumor-surrounding non-malignant samples
Variable |
Value |
|---|---|
Age, yrs, n = 204 |
59.7 ± 9.1 |
Sex, n = 204 |
|
Male (%) |
168 (82) |
Female (%) |
36 (18) |
Histological condition, n = 204 |
|
Normal (%) |
43 (21) |
Hyperplasia (%) |
77 (38) |
Mild dysplasia (%) |
44 (21) |
Severe dysplasia (%) |
40 (20) |
Degree of inflammation, n = 204 |
|
Normal (%) |
0 (0) |
Mild (%) |
27 (13) |
Moderate (%) |
92 (45) |
Severe (%) |
85 (42) |
DNA damage in samples with different histological severity from the same ESCC patient
To rule out the potential influence of genetic background, we selected samples with transitional pathological changes from 9 ESCC patients to explore the DNA damage status during esophageal carcinogenic process. Not surprisingly, both the level of oxidative DNA damage and DSBs increased with esophageal histological severity (Figure 3G–3L). Immunohistochemical analysis revealed that immunostaining of 8-OHdG and γH2AX reached significantly higher positive rates than that in normal tissues (P < 0.05) (Figure 3M–3N). These data provide strong pathological evidence that DNA damage may play critical role in the initiation of esophageal malignant transformation.
DISCUSSION
The present work was designed to evaluate the association between chronic inflammation-related DNA damage and esophageal premalignant lesions. In this study, we observed significantly positive correlation between chronic inflammation and esophageal precursor lesions. Subsequently, immunohistochemical analyses demonstrated that the levels of oxidative DNA damage and DSBs, evident as positive immunostaining of 8-OHdG and γH2AX, respectively, paralleled with esophageal histological severity. These findings suggest that chronic inflammation-related genomic instability may trigger the malignant transformation of esophageal epithelial cells.
Chronic inflammation has long been considered as an important risk factor for many human cancers [14–17]. Our present study shows a close association between chronic inflammation and esophageal precancerous lesions, providing histological basis for our further investigation on the link between chronic inflammation and esophageal carcinogenesis.
Histopathological features of chronic inflammation are predominance of macrophages and lymphocytes. A prolonged inflammatory microenvironment provides a constant supply of ROS, leading to the formation of oxidative DNA damage. According to previous investigations, the level of oxidative DNA damage is found increased in the inflamed gastric mucosae of Helicobacter pylori-infected people and in the inflamed colonic mucosae of rats [18, 19]. Similarly, our results also reveal that the level of oxidative DNA damage in esophageal epithelial cells was positively correlated with degree of chronic inflammation, providing a rationale for the hypothesis that persistent existence of chronic inflammation could trigger oxidative DNA damage on the surrounding healthy epithelial cells.
Carcinogenesis is a long and multistep process including initiation (selection of a mutated cell), promotion (selective expansion of the initiated cell), and progression as a consequence of an imbalance between cell proliferation and cell death [20]. Oxidative damage to DNA by ROS is frequently postulated to give rise to mutations in tumor-related genes involved in essential cellular processes including proliferation, cell cycle checkpoints, apoptosis, and DNA repair. Our data show that oxidative DNA damage was positively correlated with esophageal precursor lesions, implying that the accumulation of oxidative DNA damage in esophageal epithelial cells, provided lacking of timely and accurate repair, may ultimately contribute to esophageal tumorigenesis.
Genomic instability is a common characteristic of human cancers [21, 22] and plays a critical role in tumor predisposition. Of the many classes of DNA damage, DNA double-strand breaks (DSBs) is a serious lesion that can cause genomic instability, which may lead to cancer development [23]. In our present study, it is noteworthy to point out that the level of DSBs is significantly higher in dysplastic esophageal tissues compared with that in normal esophageal tissues. Furthermore, we also observe this phenomenon in samples with transitional pathological changes from the same ESCC patient. Thus, we speculate that chronic inflammation-associated genomic instability, evident as accumulation of γH2AX foci, may contribute to esophageal carcinogenesis (Figure 4). Collectively, these results provide evidence linking chronic inflammation-associated genomic instability with esophageal carcinogenesis and suggest possibilities for early detection and intervention of esophageal carcinogenesis.
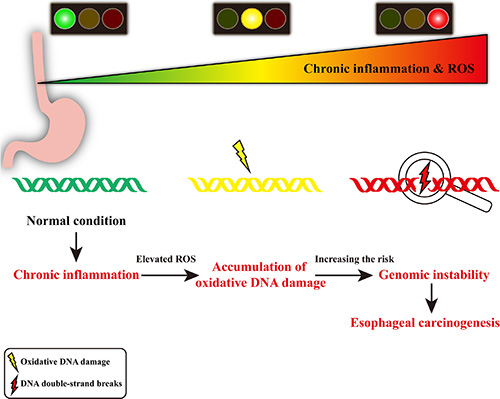
Figure 4: Schematic diagram of chronic inflammation-related DNA damage gears up esophageal carcinogenesis. In chronic inflammatory microenvironment, the infiltrating inflammatory cells may produce DNA-damaging product (such as ROS), resulting in oxidative DNA damage and DNA double-strand breaks. Elevated level of DNA double-strand breaks may lead to genomic instability, contributing to esophageal carcinogenesis.
MATERIALS AND METHODS
Esophageal mucosal samples collected from non-tumor individuals
A total of 109 endoscopic biopsy specimens of esophagus from tumor-free individuals from Chaoshan littoral were collected for examination (i.e. relationship between chronic inflammation and precursor lesions, immunostaining for 8-OHdG).
Esophageal mucosal samples collected from ESCC patients
A total of 204 esophageal tissue samples from ESCC patients with no prior radio- or chemotherapy were collected from Cancer Hospital of Shantou University Medical College from January 2008 to December 2014. For each patient, tumor-surrounding non-malignant tissues (within 2 cm from tumor) and distant non-malignant tissues (more than 5 cm away from tumor) were analyzed for DSBs status.
Additionally, we also obtained samples involving sequential pathological changes (normal-hyperplasia-dysplasia) from the same patient (9 ESCC patients) to evaluate the DNA damage status in the process of esophageal carcinogenesis.
This study was approved by Ethics Committee of Shantou University Medical College, and informed consents were obtained from patients or their families.
Grading of esophageal epithelial dysplasia
All samples were fixed, dehydrated and paraffin embedded, and presence of any epithelial changes deviating from normal esophageal epithelium were verified by two pathologists with hematoxylin and eosin staining. Hyperplasia indicated that an increase in the number of esophageal epithelial cells resulting in increased layers of the epithelium. Dysplasia was defined as nuclear atypia (enlargement, hyperchromasia, and pleomorphism), loss of normal cell polarity, but without invasion through the basement membrane (mild dysplasia: dysplastic cells within one-third layers of the epithelium; moderate dysplasia: dysplastic cells covering over one-third layers of epithelium, but confined to two-thirds layers of epithelium; severe dysplasia: dysplastic cells covering over two-thirds layers of epithelium).
Inflammation analysis
The chronic inflammation degree of esophageal epithelium was categorized into four groups according to the density and affected areas of chronic inflammatory cells (lymphocyte and macrophage) as follows: none denoted that less than 10 inflammtory cells per high power field confined to the lamina propria; mild refered to that less than 100 inflammatory cells per high power field only confined to the lamina propria and with no infiltrating into the epithelial layer; moderate signified that less than 300 inflammatory cells per high power field located in the lamina propria, and less than 20 inflammatory cells located in esophageal epithelium; severe was judged by that lymphoid follicles appeared in lamina propria with more than 300 inflammatory cells per high power field and leukocytes diffusely infiltrated into the epithelial layer with more than 20 inflammatory cells per high power field.
Immunohistochemistry
Immunohistochemistry was performed to confirm the presence of oxidative DNA damage and DNA damage response. Briefly, formalin-fixed, paraffin-embedded (FFPE) tissue blocks were sectioned at 4-μm thickness and then used to perform immunohistochemistry. Tissue sections were deparaffinized in xylene, then rehydrated through graded ethanol to water, and immersed in phosphate buffered saline (PBS). Subsequently, antigen retrieval was performed in a pressure cooker for 2 minutes at 125°C and endogenous peroxidase activity was quenched by 3% H2O2 in distilled water for 15 min. After three washes in PBS, sections were incubated overnight with anti-8-OHdG (mouse, dilution 1:300, Santa Cruz) and anti-γH2AX (mouse, dilution 1:600, Cell Signaling Technology) at 4°C in a humidified chamber followed by incubation for 30 min with goat anti-mouse/rabbit at 37°C. After washing, peroxidase substrate (DAB) was added, slides were washed in water and counterstained with hematoxylin. Slides were then washed, dehydrated and mounted using cover slips.
Images were captured using a Leica IM50 microscope at ×400, and five different fields for each index (8-OHdG and γH2AX) were selected in each sample. Due to the nuclear and cytoplasmic immunoreactivity of 8-OHdG, immunoreactivity was scored based on the percentage of positive epithelial cells (0: < 10%; 1: 11%–25%; 2: 26%–50%; 3: 51%–75%; 4: 76%–100%). The intensity was scored as negative (0), weak (1), moderate (2) and strong (3). The corresponding labeling index was computed as product of percentage of positive cells multiplied by immnostainin intensity with highest score of 12. The final evaluation of 8-OHdG expression was classified as four grades: score 0 for negative; score 1–4 for weak positivity; score 5–8 for moderate positivity; score 9–12 for strong positivity. For the quantification of γH2AX, the number of positive nuclei and total nuclei in images was counted and the corresponding immunostaining positive rates were computed as positive nuclei/total nuclei ×100%.
Statistical analysis
The statistical analyses were performed using SPSS 13.0 software package (SPSS, Chicago, IL). Spearman rank correlation analysis was used to assess the association between chronic inflammation level and esophageal histological severity, chronic inflammation level and oxidative DNA damage, oxidative DNA damage and esophageal histological severity. A two-tailed P value of or less than 0.05 was considered statistically significant. One-way analysis of variance (ANOVA) was applied for γH2AX positive rates among the multiple groups. LSD (least significance difference) and Tamhane’s T2 tests were selected to identify specific differences between each two groups for equal and unequal variance data, respectively. For multiple comparisons, P values were adjusted by Bonferroni correction.
ACKNOWLEDGMENTS AND FUNDING
This study was supported by grants from National Natural Science Foundation of China (NSFC)-Guangdong Joint Fund Key Project (Certificate U1132004), National Natural Science Foundation of China (Certificate 31171226), and Guangdong International Cooperative Technical Innovation Platform (Certificate gihz1106).
CONFLICTS OF INTEREST
The authors declare no conflicts financial interest.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108.
2. Rustgi A, El-Serag HB. Esophageal carcinoma. N Engl J Med. 2015; 372:1472–1473.
3. Glenn TF. Esophageal cancer. Facts, figures, and screening. Gastroenterol Nurs. 2001; 24:271–273; quiz 274–275.
4. Su M, Liu M, Tian DP, Li XY, Zhang GH, Yang HL, Fan X, Huang HH, Gao YX. Temporal trends of esophageal cancer during 1995–2004 in Nanao Island, an extremely high-risk area in China. Eur J Epidemiol. 2007; 22:43–48.
5. Wang GQ, Abnet CC, Shen Q, Lewin KJ, Sun XD, Roth MJ, Qiao YL, Mark SD, Dong ZW, Taylor PR, Dawsey SM. Histological precursors of oesophageal squamous cell carcinoma: results from a 13 year prospective follow up study in a high risk population. Gut. 2005; 54:187–192.
6. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001; 357:539–545.
7. Moss SF, Blaser MJ. Mechanisms of disease: Inflammation and the origins of cancer. Nat Clin Pract Oncol. 2005; 2:90–97; quiz 91 p following 113.
8. Ames BN, Gold LS, Willett WC. The causes and prevention of cancer. Proc Natl Acad Sci U S A. 1995; 92:5258–5265.
9. Kundu JK, Surh YJ. Inflammation: gearing the journey to cancer. Mutat Res. 2008; 659:15–30.
10. Lin R, Xiao D, Guo Y, Tian D, Yun H, Chen D, Su M. Chronic inflammation-related DNA damage response: a driving force of gastric cardia carcinogenesis. Oncotarget. 2015; 6:2856–2864. doi: 10.18632/oncotarget.3091.
11. Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003; 3:276–285.
12. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998; 273:5858–5868.
13. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000; 10:886–895.
14. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002; 420:860–867.
15. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005; 7:211–217.
16. Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007; 121:2373–2380.
17. Schottenfeld D, Beebe-Dimmer J. Chronic inflammation: a common and important factor in the pathogenesis of neoplasia. CA Cancer J Clin. 2006; 56:69–83.
18. Baik SC, Youn HS, Chung MH, Lee WK, Cho MJ, Ko GH, Park CK, Kasai H, Rhee KH. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res. 1996; 56:1279–1282.
19. Tardieu D, Jaeg JP, Deloly A, Corpet DE, Cadet J, Petit CR. The COX-2 inhibitor nimesulide suppresses superoxide and 8-hydroxy-deoxyguanosine formation, and stimulates apoptosis in mucosa during early colonic inflammation in rats. Carcinogenesis. 2000; 21:973–976.
20. Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer. 2007; 121:2381–2386.
21. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009; 458:719–724.
22. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
23. Jeggo PA, Lobrich M. DNA double-strand breaks: their cellular and clinical impact? Oncogene. 2007; 26:7717–7719.