
Introduction to signal transducer and activator of transcription 3
The signal transducer and activator of transcription (STAT) family is comprised of 7 members that are encoded by distinct genes. Because STAT3 is evolutionary the most conserved, it’s considered to be a very important member of the STAT family [1]. Similar to its other family members, STAT3 is present in non-stimulated cells in an inactive cytoplasmic form. Activation of STAT3 can be triggered through a multitude of factors among which interleukin-6 (IL-6) like cytokines [2], colony stimulating factors (CSF) and leptin [3], interferon (IFN) as well as IL-2 family members, and growth factors like epidermal growth factor [4]. Depending on the trigger, STAT3 activation occurs through phosphorylation on tyrosine 705 or serine 727. Phosphorylation on tyrosine 705 can be regulated by different tyrosine kinases and by members of the Janus-activated kinases (JAK) [5], whereas phosphorylation of serine 727 can be regulated by protein kinase C, mitogen-activated protein kinases and cyclin-dependent kinase 5 [6]. Phosphorylation of STAT3 results in its dimerization, which enables STAT3 to act as a transcriptional activator of various target genes. Also acetylation of lysine 685 has been described as a mode of STAT3 activation [7] and a way to enhance the stability of STAT3 dimers [6].
All transcriptional activity requires tight control, which in the case of STAT3 is performed by various negative regulators such as protein inhibitor of activated STAT proteins [8], suppressors of cytokine signaling (SOCS) proteins [9] and protein tyrosine phosphatases [10], [11]. These families of STAT3 regulating proteins interfere with STAT3 binding to DNA, hamper tyrosine kinases and remove phosphates from activated STAT3, respectively. In addition, STAT3 levels can be regulated through ubiquitination-dependent proteosomal degradation [12]. A large body of evidence has shown that STAT3 is constitutively activated in many mouse tumor models [13-16], and more importantly in human cancers including breast, liver, lung, pancreas, prostate, skin, hematological and brain cancers [17-25]. This is explained by the fact that many of the triggers that activate STAT3 are abundantly present in the tumor microenvironment (TME). Moreover, a number of genes induced by STAT3 provide a positive feedback and as such keep the STAT3 pathway continuously activated. Importantly, STAT3 activation occurs in both cancer cells and the many immune cells that infiltrate tumors, among which myeloid-derived suppressor cells (MDSCs) [6], [13], [24-30]. It has been described that STAT3 is one of the factors that allows crosstalk between the different cell types that are part of the TME. Therefore, STAT3 represents an attractive target for modulation. Although activated STAT3 is not only expressed in MDSCs, we will limit the remainder of this review to the role of STAT3 in these myeloid cells (Figure 1), as in contrast to other tumor-infiltrating immune cells like dendritic cells (DCs), macrophages and T cells, MDSCs are abundantly present in most mouse tumor models and human cancers [26], [31]. Moreover, it’s becoming increasingly clear that various cancer therapies, such as cancer vaccination, are more effective when MDSCs are depleted [32-38].
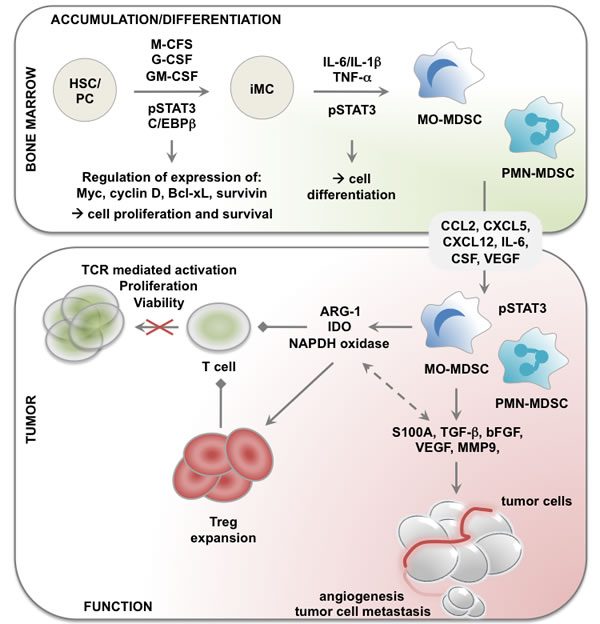
Figure 1: Role of STAT3 in accumulation, differentiation and functional regulation of MDSCs in cancer. Cytokines like M-, G- and GM-CSF stimulate myeloid cell development from hematopoietic stem cells (HSCs). Increased production of these cytokines during tumorogenesis interferes with normal myeloid development resulting in the generation of immature myeloid cells. In presence of factors like IL-6, L-1β and TNF−α, these differentiate into MDSCs. Furthermore, cancer cells secrete factors like PGE2 and CXCL12 that help in the recruitment of MDSCs to the TME. Finally, the activation of STAT3 pathway results in the expression of several factors like ARG-1, IDO, TGF-β, ROS etc. These are involved in mediating the tumor promoting function of MDSCs. The arrows "→" indicate "results in; the arrows "← →" indicate interconnectivity and the arrows "→" indicate a suppressive effect. Abbreviations: ARG-1: arginase-1; CXCL12: chemokine C-X-C motif ligand 12; G-CSF: granulocyte-colony stimulating factor; GM-CSF: granulocyte macrophage-CSF; HSC: hematopoietic stem cell; IDO: indoleamine 2,3 deoxygenase; IL: interleukin; iMC: immature myeloid cell; M-CSF: macrophage-CSF; MDSC: myeloid-derived suppressor cell; PC: progenitor cell; PGE2: prostaglandin E2; ROS: reactive oxygen species; STAT3: signal transducer and activator of transcription 3; TGF-β: transforming growth factor-β; TME: tumor microenvironment; TNF-α: tumor necrosis factor-α.
Introduction on myeloid-derived suppressor cells
Although MDSCs were described in 1970 as natural suppressor cells [39], it took until 2007 for the term MDSCs to get established. Generally the name MDSCs is used to categorize a heterogeneous mix of immature myeloid cells, which can be found in various pathological conditions, including cancer [40]. In healthy individuals, immature myeloid cells, which differentiate into mature macrophages, DCs and granulocytes, are constantly generated in the bone marrow. In cancer bearing subjects, the differentiation of immature myeloid cells is disturbed through the presence of tumor-derived factors that favor immature myeloid cell accumulation and differentiation to MDSCs both at the tumor site and secondary lymphoid organs [41]. Recruitment of MDSCs to the TME is mediated by chemokines such as chemokine C-C motif ligand 2 (CCL2), chemokine C-X-C motif ligand 5 (CXCL5) and CXCL12 [42], [43], as well as other factors such as IL-6, IL-1β, granulocyte-CSF (G-CSF) and vascular endothelial growth factor (VEGF) [44].
In mice, MDSCs are defined as CD11b and Gr-1 expressing cells [45]. Antibodies recognizing the granulocyte-specific marker Gr-1 target an epitope that is shared among the antigens Ly6C and Ly6G, two markers that have been used to divide MDSCs in monocytic (MO, Ly6ChighLy6Glow) and polymorphonuclear (PMN, Ly6ChighLy6Ghigh) cells [46]. Corresponding populations have been described in cancer patients. In general, human MDSCs are characterized by the expression of CD33, CD11b and the absence of significant levels of lineage markers and HLA-DR [47]. Human MO-MDSCs are further characterized as CD14+ but CD15− cells, while PMN-MDSCs are defined as CD14− CD15+ [26]. Several other surface markers have been put forward to distinguish MDSC subsets based on their function, among others CD40, CD49 (VLA4), CD80 (B7.1), CD115 (M-CSFR), CD124 (IL4Rα) and CCR2 [27], [48-53]. Although these markers are undoubtedly expressed on MDSCs, it’s generally accepted that they do not define specific MDSC subsets [54]. Moreover, the expression of some markers like CD80 can vary considerably depending on the cancer type and MDSC location [55]. Because of this phenotypic heterogeneity, it has frequently been suggested that the suppressive activity of MDSCs is the ultimate defining characteristic [56]. The latter is, in part, dictated by the activation of STAT3 in MDSCs. An unambiguous link between STAT3 and MDSCs is further evidenced by the fact that factors needed to phosporylate STAT3 are also associated with the activation and expansion of MDSCs. These include VEGF, granulocyte macrophage-CSF (GM-CSF), IL-6, basic fibroblast growth factor (bFGF), et cetera. Moreover, factors produced during and following activation of STAT3 are in turn essential for the accumulation and differentiation of MDSCs.
Myeloid-derived suppressor cells: a limitation to cancer immunotherapy
Cancer immunotherapy is based on the evidence that the immune system can discriminate between cancer cells and healthy cells, since the former express tumor antigens [57]. Based on this premise, cancer immunologists believe that it’s possible to stimulate tumor-specific cytotoxic T lymphocytes (CTLs) to reject and eliminate cancer cells. Several strategies have been explored of which therapeutic cancer vaccination [58-63], adoptive T-cell transfer [64], [65] and more recently blockade of inhibitory receptors such as programmed cell death-1 (PD-1) and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) [66-72] have shown promising results. Consequently, cancer immunotherapy has become a fourth treatment strategy within the clinician’s toolbox. Despite long-term tumor control in subsets of patients, it is frequently observed that in most cases where tumor-specific CTLs can be detected, they are unable to cause tumor regression. This finding hints that in these patients, mechanisms other than inhibitory immune checkpoint triggering override the function of tumor-specific T cells. Cancer cells have adapted several immune-avoiding mechanisms, for example the loss of tumor antigens or MHC I expression [73], and the recruitment of suppressive immune cells [74]. It has become increasingly clear that immunotherapy also has to interfere with the function of suppressive immune cells at the TME. Defining which suppressive immune cell types should be targeted and how, is a challenging task. In this regard, MDSCs have come to the forefront as a target population, because they are prevalent in most cancer types, both murine and human, and because they exploit a plethora of mechanisms to directly or indirectly abrogate anti-tumor immunity [75]. However, the heterogeneity of MDSCs and the diversity of inhibitory mechanisms they employ have faced us with the challenge of finding a “one fits all” strategy to deplete and/or functionally modulate them. Fortunately, the cell’s behavior is in large dictated by transcriptional programs. In the case of MDSCs, it has been suggested that the transcription factor STAT3 is a main regulator [75]. This is further highlighted by the observation that STAT3 expressed by MDSCs is implicated in their accumulation, differentiation and functionality (Figure 1).
Signal transducer and activator of transcription 3 plays a role in the accumulation and differentiation of myeloid-derived suppressor cells
Cancer-derived factors that drive the generation of MDSCs in the bone marrow include G-CSF and GM-CSF, various interleukins like IL-6 and IL-1β, prostaglandin E2 (PGE2), tumor necrosis factor-α and VEGF [42]. Many of these activate the STAT3 pathway, so it’s no surprise that STAT3 signaling has been implicated in the stimulation of myeloid cell differentiation into MDSCs. STAT3 interacts with CCAAT-enhancer-binding protein β (C/EBPβ). This transcription factor has a key role in myeloid development, as C/EBPβ-deficient bone marrow cells lose the ability to differentiate into functional MDSCs [76].
Furthermore, a correlation between C/EBPβ and accumulation of CD11b+ Gr-1+ cells in response to G-CSF was reported [77], [78]. This observation and the finding that STAT3 deficiency makes myeloid progenitors refractory to growth stimulation by G-CSF [79], suggests that STAT3 and C/EBPβ are inextricably linked in MDSC generation. This is further supported by the observation that STAT3 prolongs the binding of C/EBPβ on the myc promoter [76]. Besides myc, other cell survival and cell cycle regulating proteins like Bcl-xL, survivin, Mcl-1 and cyclin D1 are upregulated by STAT3 [6], [31], [80]. STAT3 was further linked to proteins like S100A [81] and protein kinase C βII [82], which inhibit DC differentiation from myeloid progenitor cells and thereby promote MDSC accumulation.
The studies described above clearly point towards a role for STAT3 in MDSC expansion and differentiation. In addition, there are multiple other possible mechanisms in which STAT3 can influence MDSCs. MicroRNAs (miRs) have been proven to be crucial in the regulation of myeloid cell maturation, activation, proliferation and differentiation [83], [84]. MiR155 is generally thought to be immune stimulatory by controlling lymphocyte differentiation and function [85], [86], but has recently been shown to promote the expansion of functional MDSCs [87]. In addition, miR155 has been found, together with miR21, to be the most upregulated miR during the induction of MDSCs from bone marrow. Genetic ablation of miR155 renders mice resistant to chemical-induced tumors suggesting that it also exerts its functions in immunosuppression and tumor promotion. When miR155 is absent, the suppressive functions of MDSCs are impaired through SOCS1 and an increase in SH2 (Src homology 2)-containing inositol phosphatase-1 (SHIP-1) [88]. In accordance, the study by Li et al showed a synergistic effect of miR155 on MDSCs induction via targeting of SHIP-1, phosphatase and tensin homolog, subsequently leading to STAT3 activation [87]. Moreover, miR155 can regulate inflammatory cytokine production through targeting of C/EBPβ in macrophages and MDSCs. It has been shown that the expression of C/EBPβ is inversely correlated with the amount of miR155 [89-91]. Furthermore, depletion of C/EBPβ can potentiate the transforming growth factor-β (TGF-β) response and contributes to cancer progression due to TGF-β induced endothelial-to-mesenchymal transition (EMT) [92].
The link between STAT3 activation and miR155 has been reported earlier in the context of experimental autoimmune uveitis [93] and laryngeal squamous carcinoma [94]. Moreover, it appears that these different mechanisms are interconnected. A novel axis between, S100A4, miR155, SOCS1 and matrix metalloproteinase (MMP) was unveiled in hepatocellular carcinoma. S100A4 upregulates miR155, which suppresses SOCS1, activates STAT3 signaling and in turn enhances MMP9, promoting tumor invasiveness [95]. Furthermore, the increase in miR155 correlates with upregulation of oncogenes, such as cyclin D1 and c-myc [96]. Interfering with any aspect of this axis could present a useful therapeutic approach for controlling proliferation and metastasis.
Activation and expansion of MDSCs can also be mediated by the release of tumor-derived soluble factors but recent reports have shown a role for microvesicles, namely exosomes, in MDSC biology [97], [98]. Exosomes are endosome-derived organelles with sizes of 50 to 150 nm, which are actively secreted through an exocytosis pathway to serve as mediators of intracellular crosstalk [99]. They can contain different cargo including miRs, proteins and lipids, which are then delivered to the receptor cell. Their story can be deemed identical to the miR story: exosomes were initially described to be immune stimulatory but recent research contradicts this statement, showing a role of exosomes in both inducing MDSCs [100], and inhibiting T-cell function or DC differentiation [101]. However, discrepancies in this field still exist, as the exact effects of exosomes on MDSCs are not fully elucidated yet. Chalmin et al describe that heat shock protein 72 (HSP72) expressing exosomes derived from different solid tumor cell lines, account for MDSC activation through triggering of STAT3 in a Toll-like receptor 2 (TLR2)/myeloid differentiation protein 88 (MyD88)-dependent manner through an autocrine IL-6 production, whereas tumor-derived soluble factors are responsible for MDSC expansion [102]. In the model by Xiang et al exosomes derived from the supernatants of cultured tumor cells (C-exo) induced both MDSC activation and expansion. It was suggested that this discrepancy could be attributed to the presence of PGE2 in the C-exo, while no PGE2 was detected in the exosomes used by Chalmin et al [100], [102].
Moreover, we found in a multiple myeloma mouse model that exosomes derived from bone marrow stromal cells or multiple myeloma cells themselves could activate both the STAT1 and STAT3 pathway leading to expansion and increased suppressive activity of the MDSCs [103]. This activation was independent of GM-CSF or HSP72 (unpublished data). Of note, there is a lot of controversy about the different techniques used to isolate exosomes [104]. While ultracentrifugation was long time the norm, it is nowadays acknowledged that contaminating lipids and proteins remain. These could be at the basis of contradictory data. Therefore studies concerning exosomal effects should be regarded with caution.
Signal transducer and activator of transcription 3 and its role in the tumor promoting activity of myeloid-derived suppressor cells
Several mechanisms are employed by MDSCs to promote tumor growth, including suppression of anti-tumor immune responses, stimulation of angiogenesis as well as tumor cell metastasis. These activities have been linked to activation of STAT3 in the MDSCs.
1) Inhibition of anti-tumor immune responses
Immune suppression is the most important biological characteristic of MDSCs. To that end, MDSCs deplete nutrients required by T cells for their clonal expansion, generate oxidative stress leading to reactive oxygen species (ROS) production, activate and expand regulatory T cells (Tregs), and finally inhibit T-cell trafficking [31]. Several mechanisms that are at the basis of these MDSC activities have been linked to activation of STAT3.
Expression of arginase-1 (ARG-1) is under the control of STAT3 and results in consumption of L-arginine and L-cysteine from the TME [26], [105-109]. Depletion of these amino acids results in downregulation of the CD3ζ-chain in the TCR complex and growth arrest of antigen-activated T cells [110], [111]. Moreover, Serafini et al linked the expression of ARG-1 to expansion of Tregs by MDSCs in a B-cell lymphoma model [112]. In this particular model TGF-β produced by the MDSCs had no effect on Tregs. Nonetheless, TGF-β has been linked to T-cell suppression [113], Treg expansion [25], [114] and initiation of EMT [92]. Importantly, a link between TGF-β and STAT3 was proposed based on the presence of two STAT3 binding sites in the TGF-β promoter [115]. Moreover, it was shown that TGF-β production was reduced after myeloid-specific STAT3 knock down [25]. This reduction in TGF-β was correlated to a reduction in Treg numbers. Another enzyme that is under the control of STAT3 and that depletes an essential T-cell nutrient is indoleamine 2,3 deoxygenase (IDO) [116]. STAT3-induced upregulation of IDO can be mediated by three molecular mechanisms, including binding of STAT3 to the promotor region of the IDO gene [117], and an indirect regulation of IDO expression via activation of C/EBPβ [118], [119] or non-canonical activation of NF-κB [120], [121]. Importantly, in human breast-cancer derived MDSCs non-canonical activation of NF-κB in an IL-6/STAT3-dependent fashion has been proposed as the predominant mechanism [122]. IDO depletes tryptophan thereby generating the toxic metabolite kynurenine. The mode of action of IDO is similar to that of ARG-1, suppression of TCR-mediated effector T-cell activation, growth arrest, induction of effector T-cell apoptosis and expansion of Tregs [123], [124].
Besides amino acid deprivation, STAT3 phosphorylation in MDSCs has also been linked to the activation of two subunits of NADPH oxidase (NOX2), namely P47phox and gp91phox, leading to an increased generation of intracellular ROS, another mechanism that dampens anti-tumor immunity [6], [28], [125]. It was postulated that S100A8/A9 heterodimers assist in the formation of the NADPH oxidase complex [79]. Moreover, ARG-1 can also contribute to ROS production [126], [127]. Importantly, ROS play a role in the suppression of antigen-specific T cells [128-130] and has been shown to induce T-cell apoptosis [131], much in the same way as ARG-1. The leading hypothesis states that ARG-1 is mostly expressed by PMN-MDSCs due to activation of STAT3, while MO-MDSCs mostly express inducible nitric oxide synthase (iNOS) through the activation of STAT1 and STAT6 [132], [132]. Recently, evidence contradicting this view has emerged. It was shown both in vitro and in vivo that inhibitors of iNOS suppressed VEGF release, induced STAT3 activation and ROS production [133]. Additionally, in human cells both the promotor of ARG-1 and iNOS have STAT3-binding elements, suggesting that STAT3 is not exclusively linked to ARG-1[105]. Moreover, activation of NF-κB as a result of STAT3 phosphorylation has been implicated in the regulation of iNOS expression [134]. As this study was performed on macrophages, more in depth research is needed to elucidate the molecular mechanisms that regulate the STAT3/iNOS pathway in MDSCs. Nonetheless, the studies described above demonstrate a central role for STAT3 in the active quenching of anti-tumor immunity by MDSCs.
2) Promotion of tumor cell dissemination
Immune suppression is not the only way in which MDSCs support tumor growth. They also promote tumor progression by enhancing blood vessel development, tumor cell invasion and metastasis. Angiogenesis has been linked to enhanced production of VEGF and bFGF by MDSCs. These angiogenic factors are under the control of STAT3 [135]. Moreover, STAT3 driven proteases like MMP9 and TGF-β have also been linked to angiogenesis [43]. In this regard MMP9 was shown to enhance the bioavailability of VEGF and as such support vascular stability [136]. In addition to the role in vasculogenesis, MMP also play a role in promoting tumor cell metastasis. Furthermore, MDSCs expressing active STAT3 have been implicated in the formation of pre-metastatic niches [137], [138]. These cells condition organs by creating an immunosuppressive environment that allows growth of metastatic tumor cells [139-141]. Herein, STAT3 regulated factors like bFGF, interleukins, MMP9 and S100A proteins play a role [139], [142].
3) Bidirectional link between tumor cell dissemination and immunity
It was recently shown in a mouse model that CD8+ T cells could counteract the formation of pre-metastatic niches by MDSCs by inducing MDSC apoptosis. However, activation of STAT3 compromises the ability of T cells to kill MDSCs [137], [138]. This was linked to lower granzyme B expression by CD8+ T cells and resistance of MDSCs to T-cell killing. Importantly, these mouse data are supported by data obtained in melanoma patients. Zhang et al showed a positive correlation between STAT3 activation and myeloid cell accumulation, increased IL-10, IL-6 and VEGF, while they observed an inverse correlation between STAT3 activation and CD8+ T cell numbers as well as the expression of granzyme B by T cells in melanoma draining lymph nodes [143].
The studies described above underline the role of STAT3 as a master regulator of the MDSC’s tumor promoting activity.
Signal transducer and activator of transcription 3 and its role in radiation response
STAT3 also plays a pivotal role in resistance to radiotherapy. Radiotherapy, which is currently used in cancer patients as a standard treatment, next to chemotherapy and surgery, still has certain hurdles to overcome, among which toxicity and (acquired) radiotherapy resistance. A considerable part of primary tumors are (partly) resistant to radiotherapy. A major goal in the field of radiobiology is to re-sensitize these tumors to radiation therapy. The first evidence on a role for STAT3 in radiotherapy resistance originated from a study by Otero et al in 2006 where radiation-induced apoptosis resistant peritoneal B-1 B cell subsets were used. B-1 cells possessed constitutively active STAT3. The radioresistance of B-1 cells could be conferred to radiosensitive B-2 cells by crosslinking in the presence of IL-6. Moreover, the B-1 cells became susceptible to irradiation by knocking out STAT3 [144]. Similarities exist for human cells as it was shown that downregulation of STAT3 enhanced the radiotherapy sensitivity of laryngeal squamous cell carcinoma xenografts. Furthermore, a positive correlation between the expression of STAT3 and Bcl-2 was uncovered [145]. This was further confirmed when it was shown that radiation itself induces phosphorylation of JAK2/STAT3 and increases the levels of Bcl-2 and Bcl-XL [146]. STAT3 affects various biochemical processes; therefore it’s very likely that it serves as a modulator of radioresponses in more than one way. We will discuss the interaction of STAT3 and hypoxia-inducible factor (HIF) as the hypoxic environment of the tumor is considered to be the main cause of clinical radiotherapy resistance [143]. In renal cell carcinoma, it has been shown that hypoxia activates STAT3, which consequently binds to the HIF-1α promotor and contributes to the stability and synthesis of the HIF-1α protein [147]. Inhibitors of STAT3 efficiently radiosensitized esophageal-, head and neck squamous cell carcinoma and prostate cancer cells and inhibited both hypoxia/radiation-induced activation of STAT3 and upregulation of HIF-1α and VEGF expression [148-150]. Further in vitro and in vivo data using a large spectrum of human tumors also convincingly show that JAK/STAT signaling is important in mediating resistance to radiation therapy. This is reviewed elsewhere [151]. Despite this compelling evidence, only the effect of STAT3 in the tumor cells is studied, while immune cells have been largely disregarded. However, there is evidence that downregulation of STAT3 in cancer cells impacts on the number of MO-MDSCs, while influencing the activity of PMN-MDSCs [1]. Moreover, it’s increasingly clear that immune cells play an important role in the radiation response, as reviewed elsewhere [152]. The role of MDSCs in radiation response has not been fully elucidated, but evidence has emerged that the response of myeloid cells to radiation is model- and dose- dependent and can be both pro- and anti-tumoral [153]. This raises the question whether the STAT3 status of both tumor and immune cells is important in the general radiation response?
Targeting signal transducer and activator of transcription 3 as a strategy to manipulate myeloid-derived suppressor cells
As mentioned, MDSCs have come to the forefront as a target in cancer (immune)therapy because of several reasons. Firstly, MDSCs are abundantly present in most cancer patients, irrespective of the cancer type [26]. Secondly, the presence of MDSCs correlates with cancer stage and metastatic disease [154]. Thirdly, MDSCs accelerate tumor progression by inhibiting anti-tumor immune responses, stimulating angiogenesis, tumor cell invasion and metastasis [26], [31].
Throughout this review, we showed that STAT3 is implicated in the accumulation, differentiation and function of MDSCs. Consequently, several research teams have evaluated STAT3 targeting drugs as a means to interfere with these processes and as such put a brake on tumor progression [35], [75]. A gene therapy approach has been investigated in a transgenic mouse model that spontaneously develops medulloblastoma tumors. Herein the myeloid cell LysM promotor was driving the expression of Cre recombinase to conditionally delete STAT3 using the Cre/LoxP system. STAT3 removal resulted in a reduction of PMN-MDSCs in the tumor and an increased effector T cell/Treg ratio. However, no changes in tumor incidence were reported [25].
Next to the genetic approach a list of drugs has been investigated to avoid phosphorylation of STAT3. This list includes curcumin derivatives and other JAK2/STAT3 inhibitors including AZD1480 [30], [155-158], Icariin flavone and its derivative 3,5,7-trihydroxy-4’-emthoxy-8-(3-hydroxy-3-methylbutyl)-flavone [159], tyrosine kinase inhibitors such as sunitinib [160-163], VEGF inhibiting molecules such as VEGF-trap (a VEGF receptor fused to the Fc part of human IgG1) [164], [165] and anti-VEGF antibodies (bevacizumab) [166], [167], monoclonal antibodies specific for IL-6 [168], molecules like bardoxolone methyl (CDDO-Me) [33], [169-171] and chemotherapeutics such as docetaxel [155-158] (Table 1).
Table 1: Effect of (in)direct targeting of STAT3 on myeloid-derived suppressor cells.
Agent |
Mechanism of action |
Study setting |
Tumor model |
Study finding |
Ref |
AZD1480 |
JAK2/STAT3 inhibitor |
Preclinical (in vivo) |
Melanoma model |
Decreased levels of MDSCs, but higher suppressive activity on a per cell basis |
30 |
Curcumin derivatives |
JAK2/STAT3 inhibitor |
Preclinical (in vivo) Patient study (blood samples) |
Gastric-colon carcinoma Lung cancer |
Inhibits accumulation and induces differentiation of MDSCs Decreased numbers of MDSCs, while increasing mature myeloid cells in peripheral blood |
156-157 |
Icariin flavone and its derivatives |
Inhibit STAT3 signaling and expression of S100A8 and S100A9 |
Preclinical (in vivo) |
Breast carcinoma |
Downregulates MDSC numbers |
159 |
Sunitinib |
Receptor tyrosine kinase inhibitor |
Preclinical (in vivo) Preclinical (in vivo) Patient study (blood samples) |
Renal-breast-colon carcinoma Breast carcinoma Metastatic renal carcinoma |
Eliminates MDSCs. Still debate about the location of depletion. Improves TH1 function and lowers Treg |
160-163 |
Avastin |
Anti-VEGF antibody |
Xenografts |
Renal cell carcinoma |
Reduces the number of circulating myeloid cells |
166 |
Bevacizumab |
Anti-VEGF antibody |
Patient study (blood samples) |
Renal cell carcinoma |
No effects on MDSCs in peripheral blood |
167 |
Monoclonal antibodies (specific for IL-6) |
Anti IL-6 receptor antibody |
Preclinical (in vivo) |
Skin squamous cell carcinoma |
Downregulates accumulation of MO-MDSCs |
168 |
Bardoxolone methyl (CDDO-Me) |
Inhibits JAK1 activity |
Preclinical (in vivo) Patient study (blood samples) |
Colon-lung carcinoma Lymphoma model Renal -soft tissue carcinoma |
Abrogated immune suppressive activity of MDSCs |
33, 171 |
Docetaxel |
Inhibition of pSTAT3 |
Preclinical (in vivo) |
Breast carcinoma |
Decreased MDSC numbers in the spleen |
172 |
CpG-siSTAT3 |
TLR9-targeted STAT3 silencing |
Preclinical (in vivo) Patient study (blood samples-tumor specimens) |
Melanoma model Leukemia Prostate carcinoma |
Abrogates immunosuppressive activity of MDSCs Induces potent innate anti-tumor responses |
178-180 |
Conditional STAT3 gene disruption |
Cre/LoxP system under LysM promoter |
Preclinical (in vivo) |
Medulloblastoma |
A significant reduction in the abundance of PMN-MDSCs and Tregs was observed within tumors No effect on tumor incidence in mice |
25 |
Curcumin and its derivatives are naturally occurring phenols that are used for their anti-oxidant and anti-inflammatory activities. Furthermore, these have been used to selectively inhibit the JAK2/STAT3 pathway [155-158]. Administration of Cucurbitacin B (CuB) to lung cancer patients was shown to decrease the numbers of bona fide MDSCs (Lin- HLA-DR- CD33+), while it increased the numbers of mature Lin- HLA-DR+ CD33+ myeloid cells in peripheral blood. Moreover, it was shown in vitro that CuB induced DC differentiation and increased the sensitivity of tumor cells to antigen (p53)-specific T cells [156]. Also other JAK2/STAT3 inhibitors have been tested, including AZD1480 [30], which resulted in low levels of MDSCs in tumor bearing AZD1480 treated mice. However, AZD1480 treatment did not abrogate the ability of the remaining MDSCs to suppress T cells. Moreover, when evaluated on a per cell basis, it was shown that the suppressive activity of the MDSCs was higher after treatment with AZD1480. Similar to JAK2/STAT3 inhibitors, flavanoids like Icariin and its derivative were reported to downregulate MDSC numbers [159]. These natural compounds were shown to inhibit STAT3 signaling and expression of S100A8 and S100A9, resulting in differentiation of immature myeloid cells to mature cells. Sunitinib is a small-molecule multikinase inhibitor that targets among others the VEGF receptor, platelet-derived growth factor receptor and c-kit, and as such hampers the phosphorylation of STAT3. Ko et al [160], showed that sunitinib efficiently eliminates peripheral MDSCs, whereas it did not reduce MDSCs in tumors. This was linked to high levels of GM-CSF in the tumor and STAT5 signaling in MDSCs. Nonetheless, other studies show that MDSC depletion by sunitinib is irrespective of the location [162]. Importantly, treatment of metastatic renal cell cancer patients with sunitinib reduced the level of MDSCs in peripheral blood by half and was associated with improved TH1 function (reduced IL-4 and higher IFN-γ) and lower Treg numbers [161], [163]. Although sunitinib, which affects downstream VEGF receptor signaling and as such STAT3 activation, was shown to modulate MDSC levels, other strategies that impact on VEGF receptor signaling, such as VEGF-trap [164], [165] and anti-VEGF antibodies demonstrated no effect on MDSC levels in peripheral blood of cancer patients [167]. This is an unexpected finding, since the link between VEGF and MDSC accumulation is longstanding and as it was shown that anti-VEGF antibodies successfully reduce MDSC numbers in mice [166]. Besides antibodies to capture VEGF and as such inhibit STAT3 activation upon VEGF receptor interaction, researchers have developed monoclonal antagonistic antibodies specific for the IL-6 receptor, as it’s triggering is directly linked to STAT3 activation and MDSCs. These anti-IL-6 receptor antibodies neutralize the effects of tumor-derived IL-6 and suppress expansion of cancer-associated MDSCs [168]. Another molecule that was shown to inhibit STAT3 activation in MDSCs, at least when used at high concentrations (1-5 µM), is CDDO-Me. Treatment with this synthetic triterpentoid (a methyl ester of 2-cyano-3,12-dioxooleana-1,9 (11)-dien-28-oic acid) resulted in reduced production of ROS, improved T-cell function and more importantly reduced tumor growth [33], [169-171]. Finally, the chemotherapeutic agent, docetaxel was evaluated for its direct effect on MDSCs. Low-dose treatment polarized MDSCs toward an M1-like phenotype, as deduced from the expression of CCR7, MHC II, CD11c and CD68, and reduced the number of MDSCs in the spleen. Administration of docetaxel also resulted in increased CTL responses and reduced tumor growth. These effects were partially attributed to inhibition of STAT3 activation [172]. The studies above show the potential of targeting STAT3 in MDSCs as an anti-cancer strategy. At the same time these studies demonstrate that although the aforementioned drugs act on STAT3 activation their mode of action can differ from MDSC depletion over maturation to functional modulation. Moreover, treatment of cancer bearing subjects with only these drugs was shown to be insufficient to provide a cure. As MDSCs represent a confounding factor for anti-tumor immunity and as it was found that MDSC depletion improves the outcome of cancer vaccines [32-34], [34-38], it’s not surprising that drugs such as CDDMO-Me [33] and sunitinib [37], [38], [173] have been evaluated in combination with cancer vaccination. In these studies the combination therapy showed improved curative potential when compared to either component alone. However instead of combining therapies, it would be more elegant if one drug could lead to activation of tumor-specific CTLs, while modulating MDSCs. Importantly, various studies have demonstrated that MDSCs can be reverted into stimulatory APCs under the influence of cytokines such as IL-12 [174], [175] or TLR ligands like CpG oligonucleotides [176], [177]. The latter has offered an opportunity to design a drug consisting of CpG oligonucleotides conjugated to STAT3 specific small interfering RNA (referred to as CpG-siSTAT3 conjugates) [178-180]. It has been shown in mouse cancer models and using STAT3+ PMN-MDSCs of prostate cancer patients that CpG-siSTAT3 conjugates mediate selective delivery of silencing siSTAT3 to TLR9+ myeloid cells, resulting in disruption of the STAT3 supported suppressive signaling network and stimulation of anti-tumor immunity. These findings indicate that this gene- and cell type-specific inhibitory oligonucleotides represent a novel therapeutic approach to mitigate immunosuppression in cancer patients.
In vitro generated myeloid-derived suppressor cells: a platform for drug screening
The ample evidence on the importance of STAT3 in MDSCs has instigated research into existing and novel STAT3 targeting drugs and their effect on MDSC viability and functionality (Table 1). These studies mostly use mouse MDSCs, as our knowledge of the murine system is way ahead of their human counterparts. However, studying murine MDSCs poses the technical challenge of obtaining sufficient numbers of cells at high purity from a limited number of tumors. Therefore, MDSCs from the spleen are often used as an alternative. However, spleen MDSCs are phenotypically and functionally different from tumor MDSCs [55], [181]. Consequently, to ensure reliability and potency of novel drugs, they should be evaluated on tumor rather than spleen MDSCs. To circumvent this conundrum, researchers have evaluated various in vitro culture systems to obtain MDSCs. These range from the use of cell lines to the differentiation of bone marrow cells [77], [80], [100], [101], [181-194]. In particular, ex vivo differentiation of bone marrow cells using conditioned media from GM-CSF secreting tumor cells has proven to be a successful approach [80], [175], [181], [184]. A proof-of-concept on the value of this strategy to obtain large amounts of MDSCs that resemble those found within various cancer types, including multiple myeloma, melanoma and colorectal cancer was delivered [80], [175], [181]. Since 50 to 60 million MDSCs are obtained from a single mouse without the necessity of inducing cancer, this system allows systematic and high throughput in vitro testing of anti-neoplastic treatments. Furthermore, these in vitro MDSCs offer the possibility to study the role of individual factors, including STAT3 on MDSC development [80].
Efficient in vitro systems should also be developed to generate human MDSCs, as this will surely facilitate research into MDSCs and more specifically STAT3-targeting drugs. Major advances have been made towards this goal. Human MDSCs have been differentiated from bone marrow cells or peripheral blood mononuclear cells using recombinant cytokine cocktails [77], [188], [189], or tumor-conditioned media [80], [101]. However, most human MDSC differentiation systems are still poorly efficient, most likely because they do not make use of fully pluripotent hematopoietic precursors [195]. Moreover, the endeavor of generating human MDSCs in vitro is further hampered by the lack of an in depth understanding on the nature of human MDSCs in vivo. Nonetheless, the study by Chen et al [190] in which GM-CSF and IL-6 were used to differentiate human MDSC from peripheral blood mononuclear cells, shows the applicability of these cells, as they possess hallmark immunosuppressive pathways, including STAT3 signaling.
Conclusion
Accumulation of MDSCs in cancer patients has been linked to cancer stage and overall survival in patients with a variety of different cancer types. Therefore a strategy that targets MDSCs could be used in combination with other immune stimulatory therapies. However, due to the heterogeneity of MDSCs, both in phenotype and function, it’s a challenge to pinpoint the most effective MDSC target. A large body of evidence links STAT3 activation to MDSC accumulation, differentiation and function, both direct and indirect. Because of the numerous physiological processes and signal transduction pathways that are affected by STAT3, it’s likely that this can account for therapy resistance. We believe that specific STAT3 targeting in MDSCs offers great opportunities. This view has only gained in strength as increasing numbers of drugs that counteract JAK/STAT signaling have been tested in clinical trials and even have been approved by the FDA. In future multimodal treatment, specific targeting of STAT3 in MDSCs can be the way forward. This is underscored by the data provided by the CpG-siSTAT3 conjugate studies, which show that targeted delivery of STAT3 inhibiting molecules is a successful approach [177-180]. Such an approach could encompass the use of myeloid cell-targeted lentiviral vectors [196], [197] or nanoparticles [198], which could than deliver silencing RNA for STAT3 [13], [178-180] or genes encoding negative regulators of STAT3 [8-11]. Evaluating such strategies will be greatly facilitated by the use of in vitro mouse and/or human MDSCs. Extensive in vivo testing of STAT3 targeting drugs will also be required in order to determine their safety. In fact, STAT3 expressed in MDSCs is involved in homeostasis, for instance in the gut. Consequently, systemic downregulation of STAT3 in MDSCs withholds the risk of inducing immune-related adverse events such as colitis. Various studies show that myeloid cell-specific STAT3 deficiency results in enhanced inflammation and chronic colitis as a consequence of a decrease in PMN-MDSCs and linked herewith a decrease in suppressive cytokines (e.g. IL-10), an increase in pro-inflammatory cytokines (e.g. IFN-γ) and iNOS. These studies caution against systemic, long-term STAT3 depletion and highlight the necessity to develop targeted strategies [199-201].
Conflict of interest
The authors declare no conflict of interest.
References
1. Pietra LD, Bressan A, Pezzotti AR, Serlupi-Crescenzi O: Highly conserved amino-acid sequence between murine STAT3 and a revised human STAT3 sequence. Gene. 1998; 213(1): 119–124.
2. Kishimoto T, Akira S, Narazaki M, Taga T: Interleukin-6 family of cytokines and gp130. Blood. 1995; 86(4): 1243–1254.
3. Akira S: Functional roles of STAT family proteins: lessons from knockout mice. Stem Cells. 1999; 17(3):138–146.
4. Zhong Z, Wen Z, Darnell JE: Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994; 264(5155): 95–98.
5. Hirano T, Ishihara K, Hibi M: Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000; 19(21): 2548–2556.
6. Rébé C, Végran F, Berger H, Ghiringhelli F: STAT3 activation: A key factor in tumor escape. JAK-STAT. 2014; 2(1): e23010.
7. Yuan ZL, Guan YJ, Chatterjee D, Chin YE: Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005, 307(5707): 269–273.
8. Shuai K, Liu B: Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005; 5(8): 593–605.
9. Alexander WS, Hilton DJ: The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004; 22: 503–529.
10. Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, Griffiths E, Krawczyk C, Richardson CD, Aitken K, Iscove N, Koretzky G, Johnson P, et al.: CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature. 2001; 409(6818): 349–354.
11. Schaper F, Gendo C, Eck M, Schmitz J, Grimm C, Anhuf D, Kerr IM, Heinrich PC: Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6 signal transducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression. Biochem J. 1998; 335: 557–565.
12. Daino H, Matsumura I, Takada K, Odajima J, Tanaka H, Ueda S, Shibayama H, Ikeda H, Hibi M, Machii T, Hirano T, Kanakura Y: Induction of apoptosis by extracellular ubiquitin in human hematopoietic cells: possible involvement of STAT3 degradation by proteasome pathway in interleukin 6-dependent hematopoietic cells. Blood. 2000; 95(8): 2577–2585.
13. Emeagi PU, Maenhout S, Dang N, Heirman C, Thielemans K, Breckpot K: Downregulation of Stat3 in melanoma: reprogramming the immune microenvironment as an anticancer therapeutic strategy. Gene Ther. 2013; 20(11): 1085–1092.
14. Yang CH, Fan M, Slominski AT, Yue J, Pfeffer LM: The role of constitutively activated STAT3 in B16 melanoma cells. Int J Infereron Cytokine Mediator Res. 2010; 2010(2): 1–7.
15. Liu Y, Deng J, Wang L, Lee H, Armstrong B, Scuto A, Kowolik C, Weiss LM, Forman S, Yu H: S1PR1 is an effective target to block STAT3 signaling in activated B cell-like diffuse large B-cell lymphoma. Blood. 2012; 120(7): 1458–1465.
16. Tkach M, Coria L, Rosemblit C, Rivas MA, Proietti CJ, Díaz Flaqué MC, Beguelin W, Frahm I, Charreau EH, Cassataro J, Elizalde PV, Schillaci R: Targeting Stat3 induces senescence in tumor cells and elicits prophylactic and therapeutic immune responses against breast cancer growth mediated by NK cells and CD4+ T cells. J Immunol. 2012; 189(3): 1162–1172.
17. Buettner R, Mora LB, Jove R: Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002; 8(4): 945–954.
18. Bowman T, Garcia R, Turkson J, Jove R: STATs in oncogenesis. Oncogene. 2000; 19(21): 2474–2488.
19. Krasilnikov M, Ivanov VN, Dong J, Ronai Z: ERK and PI3K negatively regulate STAT-transcriptional activities in human melanoma cells: implications towards sensitization to apoptosis. Oncogene. 2003; 22(26): 4092–4101.
20. Darnell JE: Validating Stat3 in cancer therapy. Nature Med. 2005; 11(6): 595–596.
21. Hung MH, Tai WT, Shiau CW, Chen KF: Downregulation of signal transducer and activator of transcription 3 by sorafenib: a novel mechanism for hepatocellular carcinoma therapy. World J Gastroenterol. 2014; 20(41): 15269–15274.
22. Scuto A, Kujawski M, Kowolik C, Krymskaya L, Wang L, Weiss LM, Digiusto D, Yu H, Forman S, Jove R: STAT3 inhibition is a therapeutic strategy for ABC-like diffuse large B-cell lymphoma. Cancer Res. 2011; 71(9): 3182–3188.
23. Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernández-Luna JL, Nuñez G, Dalton WS, Jove R: Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999; 10(1): 105–115.
24. Griesinger AM, Josephson RJ, Donson AM, Mulchay Levy JM, Amani V, Birks DK, Hoffman LM, Furtek SL, Reigan P, Hnadler MH, Vibhakar R, Foreman NK. Interleukin-6/STAT3 Pathway Signaling Drives an Inflammatory Phenotype in Group A Ependymoma. Cancer Immunol Res. 2015; 3(10): 1165–1174.
25. Abad C, Nobuta H, Li J, Kasai A, Yong WH, Waschek JA. Targeted STAT3 disruption in myeloid cells alters immunosuppressor cell abundance in a murine model of spontaneous medulloblastoma. JLB. 2014; 95(2): 357–367.
26. Jiang J, Guo W, Liang X: Phenotypes, accumulation, and functions of myeloid-derived suppressor cells and associated treatment strategies in cancer patients. Human Immunol. 2014; 75(11): 1128–1137.
27. Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R: Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010; 70(11): 4335–4345.
28. Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D: Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. 2004; 172(1): 464–474.
29. Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI: Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. JEM. 2008; 205(10): 2235–2249.
30. Maenhout SK, Four Du S, Corthals J, Neyns B, Thielemans K, Aerts JL: AZD1480 delays tumor growth in a melanoma model while enhancing the suppressive activity of myeloid-derived suppressor cells. Oncotarget. 2014; 5(16): 6801–6815. doi: 10.18632/oncotarget.2254.
31. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V: Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012; 12(4): 253–268.
32. Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich D: All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003; 63(15): 4441–4449.
33. Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, Meyer C, Becerra CR, Fishman M, Antonia S, Sporn MB, Liby KT, Rawal B, et al.: Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. 2010; 16(6): 1812–1823.
34. Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D: Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother. 2013; 62(5): 909–918.
35. Wesolowski R, Markowitz J, Carson WE III: Myeloid derived suppressor cells: a new therapeutic target in the treatment of cancer. J Immunother Cancer. 2013; 1(10).
36. Poschke I, Mao Y, Adamson L, Salazar-Onfray F, Masucci G, Kiessling R: Myeloid-derived suppressor cells impair the quality of dendritic cell vaccines. Cancer Immunol Immunother. 2011; 61(6): 827–838.
37. Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, Ko JS, Cohen PA, Finke JH, Storkus WJ: Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer. 2011; 129(9): 2158–2170.
38. Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T, Daemen T: Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology. 2015; 4(3): e989764.
39. Strober S. Natural suppressor (NS) cells, neonatal tolerance, and total lymphoid irradiation: exploring obscure relationships. Ann rev immunol. 1984; 2: 219–237.
40. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, Schreiber H: The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res. 2007; 67(1): 425–425.
41. Bronte V, Serafini P, Apolloni E, Zanovello P: Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunother. 2001; 24(6): 431–446.
42. Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, Abe J, Shono Y, Kitabatake M, Kakimi K, Mukaida N, Matsushima K. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008; 111(12): 5457–5466.
43. Yang L, Huang J, Ren X, Gorska AE, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008; 13(1): 23–35.
44. Poschke I, Kiessling R: On the armament and appearances of human myeloid-derived suppressor cells. Clin Immunol. 2012; 144(3): 250–268.
45. Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P: Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000; 96(12): 3838–3846.
46. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA: Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008; 111(8): 4233–4244.
47. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI: Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001; 166(1): 678–689.
48. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, Divino CM, Chen SH: Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010; 70(1): 99–108.
49. Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P, Bicciato S, Bronte V: Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006; 116(10): 2777–2790.
50. Yang R: CD80 in Immune Suppression by Mouse Ovarian Carcinoma-Associated Gr-1+CD11b+ Myeloid Cells. Cancer Res. 2006; 66(13): 6807–6815.
51. Kohanbash G, McKaveney K, Sakaki M, Ueda R, Mintz AH, Amankulor N, Fujita M, Ohlfest JR, Okada H: GM-CSF Promotes the Immunosuppressive Activity of Glioma-Infiltrating Myeloid Cells through Interleukin-4 Receptor. Cancer Res. 2013; 73(21): 6413–6423.
52. Haile LA, Gamrekelashvili J, Manns MP, Korangy F, Greten TF: CD49d is a new marker for distinct myeloid-derived suppressor cell subpopulations in mice. J Immunol. 2010; 185(1): 203–210.
53. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH: Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006; 66(2): 1123–1131.
54. Youn JI, Gabrilovich DI: The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010; 40(11): 2969–2975.
55. Maenhout SK, Lint SV, Emeagi PU, Thielemans K, Aerts JL: Enhanced suppressive capacity of tumor-infiltrating myeloid-derived suppressor cells compared with their peripheral counterparts. Int J Cancer. 2013; 134(5): 1077-1090.
56. Ostrand-Rosenberg S, Sinha P: Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. J Immunol. 2009; 182(8): 4499–4506.
57. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T: Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nature Rev Cancer. 2014; 14(2): 135–146.
58. Tuyaerts S, Aerts JL, Corthals J, Neyns B, Heirman C, Breckpot K, Thielemans K, Bonehill A: Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol Immunother. 2007; 56(10): 1513–1537.
59. Benteyn D, Heirman C, Bonehill A, Thielemans K, Breckpot K: mRNA-based dendritic cell vaccines. Expert Rev Vaccines. 2015; 14(2): 161–176.
60. González FE, Gleisner A, Falcón-Beas F, Osorio F, López MN, Salazar-Onfray F: Tumor cell lysates as immunogenic sources for cancer vaccine design. Hum Vaccin Immunother. 2014; 10(11): 3261–3269.
61. Ura T, Okuda K, Shimada M: Developments in Viral Vector-Based Vaccines. Vaccines. 2014; 2(3): 624–641.
62. Van Lint S, Renmans D, Broos K, Dewitte H, Lentacker I, Heirman C, Breckpot K, Thielemans K: The ReNAissanCe of mRNA-based cancer therapy. Expert Rev Vaccines. 2015; 14(2): 235–251.
63. Fioretti D, Iurescia S, Fazio VM, Rinaldi M: DNA Vaccines: Developing New Strategies against Cancer. J Biomed Biotechnol. 2010; 2010(938): 1–16.
64. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME: Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nature Rev Cancer. 2008; 8(4): 299–308.
65. Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nature Med. 2016; 22(1): 26–36.
66. Topalian SL, Drake CG, Pardoll DM: Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell. 2015; 27(4): 450–461.
67. Pardoll DM: The blockade of immune checkpoints in cancer immunotherapy. Nature Rev Cancer. 2012; 12(4): 525-564.
68. Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA, Xu H, Yao S, Pons A, Chen L, Pardoll DM, Brahmer JR, Topalian SL: Durable Cancer Regression Off-Treatment and Effective Reinduction Therapy with an Anti-PD-1 Antibody. Clin Cancer Res. 2013; 19(2): 462–468.
69. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, et al.: Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. N Engl J Med. 2013; 369(2): 134–144.
70. Kwek SS, Dao V, Roy R, Hou Y, Alajajian D, Simko JP, Small EJ, Fong L: Diversity of Antigen-Specific Responses Induced In Vivo with CTLA-4 Blockade in Prostate Cancer Patients. J Immunol. 2012; 189(7): 3759–3766.
71. Pico de Coana YP, Poschke I, Gentilcore G, Mao Y, Nystrom M, Hansson J, Masucci GV, Kiessling R: Ipilimumab Treatment Results in an Early Decrease in the Frequency of Circulating Granulocytic Myeloid-Derived Suppressor Cells as well as Their Arginase1 Production. Cancer Immunol Res. 2013; 1(3): 158–162.
72. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor David, et al.: Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N Engl J Med. 2015; 372(21): 2006–2017.
73. Breckpot K, Dullaers M, Bonehill A, Van Meirvenne S, Heirman C, De Greef C, van der Bruggen P, Thielemans K: Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J Gene Med. 2003; 5(8): 654–667.
74. Munn DH, Bronte V: Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol. 2016; 39: 1–6.
75. Draghiciu O, Lubbers J, Nijman HW, Daemen T: Myeloid derived suppressor cells—An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology. 2015; 4(1): e954829.
76. Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS: STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010; 116(14): 2462–2471.
77. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, Calabrese F, Basso G, Zanovello P: Tumor-Induced Tolerance and Immune Suppression Depend on the C/EBPβ Transcription Factor. Immunity. 2010; 32(6): 790–802.
78. Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno SI, Imanishi J, Akashi K, Tenen DG: C/EBPbeta is required for “emergency” granulopoiesis. Nat Immunol. 2006; 7(7): 732–739.
79. Trikha P, Carson WE III: Signaling pathways involved in MDSC regulation. BBA. 2014; 1846(1): 55–65.
80. De Veirman K, Van Ginderachter JA, Lub S, De Beule N, Thielemans K, Bautmans I, Oyajobi BO, De Bruyne E, Menu E, Lemaire M, Van Riet I, Vanderkerken K, Van Valckenborgh E: Multiple myeloma induces Mcl-1 expression and survival of myeloid-derived suppressor cells. Oncotarget. 2015; 6(12): 10532–10547. doi:10.18632/oncotarget.3300.
81. Foell D, Wittkowski H, Vogl T, Roth J: S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. JLB. 2007; 81(1): 28–37.
82. Farren MR, Carlson LM, Lee KP: Tumor-mediated inhibition of dendritic cell differentiation is mediated by down regulation of protein kinase C beta II expression. Immunol Res. 2010; 46(1): 165–176.
83. Chen CZ, Li L, Lodish HF, Bartel DP: MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004; 303(5654): 83–86
84. Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD: MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008; 9(8): 839–845.
85. Calame K: MicroRNA-155 function in B Cells. Immunity. 2007; 27(6): 825–827.
86. Turner M, Vigorito E: Regulation of B- and T-cell differentiation by a single microRNA. Biochem Soc Trans. 2008; 36(3): 531–533.
87. Li L, Zhang J, Diao W, Wang D, Wei Y, Zhang CY, Zen K: MicroRNA-155 and MicroRNA-21 Promote the Expansion of Functional Myeloid-Derived Suppressor Cells. J Immunol. 2014; 192(3): 1034–1043.
88. Chen S, Wang L, Fan J, Ye C, Dominguez D, Zhang Y, Curiel TJ, Fang D, Kuzel TM, Zhang B: Host miR155 Promotes Tumor Growth through a Myeloid-Derived Suppressor Cell-Dependent Mechanism. Cancer Res. 2015; 75(3): 519–531.
89. Worm J, Stenvang J, Petri A, Frederiksen KS, Obad S, Elmén J, Hedtjärn M, Straarup EM, Hansen JB, Kauppinen S. Silencing of microRNA-155 in mice during acute inflammatory response leads to derepression of c/ebp Beta and down-regulation of G-CSF. Nucleic Acids Res. 2009; 37(17): 5784–5792.
90. He M, Xu Z, Ding T, Kuang DM, Zheng L. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell Mol Iimmunol. 2009; 6(5): 343–352.
91. Kim S, Song JH, Kim S, Qu P, Martin BL, Sehareen WS, Haines DC, Lin PC, Sharan SK, Chang S. Loss of oncogenic miR-155 in tumor cells promotes tumor growth by enhancing C/EBP-β-mediated MDSC infiltration. Oncotarget. 2016; doi: 10.18632/oncotarget.7150
92. Johansson J, Berg T, Kurzejamska E Pang MF, Tabor V, Jansson M, Roswall P, Pietras K, Sund M, Reloga P, Fuxe J. MiR-155-mediated loss of CEBPbeta; shifts the TGF-beta; response from growth inhibition to epithelial-mesenchymal transition, invasion and metastasis in breast cancer. Oncogene. 2013; 32(50): 5614–5624.
93. Escobar T, Yu C-R, Muljo SA, Egwuagu CE: STAT3 activates miR-155 in Th17 cells and acts in concert to promote experimental autoimmune uveitis. Invest Ophthalmol Vis Sci. 2013; 54(6): 4017–4025.
94. Zhao XD, Zhang W, Liang HJ, Ji WY: Overexpression of miR -155 promotes proliferation and invasion of human laryngeal squamous cell carcinoma via targeting SOCS1 and STAT3. PLoS ONE. 2013; 8(2): e56395.
95. Yan XL, Jia YL, Chen Q, Zeng Q, Zhou JN, Fu CJ, Chen HX, Yuan HF, Li ZW, Shi L, Xu YC, Wang JX, Zhang XM et al. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology. 2013; 57(6): 2274–2286.
96. Zhang Y, Wei W, Cheng N, Wang K, Li B, Jiang X, Sun S. Hepatitis C virus-induced up-regulation of microRNA-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology; 2012; 56(5): 1631–1640.
97. Mignot G, Roux S, Théry C, Ségura E, Zitvogel L: Prospects for exosomes in immunotherapy of cancer. J Cell Mol Med. 2006; 10(2): 376–388.
98. Théry C, Zitvogel L, Amigorena S: Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002; 2(8): 569–579.
99. van Niel G, Porto-Carreiro I, Simoes S, Raposo G. Exosomes: a common pathway for a specialized function. Journal of biochemistry. 2006; 140(1): 13–21.
100. Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, Cheng Z, Shah SV, Wang GJ, Zhang L, Grizzle WE, Mobley J, Zhang HG: Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. 2008; 124(11): 2621–2633.
101. Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, Corbelli A, Fais S, Parmiani G, Rivoltini L: Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res; 2006; 66(18): 9290–9298.
102. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D, De Thonel A, Multhoff G, Hamman A: Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. 2010; 120(2): 457-471.
103. Wang J, De Veirman K, De Beule N, Maes K, De Bruyne E, Van Valckenborgh E, Vanderkerken K, Menu E: The bone marrow microenvironment enhances multiple myeloma progression by exosome-mediated activation of myeloid-derived suppressor cells. Oncotarget. 2015; 6(41): 43992-4004. doi:10.18632/oncotarget.6083.
104. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, Sahoo S, Tahara H, Wauben MH, et al.: Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. JEV. 2014; 3: 26913.
105. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, Blosser RL, Tam AJ, Bruno T, Zhang H, Pardoll D, Kim Y: STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. 2013; 123(4): 1580–1589.
106. Bronte V, Zanovello P: Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005; 5(8): 641–654.
107. Albeituni SH, Ding C, Yan J: Hampering immune suppressors: therapeutic targeting of myeloid-derived suppressor cells in cancer. Cancer J. 2013; 19(6): 490–501.
108. Wang L, Chang EWY, Wong SC, Ong SM, Chong DQY, Ling KL: Increased Myeloid-Derived Suppressor Cells in Gastric Cancer Correlate with Cancer Stage and Plasma S100A8/A9 Proinflammatory Proteins. J Immunol. 2013; 190(2): 794–804.
109. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O’Neill A, Mier J, Ochoa AC: Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005; 65(8): 3044–3048.
110. Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI: Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010; 184(6): 3106–3116.
111. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, Antonia S, Ochoa JB, Ochoa AC: Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004; 64(16): 5839–5849.
112. Serafini P, Mgebroff S, Noonan K, Borrello I: Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008; 68(13): 5439–5449.
113. Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, Castelli C, Mariani L, Parmiani G, Rivoltini L: Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007; 25(18): 2546–2553.
114. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mulé J, Kerr WG, Jove R, Pardoll D, Yu H: Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nature Med. 2005; 11(12): 1314–1321.
115. Kinjyo I, Inoue H, Hamano S, Fukuyama S, Yoshimura T, Koga K, Takaki H, Himeno K, Takaesu G, Kobayashi T, Yoshimura A: Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J Exp Med. 2006; 203(4): 1021–1031.
116. Yu J, Wang Y, Yan F, Li H, Ren X: Response to comment on “Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer”. J Immunol. 2013; 190(11): 5341–5342.
117 Wang Y, Yang BH, Li H, Cao S, Ren XB, Yu JP. IDO⁺ DCs and signalling pathways. Curr cancer drug targets. 2013; 13(3): 278–288.
118. Babcock TA, Carlin JM. Transcriptional activation of indoleamine dioxygenase by Interleukin 1 and tumor necrosis factor alpha in interferon-treated epithelial cells. Cytokine. 2000; 12(6): 588–594.
119. Lechner MG, Megiel C, Russel SM, Bingham B, Arger N, Woo T, Epstein AL. Functional characterization of human Cd33+ And Cd11b+ myeloid-derived suppressor cell subsets induced from peripheral blood mononuclear cells co-cultured with a diverse set of human tumor cell lines. J Transl Med. 2011; 9(1): 90.
120. Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, Servillo G, Brunacci C, Calvitti M, Bicciato S, Mazza EM, Boon L, Grassi F et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nature immunol. 2011; 12(9): 870–878.
121. Pine R. Convergence of TNFalpha and IFNgamma signalling pathways through synergistic induction of IRF-1/ISGF-2 is mediated by a composite GAS/kappaB promoter element. Nucleic Acids Res. 1997; 25(21): 4346–4354.
122. Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, Yan C, Yan F, Ren X. Noncanonical NF- B Activation Mediates STAT3-Stimulated IDO Upregulation in Myeloid-Derived Suppressor Cells in Breast Cancer. J Immunol. 2014; 193(5): 2574–2586.
123. Curti A, Pandolfi S, Valzasina B, Aluigi M, Isodori A, Ferri E, Salvestrini V, Bonanno G, Rutella S, Durelli I, Horenstein AL, Fiore F, Massaia M et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood. 2007; 109(7): 2871–2877.
124. Yu J, Sun J, Wang SE, Li H, Cao S, Cong Y, Liu J, Ren X. Upregulated expression of indoleamine 2, 3-dioxygenase in primary breast cancer correlates with increase of infiltrated regulatory T cells in situ and lymph node metastasis. Clin Dev Immunol. 2011; 2011: 469135.
125. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI: Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009; 182(9): 5693–5701.
126. Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, Colombo MP, Zanovello P: IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003; 170(1): 270–278.
127. Xia Y, Roman LJ, Masters BS, Zweier JL: Inducible nitric-oxide synthase generates superoxide from the reductase domain. J Biol Chem. 1998; 273(35): 22635–22639.
128. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI: Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004; 172(2): 989–999.
129. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI: Mechanism of All-Trans Retinoic Acid Effect on Tumor-Associated Myeloid-Derived Suppressor Cells. Cancer Res. 2007; 67(22): 11021–11028.
130. Ando T, Mimura K, Johansson CC, Hanson MG, Mougiakakos D, Larsson C, Martins da Palma T, Sakurai D, Norell H, Li M, Nishimura MI, Kiessling R: Transduction with the antioxidant enzyme catalase protects human T cells against oxidative stress. J Immunol. 2008; 181(12): 8382–8390.
131. Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P: Control of Bcl-2 expression by reactive oxygen species. Proc Natl Acad Sci U.S.A. 2003; 100(25): 15035–15040.
132. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009; 9(3): 162–174.
133. Jayaraman P, Parikh F, Lopez-Rivera E, Hailemichael Y, Clark A, Ma G, Cannan D, Ramacher M, Kato M, Overwijk WW, Chen SH, Umansky VY, Sikora AG. Tumor-Expressed Inducible Nitric Oxide Synthase Controls Induction of Functional Myeloid-Derived Suppressor Cells through Modulation of Vascular Endothelial Growth Factor Release. J Immunol. 2012; 188(11): 5365–5376.
134. Lee KC, Chang HH, Chung YH, Lee TY. Andrographolide acts as an anti-inflammatory agent in LPS-stimulated RAW264.7 macrophages by inhibiting STAT3-mediated suppression of the NF-κB pathway. J Ethnopharmacol. 2011; 135(3): 678–684.
135. Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell–dependent tumor angiogenesis in mice. J Clin Invest. 2008; 118(10): 3367–3377.
136. Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008; 13(3): 193–205.
137. Zhang W, Zhang C, Li W, Deng J, Herrmann A, Priceman SJ, Liang W, Shen S, Pal SK, Hoon DSB, Yu H: CD8+ T-cell immunosurveillance constrains lymphoid premetastatic myeloid cell accumulation. Eur J Immunol. 2015; 45(1): 71–81.
138. Auphan-Anezin N, Schmitt-Verhulst A-M: Silence STAT3 in the procancer niche… and activate CD8+ T cells to kill premetastatic myeloid intruders. Eur J Immunol. 2015; 45(1): 44–48.
139. Hiratsuka S, Watanabe A, Aburatani H, Maru Y: Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006; 8(12): 1369–1375.
140. Kaplan RN, Rafii S, Lyden D: Preparing the “soil”: the premetastatic niche. Cancer Res. 2006; 66(23): 11089–11093.
141. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, et al.: VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005; 438(7069): 820–827.
142. Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, Geng Y, Gray JW, Moses HL, Yang L: Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010; 70(15): 6139–6149.
143. Thomlinson RH, Gray LH: The histological structure of some human lung cancers and the possible implications for radiotherapy. BJC. 1955; 9(4): 539–549.
144. Otero DC, Poli V, David M, Rickert RC: Cutting edge: inherent and acquired resistance to radiation-induced apoptosis in B cells: a pivotal role for STAT3. J Immunol. 2006; 177(10): 6593–6597.
145. Li X: Silencing STAT3 with short hairpin RNA enhances radiosensitivity of human laryngeal squamous cell carcinoma xenografts in vivo. Exp Ther Med. 2010; 1(6): 947-953.
146. You S, Li R, Park D, Xie M, Sica GL, Cao Y, Xiao ZQ, Deng X: Disruption of STAT3 by Niclosamide Reverses Radioresistance of Human Lung Cancer. Mol Cancer Ther. 2014; 13(3): 606–616.
147. Niu G, Briggs J, Deng J, Ma Y, Lee H, Kortylewski M, Kay H, Cress WD, Jove R, Yu H. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol Cancer Res. 2008; 6(7): 1099–1105.
148. Singh-Gupta V, Zhang H, Banerjee S, Kong D, Raffoul JJ, Sarkar FH, Hillman GG. Radiation-induced HIF-1α cell survival pathway is inhibited by soy isoflavones in prostate cancer cells. Int J Cancer. 2009; 124(7): 1675–1684.
149. Adachi M, Cui C, Dodge CT, Bhayani MK, Lai SY. Targeting STAT3 inhibits growth and enhances radiosensitivity in head and neck squamous cell carcinoma. Oral Oncology.2012; 48(12): 1220–1226.
150. Zhang C, Yang X, Zhang Q, Guo Q, He J, Qin Q, Zhu H, Liu J, Zhan L, Lu J, Liu Z, Xu L, Ma J, et al. STAT3 inhibitor NSC74859 radiosensitizes esophageal cancer via the downregulation of HIF-1α. Tumor Biol. 2014; 35(10): 9793–9799.
151. Spitzner M, Ebner R, Wolff H, Ghadimi B, Wienands J, Grade M: STAT3: A Novel Molecular Mediator of Resistance to Chemoradiotherapy. Cancers. 2014; 6(4): 1986–2011.
152. Demaria S, Formenti SC: Radiotherapy effects on anti-tumor immunity: implications for cancer treatment. Front Oncol. 2013; 3: 123.
153. Vatner RE, Formenti SC: Myeloid-Derived Cells in Tumors: Effects of Radiation. YSRAO. 2015; 25(1): 18–27.
154. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ: Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009; 58(1): 49–59.
155. Bill MA, Fuchs JR, Li C, Yui J, Bakan C, Benson DM, Schwartz EB, Abdelhamid D, Lin J, Hoyt DG, Fossey SL, Young GS, Carson WE III, et al.: The small molecule curcumin analog FLLL32 induces apoptosis in melanoma cells via STAT3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Mol Cancer. 2010; 9: 165.
156. Lu P, Yu B, Xu J: Cucurbitacin B regulates immature myeloid cell differentiation and enhances antitumor immunity in patients with lung cancer. Cancer Biother Radiopharm. 2012; 27(8): 495–503.
157. Tu SP, Jin H, Shi JD, Zhu LM, Suo Y, Lu G, Liu A, Wang TC, Yang CS: Curcumin induces the differentiation of myeloid-derived suppressor cells and inhibits their interaction with cancer cells and related tumor growth. Cancer Prev Res. 2012; 5(2): 205–215.
158. Lin L, Deangelis S, Foust E, Fuchs J, Li C, Li PK, Schwartz EB, Lesinski GB, Benson D, Lü J, Hoyt D, Lin J: A novel small molecule inhibits STAT3 phosphorylation and DNA binding activity and exhibits potent growth suppressive activity in human cancer cells. Mol Cancer. 2010; 9: 217.
159. Zhou J, Wu J, Chen X, Fortenbery N, Eksioglu E, Kodumudi KN, Pk E-B, Dong J, Djeu JY, Wei S: Icariin and its derivative, ICT, exert anti-inflammatory, anti-tumor effects, and modulate myeloid derived suppressive cells (MDSCs) functions. Int Immunopharmacol. 2011; 11(7): 890–898.
160. Ko JS, Rayman P, Ireland J, Swaidani S, Li G, Bunting KD, Rini B, Finke JH, Cohen PA: Direct and Differential Suppression of Myeloid-Derived Suppressor Cell Subsets by Sunitinib Is Compartmentally Constrained. Cancer Res. 2010; 70(9): 3526–3536.
161. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, Dreicer R, Bukowski R, Finke JH: Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009; 15(6): 2148–2157.
162. Kodera Y, Katanasaka Y, Kitamura Y, Tsuda H, Nishio K, Tamura T, Koizumi F: Sunitinib inhibits lymphatic endothelial cell functions and lymph node metastasis in a breast cancer model through inhibition of vascular endothelial growth factor receptor 3. Breast Cancer Res. 2011; 13(3): R66.
163. Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, Wood L, Elson P, Garcia J, Dreicer R, Bukowski R: Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008; 14(20): 6674–6682.
164. Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, Loffe E, Huang T, Rasziejewski C, et al.: VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U.S.A. 2002; 99(17): 11393–11398.
165. Fricke I, Mirza N, Dupont J, Lockhart C, Jackson A, Lee JH, Sosman JA, Gabrilovich DI: Vascular endothelial growth factor-trap overcomes defects in dendritic cell differentiation but does not improve antigen-specific immune responses. Clin Cancer Res. 2007; 13(16): 4840–4848.
166. Kusmartsev S, Eruslanov E, Kübler H, Tseng T, Sakai Y, Su Z, Kaliberov S, Heiser A, Rosser C, Dahm P, Siemann D, Vieweg J: Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol. 2008; 181(1): 346–353.
167. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, Ochoa AC: Arginase I-Producing Myeloid-Derived Suppressor Cells in Renal Cell Carcinoma Are a Subpopulation of Activated Granulocytes. Cancer Res. 2009; 69(4): 1553–1560.
168. Sumida K, Wakita D, Narita Y, Masuko K, Terada S, Watanabe K, Satoh T, Kitamura H, Nishimura T: Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur J Immunol. 2012; 42(8): 2060–2072.
169. Liby KT, Yore MM, Sporn MB: Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007; 7(5): 357–369.
170. Konopleva M, Zhang W, Shi YX, McQueen T, Tsao T, Abdelrahim M, Munsell MF, Johansen M, Yu D, Madden T, Safe SH, Hung MC, Andreeff M: Synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces growth arrest in HER2-overexpressing breast cancer cells. Mol Cancer Ther. 2006; 5(2): 317–328.
171. Ahmad R, Raina D, Meyer C, Kufe D: Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1)-->signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res. 2008; 68(8): 2920–2926.
172. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY: A Novel Chemoimmunomodulating Property of Docetaxel: Suppression of Myeloid-Derived Suppressor Cells in Tumor Bearers. Clin Cancer Res. 2010; 16(18): 4583–4594.
173. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH: The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009; 69(6): 2514–2522.
174. Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, Leonardi AJ, Morgan RA, Wang E, Marincola FM, Trinchiere G, Rosenberg SA, Restifo NP: IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 2011; 121(12): 4746–4757.
175. Liechtenstein T, Perez-Janices N, Blanco-Luquin I, Goyvaerts C, Schwarze J, Dufait I, Lanna A, Ridder MD, Guerrero-Setas D, Breckpot K, Escors D: Anti-melanoma vaccines engineered to simultaneously modulate cytokine priming and silence PD-L1 characterized using ex vivo myeloid-derived suppressor cells as a readout of therapeutic efficacy. Oncoimmunology. 2014; 3(7): e945378.
176. Zoglmeier C, Bauer H, Norenberg D, Wedekind G, Bittner P, Sandholzer N, Rapp M, Anz D, Endres S, Bourquin C: CpG Blocks Immunosuppression by Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. Clin Cancer Res. 2011; 17(7): 1765–1775.
177. Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J Immunol. 2012; 188(4): 1592–1599.
178. Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, Lee H, Scuto A, Liu Y, Yang C,Deng J, Soifer HS, Raubitscheke A, et al.: In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009; 27(10): 925–932.
179. Zhang Q, Hossain DMS, Nechaev S, Kozlowska A, Zhang W, Liu Y, Kowolik CM, Swiderski P, Rossi JJ, Forman S, Pal S, Bhatia R, Raubitscheke A, et al.: TLR9-mediated siRNA delivery for targeting of normal and malignant human hematopoietic cells in vivo. Blood. 2013; 121(8): 1304–1315.
180. Hossain DMS, Pal SK, Moreira D, Duttagupta P, Zhang Q, Won H, Jones J, D’Apuzzo M, Forman S, Kortylewski M: TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin Cancer Res. 2015; 21(16): 3771–3782.
181. I Dufait I, Schwarze JK, Liechtenstein T, Leonard W, Jiang H, Escors D, De Ridder M, Breckpot K: Ex vivo generation of myeloid-derived suppressor cells that model the tumor immunosuppressive environment in colorectal cancer. Oncotarget. 2015; 6(14): 12369–12382. doi: 10.18632/oncotarget.3682.
182. Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH: GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat. 2009; 123(1): 39–49.
183. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI: Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008; 181(8): 5791–5802.
184. Liechtenstein T, Perez-Janices N, Gato M, Caliendo F, Kochan G, Blanco-Luquin I, Van der Jeught K, Arce F, Guerrero-Setas D, Fernandez-Irigoyen J, Santamaria E, Breckpot K, Escors D: A highly efficient tumor-infiltrating MDSC differentiation system for discovery of anti-neoplastic targets, which circumvents the need for tumor establishment in mice. Oncotarget. 2014; 5(17): 7843–7857. doi:10.18632/oncotarget.2279.
185. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, Tolar J, Ochoa AC, Blazar BR: Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010; 116(25): 5738–5747.
186. Lutz M, Kukutsch N, Menges M, Rössner S, Schuler G: Culture of bone marrow cells in GM-CSF plus high doses of lipopolysaccharide generates exclusively immature dendritic cells which induce alloantigen- specific CD4 T cell anergy in vitro. Eur J Immunol. 2000; 30(4): 1048-1052.
187. Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, Restifo NP: Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999; 162(10): 5728–5737.
188. Lechner MG, Liebertz DJ, Epstein AL: Characterization of Cytokine-Induced Myeloid-Derived Suppressor Cells from Normal Human Peripheral Blood Mononuclear Cells. J Immunol. 2010; 185(4): 2273–2284.
189. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P: Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011; 118(20): 5498–5505..
190. Chen MF, Kuan FC, Yen TC, Lu MS, Lin PY, Chung YH, Chen WC, Lee KD: IL-6-stimulated CD11b+ CD14+ HLA-DR- myeloid-derived suppressor cells, are associated with progression and poor prognosis in squamous cell carcinoma of the esophagus. Oncotarget. 2014; 5(18): 8716–8728. doi:10.18632/oncotarget.2368.
191. Zhou Z, French DL, Ma G, Eisenstein S, Chen Y, Divino CM, Keller G, Chen SH, Pan PY: Development and Function of Myeloid-Derived Suppressor Cells Generated from Mouse Embryonic and Hematopoietic Stem Cells. Stem Cells. 2010; 28(3): 620-32.
192. Cheng P, Kumar V, Liu H, Youn JI, Fishman M, Sherman S, Gabrilovich D: Effects of notch signaling on regulation of myeloid cell differentiation in cancer. Cancer Res. 2014; 74(1): 141–152.
193. Xiao G: Soluble NKG2D ligand promotes MDSC expansion and skews macrophage to the alternatively activated phenotype. J Hematol Oncol. 2015; 8: 1-10.
194. Rössner S, Voigtländer C, Wiethe C, Hänig J, Seifarth C, Lutz MB: Myeloid dendritic cell precursors generated from bone marrow suppress T cell responses via cell contact and nitric oxide production in vitro. Eur J Immunol. 2005; 35(12): 3533–3544.
195. Escors D, Liechtenstein T, Perez-Janices N, Schwarze JK, Dufait I, Goyvaerts C, Lanna A, Arce F, Blancp-Luquin I, Kochan G, Guerrero-Setas D, Breckpot K. Assessing T-cell responses in anticancer immunotherapy: Dendritic cells or myeloid-derived suppressor cells? Oncoimmunology. 2013; 2(10): e26148.
196. Goyvaerts C, De Groeve K, Dingemans J, Van Lint S, Robays L, Heirman C, Reiser J, Zhang XY, Thielemans K, De Baetselier P, Raes G, Breckpot K: Development of the Nanobody display technology to target lentiviral vectors to antigen-presenting cells. Gene Ther. 2012; 19(12): 1133–1140.
197. Goyvaerts C, Dingemans J, De Groeve K, Heirman C, Van Gulck E, Vanham G, De Baetselier P, Thielemans K, Raes G, Breckpot K: Targeting of human antigen-presenting cell subsets. J Virol. 2013; 87(20): 11304–11308.
198. Amoozgar Z, Goldberg MS: Targeting myeloid cells using nanoparticles to improve cancer immunotherapy. Adv Drug Deliv Rev. 2015; 91: 38–51.
199. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Förster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999; 10(1): 39–49.
200. Reindl W, Weiss S, Lehr HA, Förster I. Essential crosstalk between myeloid and lymphoid cells for development of chronic colitis in myeloid-specific signal transducer and activator of transcription 3-deficient mice. Immunol. 2007; 120(1): 19–27.
201. Däbritz J, Judd LM, Chalinor HV, Menheniott TR, Giraud AS. Altered gp130 signalling ameliorates experimental colitis via myeloid cell-specific STAT3 activation and myeloid-derived suppressor cells. Sci rep. 2016; 6: 20584.