
INTRODUCTION
Recent work has highlighted the instructive roles of bioelectric signals in large-scale pattern formation during embryogenesis and regeneration [1–7]. We are beginning to understand how these ionic signals function as a layer of physiological control and how they are integrated mechanistically with biochemical and genetic pathways [8–12]. Indeed, because bioelectrical states regulate differentiation, migration, and proliferation [13–17], these physiological circuits are an ideal candidate for an important aspect of the patterning cues that go awry in cancer [18–23]. Bioelectricity has long been implicated in neoplasm [24, 25], and recent molecular efforts have focused on ion channels as important cancer targets [26–33], and ion channel drugs as a promising class of therapies [13, 34–38].
Importantly however, it is beginning to be seen that modulating carcinogenesis and metastasis is not as simple as targeting individual ion channel genes for loss- or gain-of function applications. The key parameter can be a complex, non-cell-autonomous physiological state not intrinsically limited to any one specific channel or pump [3, 39–43]. Thus, it is important to exploit amenable model systems to understand how aspects of cancer are regulated by bioelectrical states of tissues in vivo. However, to fully understand and exploit the control of cell functions via bioelectrical signaling requires the development of new techniques that allow ion flux to be influenced in any cell/tissue of interest with much greater spatio-temporal control than has been possible to date with pharmacological or genetic approaches.
One exciting candidate for achieving improved spatio-temporal control of ion flux is optogenetics [44–47]. Optogenetics is the expression of light-gated ion translocators and enzymes, with different kinetics and different wavelengths of activation, to control downstream processes, such as ion flux or enzyme activity. It has primarily been used as a precise tool to control neural and muscle excitation/inhibition, and to regulate biochemical processes [44, 45]. Extending the use of optogenetics to non-excitable cells [48], our lab has successfully initiated Xenopus tail regeneration by hyperpolarizing cells using the light-dependent H+ pump, Archaerhodopsin, thereby reversing the normal, age-dependent loss of regenerative ability [49]. Building on this work and recent data on the bioelectric control of tumorigenesis [43, 50], we investigated here the potential applicability of optogenetics to hyperpolarization-induced tumor suppression.
Xenopus laevis is a model system that facilitates the investigation of the role of bioelectric signals in morphogenesis and cellular regulation [12, 51–55]. The organism also provides a powerful model system for studying cancer [4, 50, 56–60] due to its tractability for optical, molecular, and electrophysiological studies, and to the many conserved pathways it shares with humans [61–66]. Moreover, this model system offers well characterized tumor-inducing molecular reagents (the human oncogenes Gli1, Xrel3, p53Trp248 and KRASG12D), whose expression results in induced tumor-like structures (ITLSs) with many of the hallmarks of mammalian tumors: increased mitotic activity, induced vasculogenesis, increased hypoxia, acidic microenvironment, disorganization of normal cell architecture, and ability to trigger an innate immune response [3, 4, 50, 67, 68].
In addition to exhibiting other classic tumor characteristics, these tumor-like structures maintain a depolarized membrane voltage. This bioelectric signature can be used to detect prospective ITLS regions before they become morphologically apparent [42]. Moreover, we have shown that this depolarization is not only a signature but is functionally required for tumorigenesis, by demonstrating that artificially hyperpolarizing oncogene-expressing cells, by expressing any of several ion channels, significantly reduces the incidence of ITLS formation despite high levels of otherwise-sufficient oncogene expression in the same tissue [3, 4].
Given the importance of bioelectric signaling, and the need for high-resolution manipulation both for the clinic and for probing the basic biology of this process, we explored whether optogenetics can be used to improve our ability to regulate, in both time and space, the bioelectric signaling that is disrupted during cancer. We hypothesized that spatio-temporally-controllable light-gated ion translocators would enable precise control over ion flux (and therefore membrane voltage (Vmem)) in a tissue of interest, and thus we sought to extend optogenetics to the biophysical control of oncogene-dependent tumorigenesis.
To test the ability of optogenetic tools to alter endogenous Vmem and thereby suppress ITLS growth and promote tissue normalization, we used Archaerhodopsin (Arch) [69], a light-gated H+ transporter that hyperpolarizes cells [49, 70]. We confirm here that microinjection of KRASG12D [71] mRNA, a tumor-inducing KRAS mutant, results in the formation of the ITLSs that we have previously shown to exhibit classic hallmarks of tumors, including histopathology, increased proliferation, lack of differentiation, attraction of vasculature, etc. [3, 4, 41]. We then demonstrate that co-injection of Arch mRNA and subsequent expression of Arch in KRASG12D-expressing cells, followed by a 24-hour light-activation of the Arch transporter, reduced ITLS incidence by 32%. We also show that suppression and normalization of ITLSs are not specific to Arch, but can also be accomplished by injection of a different optogenetic reagent, channelrhodopsin-2 (ChR2D156A) [72]. Most interestingly, by delaying the activation of ChR2D156A until ~ stage 35, we were able to convert fully developed ITLSs into normal cells. Thus we demonstrate the utility of optogenetics to suppress ITLS formation and to promote normalization of existing ITLSs into wildtype tissue.
RESULTS
Injection of KRASG12D results in ITLS formation
To study the usability of Vmem-altering optogenetic tools in oncogene-mediated tumorigenesis, we expressed a human oncogene in Xenopus embryos. Injection of the oncogene KRASG12D [71] into Xenopus embryos (1 blastomere at the 16-cell stage) induced ITLSs (Figure 1A), which have previously been shown to exhibit many of the defining hallmarks of their mammalian counterparts.
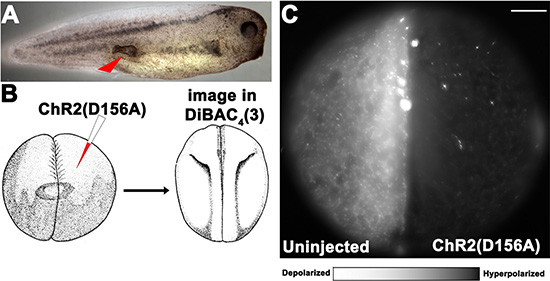
Figure 1: Optogenetic modulation of Vmem to control ITLSs is achieved using a KRASG12D oncogene and light-sensitive ion channels in Xenopus laevis embryos. (A) ITLSs were generated by injecting KRASG12D mRNA into a single blastomere of 16 cell stage embryos. Injected embryos were raised in 0.1 × MMR before they were scored for the presence of ITLSs and imaged using bright field microscopy between stages 28 and 35. (B) Schematic of optogenetic Vmem modulation using a Channelrhodopsin-2 mutant (ChR2D156A) channel: ChR2D156A mRNA was injected into 1 cell of a 2-cell embryo, allowing the uninjected side to serve as an internal control. Embryos were raised to stage 18 in 0.1XMMR. (C) At stage 18, embryos were soaked in 1.9 μM DiBAC4(3) solution in 0.1 × MMR, and imaged using a DiBAC4 (3) filter set (470/20; BS 485; EM 517/23). The un-injected left half of the embryo was highly fluorescent, indicating relative depolarization compared to the right half of the embryo, which is expressing ChR2D156A. Scale bar = 150 μm.

Figure 2: Set up used to deliver spatio-temporally precise light stimulation of optogenetic ion-translocators expressed in Xenopus embryo ITLSs. We customized a Nikon AZ100 dissection scope for in vivo optogenetics [87] by replacing the epifluorescence illumination source and light guide with a Spectra4 LED illuminator connected to the scope via fiber optic cable (the black light guide that passes in front of the vacuum source). The light is passed through a pinhole to set spot size diameter (located behind the oculars, not visible in this image), then enters the scope and goes through an 80/20 splitter that allows the user to view the specimen on the monitor even while the LED is on. Finally, the light passes through the 5 × objective lens which further reduces spot diameter and aims the spot at the sample. An automated Ludl MAC6000 XY stage (that can also be manipulated manually by a joystick) allows multiple embryos to be exposed repeatedly to the activating wavelength of light. Up to thirty embryos are loaded into a slide-mounted PDMS “chip” designed to use microfluidics to hold embryos in place [100]; the chip is held to the slide by a vacuum and 0.1 × MMR is circulated by a peristaltic pump. The optogenetics components and the microscope are all controlled by NIS Elements.
ChR2D156A activity alters membrane voltage of Xenopus embryonic cells
To allow modulation of Vmem via light activation of a channelrhodopsin-2 (ChR2D156A) channel, ChR2D156A mRNA was injected into 1 cell of a 2-cell embryo, allowing the uninjected side to serve as an internal control (Figure 1). ChR2D156A is a non-specific cation channel that, at physiological pH, passes mostly protons, but there is also significant Na+ and K+ flux [45] [73]; it was selected because of the low incidence of side effects in Xenopus embryos [48]. Because of the extremely low ion concentration of the surrounding medium, light activation of this channel is predicted to hyperpolarize those cells due to efflux of cations; Table 1 gives the internal and external ion concentrations. Injected embryos, raised to stage 18, were exposed to blue light then imaged using the membrane voltage-sensitive dye DiBAC4 (3), a semi-quantitative method that has been extensively used to monitor relative resting-potential differences among cells in vivo [49, 54, 74–78]. As predicted, we observed that the injected right half of the embryo was much dimmer, indicating relative hyperpolarization compared to the uninjected left side.
Table 1: Ion concentrations in Xenopus embryonic cells and their medium (from [102])
Ion |
External medium [mM] |
Intracellular [mM] |
|---|---|---|
Na+ |
9.9 |
38 |
K+ |
0.2 |
51 |
Ca++ |
0.3 |
5 |
Mg++ |
0.2 |
12 |
H+ |
1.60E–05 |
1.80E–05 |
Cl– |
11.1 |
30 |
Arch and ChR2D156A activities reduce KRASG12D-induced ITLS incidence
Co-injection of mRNAs for KRASG12D and either Arch or ChR2D156A into one cell of a 16-cell stage embryo resulted in the expression of the corresponding proteins as early as 4 hours post injection (data not shown). Embryos expressing light-activated ion channels were exposed to light for 24 hours: arch-expressing embryos were stimulated by green light, 555 nm, irradiance of 1 mW/mm2 for 500 ms followed by 1.5s in darkness, while ChR2D156A-expressing cells were stimulated by blue light: 470 nm light of 2.4 mW/mm2 for 10 ms every 30 seconds (Figure S2, 3A; [48]). To study ITLS prevention, embryos were exposed to the light beginning four hours post injection, or, approximately stage 9; to induce normalization of tumors, exposure was from stage 28 to stage 35. Two sets of controls were used for comparison: embryos injected with only KRASG12D and un-stimulated embryos expressing the oncogene and either Arch or ChR2D156A (light by itself does not affect Vmem of cells that do not express light-gated channels, and tumor incidence does not vary among KRAS-only injected embryos kept in dark, ambient light, or blue/red optogenetic exposure [data not shown]).
Consistent with our previous finding that hyperpolarization suppresses ITLS formation, our experimental data show that Arch activity significantly reduces the number of embryos that develop KRASG12D ITLSs by 32% compared to KRASG12D-expressing embryos lacking Arch (χ2 = 7.1, p = 0.007) (Figure 3B). Similarly, the activity of ChR2D156A was able to significantly lower KRASG12D ITLS incidence by 31.4% (χ2 = 6.8, p = 0.009) (Figure 3B). Together, these data show that light-activated Arch and ChR2D156A can reduce ITLS formation by KRASG12D in non-excitable tissues, suggesting that light-dependent activity of optogenetic reagents is a Vmem-altering modality with efficacy in the suppression of ITLS formation.
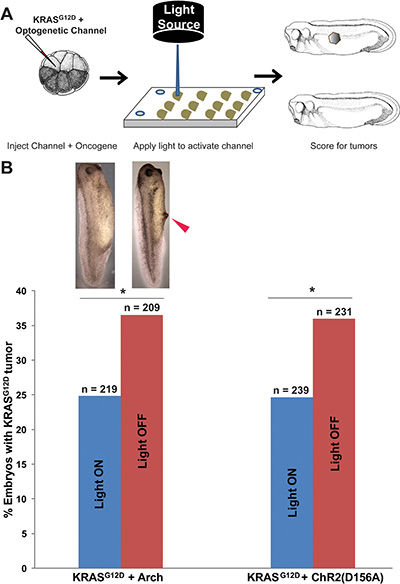
Figure 3: Expression of optogenetic channels Arch and ChR2D156A prior to ITLS appearance suppresses the formation of KRASG12D ITLSs. (A) Schematic of experimental design for ITLS suppression: mRNAs for KRASG12D and ChR2D156A or Arch were co-injected into a single blastomere of 16-cell stage embryos; between 4 and 28 hours post injection (a total exposure of 24 hours), injected embryos were exposed to 580 nm wavelength of light, 1 mw/mm2 irradiance (stimulating Arch) or 450 nm wavelength of light with 2.4 mw/mm2 irradiance (stimulating ChR2D156A). Arch and ChR2D156A experiments were done separately to test the hypothesis that ITLS suppression is due to changes in membrane voltage as opposed to channel or ion specific properties. Light-stimulated embryos were raised to stage 35 and scored for ITLSs to assess the efficacy of hyperpolarizing optogenetic channels as ITLS suppressing reagents. (B) The two different hyperpolarizing translocators – based on active pumping of H+ (Arch) and passive diffusion of positive, monovalent cations (ChR2D156A) – both resulted in suppression of ITLSs, demonstrating that this effect is likely due to a change in Vmem and is not tied to one channel protein. 32% and 31% fewer embryos with ITLS were observed among Arch and ChR2D156A injected embryos, respectively (Arch: c2 = 7.1, *p = 0.007; ChR2D156A: χ2 = 6.8, *p = 0.009. χ2 values are for comparisons of ITLS incidence in light stimulated versus un-stimulated embryos).
Light-initiated ChR2D156A activity normalizes fully developed KRASG12D ITLSs
We next tested the utility of optogenetics in the conversion of existing ITLSs into normal tissue. Similar to the suppression experiments, ChR2D156A was co-expressed with KRASG12D; in these experiments, however, stimulation of the light-dependent channel was delayed until stages between 28–35 when ITLSs were fully developed. Tadpoles were then scored for presence or absence of ITLSs when they reached stages 45–47 (Figure 4A). Stimulation of ChR2D156A-expressing cells within the tumors resulted in 31% more embryos having normalized their tumors – compared to their injected but un-stimulated counterparts (χ2 = 8.6, *p = 0.003) (Figure 4B). These results demonstrate that ChR2D156A increases the number of oncogene-induced ITLSs that are normalized.
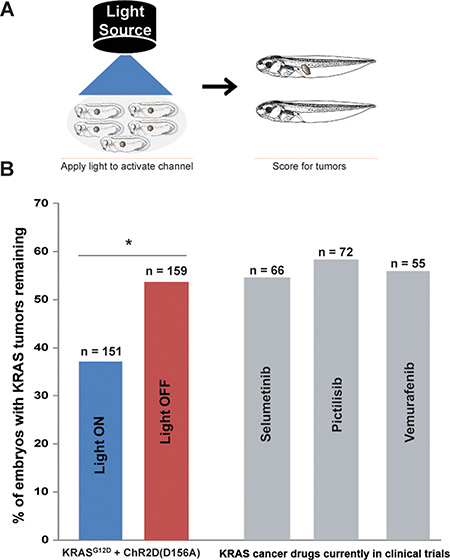
Figure 4: After ITLS formation, activation of ChR2D156A normalizes KRASG12D ITLSs. (A) Schematic of experimental design for ITLS normalization: ChR2D156A expressing embryos (stages 28–35) with KRASG12D ITLSs were subjected to 450 nm wavelength of light with irradiance of 0.5 mw/mm2. Light-stimulated embryos were raised to stage 45–47 and scored for ITLSs to assess the ability of optogenetic Vmem modulation to normalize ITLSs. (B) Light activation of ChR2D156A in ITLSs (blue bar) resulted in 31% more embryos with normalized ITLSs compared to their un-stimulated counterparts (red bar) (χ2 = 8.6, *p = 0.003). By comparison, treatment with the highly-selective MEK 1 inhibitor (Selumetinib), a potent PI3K inhibitor (Pictillisib), or a potent inhibitor of oncogenic B-RAF kinase activity (Vemurafenib), resulted in the same KRASG12D tumor incidence as in the unstimulated control.
Moreover, optogenetic stimulation had a larger effect than several drugs considered promising anti-tumor therapies in human cancer medicine: the highly-selective MEK 1 inhibitor Selumetinib; the potent Pi3K inhibitor Pictillisib; and the inhibitor of oncogenic B-RAF kinase activity Vemurafenib (grey bars, Figure 4B).
ChR2D156A activity regulates muscle marker expression in KRAS-induced ITLSs
To determine whether there is a link between ChR2D156A activation and the expressions of tumor-specific transcriptional markers, we examined chd15 (a satellite cell marker expressed in differentiating myoblasts) and myod1 (a developing muscle cell marker expressed in newly formed somites and involved in differentiation). We chose these two markers because the KRASG12D induced rhabdomyosarcoma arises from skeletal muscle tissue, and because these markers signal the presence of rhabdomyosarcoma in human samples [79–81]. As expected from comparison with KRASG12D-induced human tumors, ITLSs observed growing in a transgenic line of animals that drive muscle-specific GFP fluorescence [82] revealed the presence of ectopic muscle cells (Figure 5). Using qPCR, we next found that both muscle markers were significantly up-regulated in tadpoles with KRASG12D tumors (Figure 6, green bars) compared to uninjected controls (Figure 6, blue bars); however, the expressions of myod1 and cdh15 became normalized to control levels when ChR2D156A was activated in tadpoles (Figure 6, red bars). Taken together, these data show that optogenetic modulation of ion flux and resting potential reduces the expression of tumor markers as well as normalizes the tissue structure.
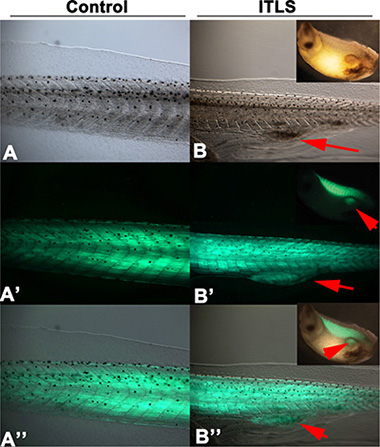
Figure 5: Muscle specific expression of GFP3 controlled by the cardiac actin (Car) promoter is present in KRASG12D ITLS. Tadpoles injected with Tol2-CarPr-GFP3 [101] at the 2-cell stage display normal muscle specific localization of GFP3 within the somites (A, A’, A” showing transmitted light, GFP fluorescence, and both, respectively). In contrast, Tol2-CarPr-GFP3 tadpoles also injected with KRASG12D reveal a strong GFP3 signal in ITLS’s, confirming the presence of ectopic muscle in the KRAS-induced tumor-like structures.
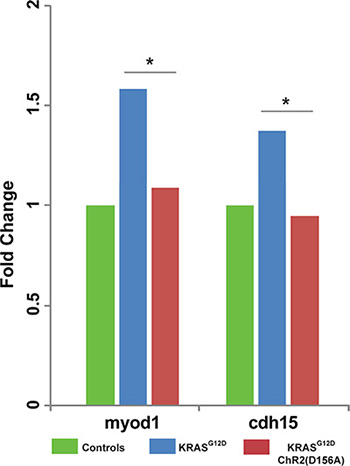
Figure 6: Light activation of ChR2D156A results in downregulated expression of ectopic muscle markers. Human tumors associated with KRAS mutations express the muscle markers myod1 and cdh15. Using qPCR (see Materials and Methods for details), we found that expression of myod1 and cdh15 was likewise up-regulated in tadpoles with KRASG12D-induced ITLSs. Moreover, activation of ChR2D156A in these tadpoles significantly lowered myod1 and cdh15 expressions, down to baseline levels (χ2 test compared to controls, *p < 0.01).
DISCUSSION
Given the instructive role of bioelectric parameters in orchestrating cell behavior towards adaptive pattern formation [53, 83–85], our lab is interested in using modulation of resting potential in vivo as an approach to understanding misregulation of developmental signaling as occurs in cancer, birth defects, and other disease conditions. Previous work has shown that tumor-like structures induced in Xenopus laevis embryos by mammalian oncogenes (Figure 1A) exhibit many of the properties of tumors: overproliferation, expression of known tumor markers, attraction of vasculature, and histological disorganization [3, 40]. They also possess a depolarized potential relative to adjacent cells that are not part of the ITLS – a property long-known to be associated with cancer cells [24, 86], that was recently shown to be an instructive factor, not merely a read-out; actively preventing this depolarization can prevent the appearance of ITLSs [4]. In this study, we extend our ability to control tumorigenic pathways by expanding the use of optogenetics to this new system, and we exploit the temporal and cellular resolution afforded by light-gated ion channels to remove ITLSs after they have already been formed.
We have previously demonstrated the functionality of Arch in Xenopus embryonic and larval tissue: light activation of this hyperpolarizing H+ pump restores tail regeneration capability at non-regenerative stages [49]. Here, we show that light activation of another light-gated channel (ChR2D156A) also induces hyperpolarization in non-excitable Xenopus cells (Figure 1C). This differs from mammalian neurons where high Na+ concentration in the medium favors influx of the cation through channels, resulting in depolarization; Xenopus embryos grow in very-low extracellular Na+ medium, thus opening of cation channels results in hyperpolarization [87].
Channelrhodopsin variant D156A was chosen because of many optogenetic constructs tested [48] it caused the fewest unwanted side effects on embryogenesis. While ChR2 does exhibit some desensitization under illumination, this was not a problem in this application because high spiking rates are not used to encode information in this kind of developmental bioelectricity context [45]. Moreover, exposure of ChR2D156A was limited to 10 ms pulses, followed by 30s of darkness, i.e. a regimen than minimizes desensitization [88] and the shift away from a mixture of positive cations to primarily proton flux [89]. Thus, we are confident that stimulation of ChR2D156A led to the hyperpolarization we measured due to efflux of a mixture of positive cations. We found that the surface expression of Arch and ChR2D156A coupled with the non-toxic, high temporal-resolution light regimen, makes the optogenetic approach (Figure 2) suitable for targeting tumorigenesis in vivo.
We successfully exploited light-induced hyperpolarization of Arch expressing cells to decrease ITLS incidence among embryos expressing the KRASG12D oncogene (Figure 3A, 3B). Importantly, compared to expressing constitutively-open hyperpolarizing channels, the light regimen was delivered for only 24 hours, was temporally more precise and convenient, and the technique is equally effective at suppressing ITLSs. To rule out any Arch H+-pump-specific ITLS suppression effects, we also employed light activation of the ChR2D156A non-specific cation channel in similar experiments (Figure 3A), thereby generating hyperpolarizing current by a different protein and a different mechanism, finding that passive cation efflux, like active H+-efflux, reduced the number of embryos with KRASG12D-induced ITLSs (Figure 3B). The results obtained using the two distinct optogenetic channels that are not restricted to one type of ion flux, are consistent with Vmem per se being the instructive factor in regulating oncogene-mediated tumorigenesis [4, 10]. We also demonstrate for the first time that light-gated channels expressed in cells of tadpoles with KRAS ITLSs can be non-invasively activated for a duration sufficient to significantly increase the frequency of ITLS normalization, an exciting finding suggesting the possibility of remission-induction in addition to prevention (Figure 3, 4). Moreover, we show that the performance of this non-invasive optogenetic stimulation is superior to other anti-tumor agents we tested, including those that have been shown to be promising in human cancer medicine: Selumetinib, a highly-selective MEK 1 inhibitor; Pictillisib, a potent Pi3K inhibitor; and Vemurafenib, a potent inhibitor of oncogenic B-RAF kinase activity, (grey bars, Figure 4B). While these are the first data testing these compounds in the Xenopus tumor assay, we cannot rule out that subsequent research could identify a treatment regime that would allow these compounds to be more efficacious.
Our qPCR data (Figure 6) indicate that light-induced hyperpolarization of tumors also involves normalization of the expression of tumor markers, although it must be kept in mind that mRNA data do not necessarily reflect a linear correspondence to the presence of protein. Together, the data show that manipulation of bioelectric cell state, a powerful, tractable regulator of cancer cell normalization and reprogramming, is possible using optogenetics, thus introducing a new class of biomedical strategies for tumor treatment. The recent development of pharmacological approaches to render existing ion channels light-sensitive [90–92] suggests a next-generation approach that would not require introduction of transgenes (optogenetic channels) into the target tissue.
In summary, we report here the first use of optogenetics as a temporally precise regulator of Vmem to suppress and normalize oncogene-induced ITLSs. More broadly, this tool will advance the study of resting potential as another important component of the microenvironment that is so crucial for cancer initiation and progression [93–97]. Our implementation of light-gated bioelectrical signaling in vivo highlights another opportunity for the optogenetic toolbox to extend beyond excitable cells. Moreover, our data suggest a light-based therapeutics strategy that couples gene therapy with optogenetics to counteract tumorigenesis and promote regression in vivo.
MATERIALS AND METHODS
Animal husbandry
Xenopus laevis eggs were fertilized in vitro, and embryos were cultured according to standard protocols [98], in 0.1 × Modified Marc’s Ringers (MMR; pH 7.8) with 0.1% Gentamicin. Xenopus embryos were housed at 14–18°C and staged according to Nieuwkoop and Faber [99]. All experimental procedures involving the use of animals for experimental purposes were approved by the Institutional Animal Care and Use Committees (IACUC) and Tufts University Department of Lab Animal Medicine (DLAM) under the protocol number M2014–79.
Microinjection
Fertilized Xenopus embryos were transferred into mesh-bottomed dishes with 3% Ficoll and injected with capped, synthetic mRNAs (made using the Ambion Message Machine kit) dissolved in water at the stages indicated. The doses per cell were KRASG12D [71] 40pg; Arch [69], 60pg; and ChR2D156A [72] 50pg. Two hours after injection, embryos were transferred into 0.75 × MMR for 45 minutes before they were washed and cultured in 0.1 × MMR until desired stage was reached. Injected embryos were stimulated with the appropriate wavelength of light and irradiance before or after ITLSs fully form (stages 28–35). Embryos were scored for the presence of ITLSs using bright field microscopy as described in [3, 41, 42].
Light stimulation
During ITLS suppression experiments, an optogenetic set up (Figure 2) was used so that each embryo, in a microfluidic chip placed on a motorized stage, individually received a light regimen with the following parameters: Arch – 580 nm wavelength, 1 mw/mm2 irradiance, 500 ms on, 1.50s off; Ch2R(D156A) – 450 nm wavelength, 2.4 mw/mm2 irradiance, 10 ms on, 30s off. For normalization experiments involving Ch2R (D156A), a petridish of embryos with KRASG12D ITLS were placed beneath an array of six LEDs delivering 450 nm of light with an irradiance of 0.5 mw/mm2.
Vmem imaging
DiBAC4(3) (bis-(1, 3-dibutylbarbituric acid) trimethine oxonol) (DiBAC; Biotium, Inc, Hayward, CA, USA) was used to measure relative polarization. Light-stimulated embryos were transferred into a DiBAC4(3) solution (1.9 mM stock in DMSO used at 1:1000 in 0.1 × MMR), and imaged while still in the DiBAC4(3) solution. An Olympus BX-61 equipped with a Hamamatsu ORCA AG CCD camera, controlled by MetaMorph software, was used for imaging. DiBAC4(3) filters were: EX 470/20; BS 485; EM 517/23 (Chroma filter set 41001).
RNA extraction and cDNA synthesis
RNA extraction was achieved using RNeasy Mini Kit (50) (QIAGEN 74104). Tadpoles (collected n = 10 per Eppendorf tube, three biological replicates) were put into 10 volumes of RNAlater solution. RNAlater-stabilized tissues were then removed from the reagent using forceps and placed into 2 ml Eppendorf tubes with 600 ul of B-ME containing Buffer RLT for disruption and homogenization. Disruption and homogenization of tissue was achieved using MICROSONTM XL 2000 ULTRASONIC LIQUID PROCESSOR. The lysate was then centrifuged for 3 minutes at full speed and the supernatant (lysate) was put into a new microcentrifuge tube. After adding and pipet-mixing 1 volume of 70% Ethanol into the lysate, 700 ul of the resultant sample was transferred to RNeasy spin column. Following the steps outlined in the RNeasy MiniHandbook 04/2006, we completed series of treatments with buffer RW1, RPE and RNase-free water to obtain total RNA. RNA yield and quality were assessed by spectrophotometry (ND-1000, NanoDrop) and gel electrophoresis, respectively, to assess integrity of 28S and 18S RNA.
Reverse transcription was performed using ThermoScript RT-PCR System (Life Technologies). Each in vitro reverse transcription reaction was performed using 1 μg of total RNA and 50 μg of oligo(dT)20 primers (Life Technologies). RNA and primers were mixed, denatured for 5 min at 65°C, and placed on ice before adding the reaction mix according to the manufacturer’s instructions. Reverse transcription reaction was carried out at 50°C for 45 min. The reaction was terminated by incubating at 85°C for 5 min, followed by RNA degradation using 1 μg of RNase H for 20 min at 37°C. The complementary DNA (cDNA) was stored at −20°C until use. The quality and quantity of cDNA were validated using Advantage 2 PCR kit (Clontech) on cDNA samples using Orinithine Decarboxylase (ODC) primers.
Quantitative real-time PCR
Primers were designed using Genius for myogenic differentiation 1 (Myod1) and myogenic differentiation 1 (Cdh15). ODC, a widely used endogenous control for Xenopus, was used to normalize target gene expression. The PCR specificity was verified by BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) using the National Center for Biotechnology Information X. laevis reference sequence. Desalted primers were obtained from Invitrogen by Life Technologies as follows: MyoD1-Forward CCGAGGGCAGTCCCTGTT; MydoD1-Reverse TGGGA CAGTTGAGTGCAGG; Cdh15-Forward ACAATCGT CCAGTGTTTGTGC; Cdh15-Reverse GTTCAGCATT GTCTGTCCTTGG. For each primer pair, standard curve primer analysis was performed using serial dilutions of cDNA from control embryos [1 (undiluted), 10−1, 10−2, 10−3]. Formation of primer-dimer and amplification specificity was assessed by efficiency and melt curve analysis. The cDNA from validated RNA was used to perform RT-qPCR assays. For each biological sample, three technical replicates were run in each RT-qPCR experiment. Each treatment contained five biological replicates. Triplicate negative controls lacking template were also run for each cDNA sample for each reaction. PCRs were assembled manually. Samples were prepared by adding 1 μl of cDNA (diluted 1:5 in ddH2O), 10 μl of 2 × Power SYBR Green PCR Master Mix (Applied Biosystems), and 0.5 μl of each primer (diluted to 10 μM) in a final volume of 20 μl. Reactions were incubated in 96-well MicroAmp Optical Reaction plates at 95°C for 10 min followed by 40 cycles at 95°C for 15 s and at 60°C for 1 min in a StepOnePlus qPCR instrument (Applied Biosystems). The RT-qPCR data were analyzed using the StepOne software v.2.3, and ΔΔCT values were calculated (Applied Biosystems). Fold change of target genes relative to the amount of the control gene ODC was calculated as 2^-ΔΔCT.
Statistics
Following the appropriate light regimens, stimulated construct-expressing embryos were compared – for ITLS incidence – to their expressing but un-stimulated counterparts using a χ2 test (α = 0.01).
Pharmacological agents
The stages selected for the antineoplastic drugs mirror those of the light treatment. Embryos with ITLS between St. 28–35 were selected, treated with the drugs, and scored for ITLS between St. 45–47. Concentration of the drugs and their effects on control embryos vs embryos with ITLS are given below. Following the manufacture’s instruction we prepared stock solutions in DMSO of Selumetinib, Pictilisib, and Vemurafenib in concentration of 100 mM, 50 mM, and 25 mM, respectively. Embryos were then exposed in 0.1 × MMR for the stages indicated to: Selumetinib –100 nMm, Pictillisib –1 μM, and Vemurafenib –1 μM. All three compounds were obtained from Selleckchem.
ACKNOWLEDGMENTS AND FUNDING
We thank Leonard Zon for the KRAS mutant construct, Edward Boyden for Arch construct, and Christian Bamann for ChR2D156A construct. We thank Amber Currier, Erin Switzer, for frog husbandry, Amanda Allen and Rakela Lubonja for general lab assistance, and Joan Lemire and Jean-Francois Pare for molecular biology assistance. This work was supported by a grant from The G. Harold and Leila Y. Mathers Charitable Foundation.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Adams DS, Levin M. Endogenous voltage gradients as mediators of cell-cell communication: strategies for investigating bioelectrical signals during pattern formation. Cell Tissue Res. 2013; 352:95–122.
2. Adams DS. A new tool for tissue engineers: ions as regulators of morphogenesis during development and regeneration. Tissue engineering. 2008; 14:1461–1468.
3. Chernet BT, Levin M. Transmembrane voltage potential of somatic cells controls oncogene-mediated tumorigenesis at long-range. Oncotarget. 2014; 5:3287–3306. doi: 10.18632/oncotarget.1935.
4. Chernet BT, Levin M. Transmembrane voltage potential is an essential cellular parameter for the detection and control of tumor development in a Xenopus model. Dis Model Mech. 2013; 6:595–607.
5. Tseng A, Levin M. Cracking the bioelectric code: Probing endogenous ionic controls of pattern formation. Communicative & Integrative Biology. 2013; 6:1–8.
6. Levin M. Bioelectric mechanisms in regeneration: Unique aspects and future perspectives. Semin Cell Dev Biol. 2009; 20:543–556.
7. Levin M. Large-scale biophysics: ion flows and regeneration. Trends Cell Biol. 2007; 17:261–270.
8. Levin M. Reprogramming cells and tissue patterning via bioelectrical pathways: molecular mechanisms and biomedical opportunities. Wiley Interdiscip Rev Syst Biol Med. 2013.
9. Pullar CE. (2011). The physiology of bioelectricity in development, tissue regeneration, and cancer. Biological effects of electromagnetics series. (Boca Raton: CRC Press).
10. Chernet B, Levin M. Endogenous Voltage Potentials and the Microenvironment: Bioelectric Signals that Reveal, Induce and Normalize Cancer. J Clin Exp Oncol. 2013; S1.
11. Levin M. Endogenous bioelectrical networks store non-genetic patterning information during development and regeneration. The Journal of physiology. 2014; 592:2295–2305.
12. Pai VP, Martyniuk CJ, Echeverri, K., Sundelacruz, S., Kaplan, DL, Levin, M. Genome-wide analysis reveals conserved transcriptional responses downstream of resting potential change in Xenopus embryos, axolotl regeneration, and human mesenchymal cell differentiation. Regeneration. 2015; in minor revision.
13. Rao VR, Perez-Neut M, Kaja S, Gentile S. Voltage-gated ion channels in cancer cell proliferation. Cancers (Basel). 2015; 7:849–875.
14. Sundelacruz S, Levin M, Kaplan DL. Role of membrane potential in the regulation of cell proliferation and differentiation. Stem cell reviews and reports. 2009; 5:231–246.
15. Blackiston DJ, McLaughlin KA, Levin M. Bioelectric controls of cell proliferation: ion channels, membrane voltage and the cell cycle. Cell Cycle. 2009; 8:3519–3528.
16. Urrego D, Tomczak AP, Zahed F, Stuhmer W, Pardo LA. Potassium channels in cell cycle and cell proliferation. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2014; 369:20130094.
17. Becchetti A. Ion channels and transporters in cancer. 1. Ion channels and cell proliferation in cancer. American journal of physiology Cell physiology. 2011; 301:C255–265.
18. Kasemeier-Kulesa JC, Teddy JM, Postovit LM, Seftor EA, Seftor RE, Hendrix MJ, Kulesa PM. Reprogramming multipotent tumor cells with the embryonic neural crest microenvironment. Dev Dyn. 2008; 237:2657–2666.
19. Hess AR, Margaryan NV, Seftor EA, Hendrix MJ. Deciphering the signaling events that promote melanoma tumor cell vasculogenic mimicry and their link to embryonic vasculogenesis: role of the Eph receptors. Dev Dyn. 2007; 236:3283–3296.
20. Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer. 2007; 7:246–255.
21. Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nature reviews Cancer. 2003; 3:411–421.
22. Tarin D. Clinical and Biological Implications of the Tumor Microenvironment. Cancer Microenviron. 2012.
23. Maffini MV, Calabro JM, Soto AM, Sonnenschein C. Stromal regulation of neoplastic development: age-dependent normalization of neoplastic mammary cells by mammary stroma. The American journal of pathology. 2005; 167:1405–1410.
24. Binggeli R, Weinstein R. Membrane potentials and sodium channels: hypotheses for growth regulation and cancer formation based on changes in sodium channels and gap junctions. J Theor Biol. 1986; 123:377–401.
25. Burr HS, Strong LC, Smith GM. Bioelectric correlates of methylcolantherene-induced tumors in mice. Yale J Biol Med. 1938; 10:539–544.
26. Yang M, Brackenbury WJ. Membrane potential and cancer progression. Front Physiol. 2013; 4:185.
27. Chen J, Luan Y, Yu R, Zhang Z, Zhang J, Wang W. Transient receptor potential (TRP) channels, promising potential diagnostic and therapeutic tools for cancer. Biosci Trends. 2014; 8:1–10.
28. Lang F, Stournaras C. Ion channels in cancer: future perspectives and clinical potential. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2014; 369:20130108.
29. Onkal R, Djamgoz MB. Molecular pharmacology of voltage-gated sodium channel expression in metastatic disease: clinical potential of neonatal Nav1.5 in breast cancer. Eur J Pharmacol. 2009; 625:206–219.
30. Fraser SP, Pardo LA. Ion channels: functional expression and therapeutic potential in cancer. Colloquium on Ion Channels and Cancer. EMBO reports. 2008; 9:512–515.
31. Brackenbury WJ, Isom LL. Voltage-gated Na+ channels: potential for beta subunits as therapeutic targets. Expert opinion on therapeutic targets. 2008; 12:1191–1203.
32. Huang X, He Y, Dubuc AM, Hashizume R, Zhang W, Reimand J, Yang H, Wang TA, Stehbens SJ, Younger S, Barshow S, Zhu S, Cooper MK, et al. EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nat Neurosci. 2015; 18:1236–46. doi: 10.1038/nn.4088.
33. Djamgoz MB, Coombes RC, Schwab A. Ion transport and cancer: from initiation to metastasis. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2014; 369:20130092.
34. Arcangeli A, Pillozzi S, Becchetti A. Targeting ion channels in leukemias: a new challenge for treatment. Current medicinal chemistry. 2012; 19:683–696.
35. Arcangeli A, Crociani O, Lastraioli E, Masi A, Pillozzi S, Becchetti A. Targeting ion channels in cancer: a novel frontier in antineoplastic therapy. Current medicinal chemistry. 2009; 16:66–93.
36. Frede J, Fraser SP, Oskay-Ozcelik G, Hong Y, Ioana Braicu E, Sehouli J, Gabra H, Djamgoz MB. Ovarian cancer: Ion channel and aquaporin expression as novel targets of clinical potential. Eur J Cancer. 2013; 49:2331–2344.
37. Perez-Neut M, Rao VR, Gentile S. hERG1/Kv11.1 activation stimulates transcription of p21waf/cip in breast cancer cells via a calcineurin-dependent mechanism. Oncotarget. 2015; doi: 10.18632/oncotarget.3797.
38. Perez-Neut M, Shum A, Cuevas BD, Miller R, Gentile S. Stimulation of hERG1 channel activity promotes a calcium-dependent degradation of cyclin E2, but not cyclin E1, in breast cancer cells. Oncotarget. 2015; 6:1631–1639. doi: 10.18632/oncotarget.2829.
39. Blackiston D, Adams DS, Lemire JM, Lobikin M, Levin M. Transmembrane potential of GlyCl-expressing instructor cells induces a neoplastic-like conversion of melanocytes via a serotonergic pathway. Disease models & mechanisms. 2011; 4:67–85.
40. Lobikin M, Chernet B, Lobo D, Levin M. Resting potential, oncogene-induced tumorigenesis, and metastasis: the bioelectric basis of cancer in vivo. Physical biology. 2012; 9:065002.
41. Chernet BT, Fields C, Levin M. Long-range gap junctional signaling controls oncogene-mediated tumorigenesis in Xenopus laevis embryos. Front Physiol. 2015; 5:519.
42. Chernet BT, Levin M. Transmembrane voltage potential is an essential cellular parameter for the detection and control of tumor development in a Xenopus model. Disease models & mechanisms. 2013; 6:595–607.
43. Chernet B, Levin M. Endogenous Voltage Potentials and the Microenvironment: Bioelectric Signals that Reveal, Induce and Normalize Cancer. J Clin Exp Oncol. 2013; Suppl 1.
44. Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci. 2011; 34:389–412.
45. Knopfel T, Lin MZ, Levskaya A, Tian L, Lin JY, Boyden ES. Toward the second generation of optogenetic tools. J Neurosci. 2010; 30:14998–15004.
46. Liu X, Tonegawa S. Optogenetics 3.0. Cell. 2010; 141:22–24.
47. Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, Goshen I, Thompson KR, Deisseroth K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010; 141:154–165.
48. Adams DS, Lemire JM, Kramer RH, Levin M. Optogenetics in Developmental Biology: using light to control ion flux-dependent signals in Xenopus embryos. The International journal of developmental biology. 2014; 58:851–861.
49. Adams DS, Tseng AS, Levin M. Light-activation of the Archaerhodopsin H(+)-pump reverses age-dependent loss of vertebrate regeneration: sparking system-level controls in vivo. Biology open. 2013; 2:306–313.
50. Lobikin M, Chernet B, Lobo D, Levin M. Resting potential, oncogene-induced tumorigenesis, and metastasis: the bioelectric basis of cancer in vivo. Phys Biol. 2012; 9:065002.
51. Pai VP, Aw S, Shomrat T, Lemire JM, Levin M. Transmembrane voltage potential controls embryonic eye patterning in Xenopus laevis. Development. 2012; 139:313–323.
52. Tseng AS, Levin M. Transducing bioelectric signals into epigenetic pathways during tadpole tail regeneration. Anatomical record. 2012; 295:1541–1551.
53. Levin M. Molecular bioelectricity in developmental biology: new tools and recent discoveries: control of cell behavior and pattern formation by transmembrane potential gradients. Bioessays. 2012; 34:205–217.
54. Pai VP, Lemire JM, Pare JF, Lin G, Chen Y, Levin M. Endogenous Gradients of Resting Potential Instructively Pattern Embryonic Neural Tissue via Notch Signaling and Regulation of Proliferation. The Journal of Neuroscience. 2015; 35:4366–4385.
55. Blackiston DJ, Anderson GM, Rahman N, Bieck C, Levin M. A Novel Method for Inducing Nerve Growth via Modulation of Host Resting Potential: Gap Junction-Mediated and Serotonergic Signaling Mechanisms. Neurotherapeutics. 2015; 12:170–184.
56. Nutt LK. The Xenopus oocyte: a model for studying the metabolic regulation of cancer cell death. Semin Cell Dev Biol. 2012; 23:412–418.
57. Nedelkovska H, Robert J. Hsp72 mediates stronger antigen-dependent non-classical MHC class Ib anti-tumor responses than hsc73 in Xenopus laevis. Cancer Immun. 2013; 13:4.
58. Cross MK, Powers MA. Learning about cancer from frogs: analysis of mitotic spindles in Xenopus egg extracts. Disease models & mechanisms. 2009; 2:541–547.
59. Goyos A, Robert J. Tumorigenesis and anti-tumor immune responses in Xenopus. Front Biosci (Landmark Ed). 2009; 14:167–176.
60. Wallingford JB. Tumors in tadpoles: the Xenopus embryo as a model system for the study of tumorigenesis. Trends Genet. 1999; 15:385–388.
61. Kaltenbrun E, Tandon P, Amin NM, Waldron L, Showell C, Conlon FL. Xenopus: An emerging model for studying congenital heart disease. Birth defects research Part A, Clinical and molecular teratology. 2011; 91:495–510.
62. Pratt KG, Khakhalin AS. Modeling human neurodevelopmental disorders in the Xenopus tadpole: from mechanisms to therapeutic targets. Disease models & mechanisms. 2013; 6:1057–1065.
63. Robert J, Cohen N. The genus Xenopus as a multispecies model for evolutionary and comparative immunobiology of the 21st century. Dev Comp Immunol. 2011; 35:916–923.
64. Schmitt SM, Gull M, Brandli AW. Engineering Xenopus embryos for phenotypic drug discovery screening. Advanced drug delivery reviews. 2014; 69–70:225–246.
65. Haynes-Gimore N, Banach M, Brown E, Dawes R, Edholm ES, Kim M, Robert J. Semi-solid tumor model in Xenopus laevis/gilli cloned tadpoles for intravital study of neovascularization, immune cells and melanophore infiltration. Dev Biol. 2015; 408:205–12.
66. Hardwick LJ, Philpott A. An oncologists friend: How Xenopus contributes to cancer research. Dev Biol. 2015; 408:180–187.
67. Yang S, Lockwood A, Hollett P, Ford R, Kao K. Overexpression of a novel Xenopus rel mRNA gene induces tumors in early embryos. J Biol Chem. 1998; 273:13746–13752.
68. Wallingford JB, Seufert DW, Virta VC, Vize PD. p53 activity is essential for normal development in Xenopus. Curr Biol. 1997; 7:747–757.
69. Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, Henninger MA, Belfort GM, Lin Y, Monahan PE, Boyden ES. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010; 463:98–102.
70. Adams DS, Lemire JM, Kramer RH, Levin M. Optogenetics in Developmental Biology: using light to control ion flux-dependent signals in Xenopus embryos. Int J Dev Biol. 2014; 58:851–61.
71. Le X, Langenau DM, Keefe MD, Kutok JL, Neuberg DS, Zon LI. Heat shock-inducible Cre/Lox approaches to induce diverse types of tumors and hyperplasia in transgenic zebrafish. Proc Natl Acad Sci U S A. 2007; 104:9410–9415.
72. Bamann C, Gueta R, Kleinlogel S, Nagel G, Bamberg E. Structural guidance of the photocycle of channelrhodopsin-2 by an interhelical hydrogen bond. Biochemistry. 2010; 49:267–278.
73. Lorenz-Fonfria VA, Heberle J. Channelrhodopsin unchained: structure and mechanism of a light-gated cation channel. Biochim Biophys Acta. 2014; 1837:626–642.
74. Adams DS, Levin M. Measuring resting membrane potential using the fluorescent voltage reporters DiBAC4 and CC2-DMPE. Cold Spring Harbor protocols. 2012; 2012:459–464.
75. Adams DS, Levin M. General principles for measuring resting membrane potential and ion concentration using fluorescent bioelectricity reporters. Cold Spring Harbor protocols. 2012; 2012:385–397.
76. Oviedo NJ, Nicolas CL, Adams DS, Levin M. Live Imaging of Planarian Membrane Potential Using DiBAC4. CSH Protoc. 2008; 2008:pdb prot5055.
77. Beane WS, Morokuma J, Adams DS, Levin M. A Chemical genetics approach reveals H,K-ATPase-mediated membrane voltage is required for planarian head regeneration. Chemistry & Biology. 2011; 18:77–89.
78. Adams DS, Masi A, Levin M. H+ pump-dependent changes in membrane voltage are an early mechanism necessary and sufficient to induce Xenopus tail regeneration. Development. 2007; 134:1323–1335.
79. Sebire NJ, Malone M. Myogenin and MyoD1 expression in paediatric rhabdomyosarcomas. J Clin Pathol. 2003; 56:412–416.
80. Cessna MH, Zhou H, Perkins SL, Tripp SR, Layfield L, Daines C, Coffin CM. Are myogenin and myoD1 expression specific for rhabdomyosarcoma? A study of 150 cases, with emphasis on spindle cell mimics. Am J Surg Pathol. 2001; 25:1150–1157.
81. Davicioni E, Anderson MJ, Finckenstein FG, Lynch JC, Qualman SJ, Shimada H, Schofield DE, Buckley JD, Meyer WH, Sorensen PH, Triche TJ. Molecular classification of rhabdomyosarcoma—genotypic and phenotypic determinants of diagnosis: a report from the Children’s Oncology Group. Am J Pathol. 2009; 174:550–564.
82. Lobikin M, Pare JF, Kaplan DL, Levin M. Selective depolarization of transmembrane potential alters muscle patterning and muscle cell localization in Xenopus laevis embryos. Int J Dev Biol. 2015; 59:303–11.
83. McCaig CD, Song B, Rajnicek AM. Electrical dimensions in cell science. J Cell Sci. 2009; 122:4267–4276.
84. McCaig CD, Rajnicek AM, Song B, Zhao M. Controlling cell behavior electrically: current views and future potential. Physiol Rev. 2005; 85:943–978.
85. Levin M. Morphogenetic fields in embryogenesis, regeneration, and cancer: non-local control of complex patterning. Bio Systems. 2012; 109:243–261.
86. Arcangeli A, Carla M, Bene M, Becchetti A, Wanke E, Olivotto M. Polar/apolar compounds induce leukemia cell differentiation by modulating cell-surface potential. PNAS. 1993; 90:5858–5862.
87. Adams DS, Lemire JM, Kramer RH, Levin M. Optogenetics in Developmental Biology: using light to control ion flux-dependent signals in Xenopus embryos. Int J Dev Biol. 2014.
88. Lin JY, Lin MZ, Steinbach P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J. 2009; 96:1803–1814.
89. Berndt A, Prigge M, Gradmann D, Hegemann P. Two open states with progressive proton selectivities in the branched channelrhodopsin-2 photocycle. Biophys J. 2010; 98:753–761.
90. Fortin DL, Dunn TW, Kramer RH. Engineering light-regulated ion channels. Cold Spring Harb Protoc. 2011; 2011:579–585.
91. Fortin DL, Dunn TW, Fedorchak A, Allen D, Montpetit R, Banghart MR, Trauner D, Adelman JP, Kramer RH. Optogenetic photochemical control of designer K+ channels in mammalian neurons. J Neurophysiol. 2011; 106:488–496.
92. Caporale N, Kolstad KD, Lee T, Tochitsky I, Dalkara D, Trauner D, Kramer R, Dan Y, Isacoff EY, Flannery JG. LiGluR restores visual responses in rodent models of inherited blindness. Mol Ther. 2011; 19:1212–1219.
93. Bizzarri M, Cucina A. Tumor and the microenvironment: a chance to reframe the paradigm of carcinogenesis? Biomed Res Int. 2014; 2014:934038.
94. D’Anselmi F, Masiello MG, Cucina A, Proietti S, Dinicola S, Pasqualato A, Ricci G, Dobrowolny G, Catizone A, Palombo A, Bizzarri M. Microenvironment promotes tumor cell reprogramming in human breast cancer cell lines. PloS one. 2013; 8:e83770.
95. Bizzarri M, Pasqualato A, Cucina A, Pasta V. Physical forces and non linear dynamics mould fractal cell shape. Quantitative morphological parameters and cell phenotype. Histol Histopathol. 2013; 28:155–174.
96. Bissell MJ, Labarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005; 7:17–23.
97. Kenny PA, Bissell MJ. Tumor reversion: correction of malignant behavior by microenvironmental cues. International journal of cancer. 2003; 107:688–695.
98. Sive HL, Grainger RM, Harland RM. (2000). Early Development of Xenopus Laevis. (New York: Cold Spring Harbor Laboratory Press).
99. Nieuwkoop PD, Faber J. (1967). Normal Table of Xenopus laevis (Daudin). (Amsterdam: North-Holland).
100. Zhu F, Skommer J, Huang Y, Akagi J, Adams D, Levin M, Hall CJ, Crosier PS, Wlodkowic D. Fishing on chips: Up-and-coming technological advances in analysis of zebrafish and Xenopus embryos. Cytometry A. 2014; 85:921–932.
101. Kawakami K, Takeda H, Kawakami N, Kobayashi M, Matsuda N, Mishina M. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev Cell. 2004; 7:133–144.
102. Gillespie JI. The distribution of small ions during the early development of Xenopus laevis and Ambystoma mexicanum embryos. J Physiol. 1983; 344:359–377.