
INTRODUCTION
Prostate cancer (PCa) is the second most common type of solid cancer and the sixth leading cause of cancer death in men worldwide [1]. In the United States in 2015, there were an estimated 220,800 new cases of PCa and 27,540 deaths from this disease (American Cancer Society: Cancer Facts and Figures 2015. Atlanta, GA: American Cancer Society, 2015). Prostate cancer has a clear familial clustering feature [2-4].The risk of developing PCa rises two to five fold with increasing numbers of affected relatives [5]. Although data from twin studies showed that the genetic contribution to PCa risk is approximately 40% [6, 7], only a few mutations in genes such as BRCA1, BRCA2, and HOXB13 definitely account for a small proportion of cases of hereditary prostate cancer [8-10]. Based on a large number of family studies, linkage analyses have identified approximately twenty chromosomal regions harboring potential PCa susceptible loci. However, only a few overlap across studies suggests that PCa exhibits locus heterogeneity [11-13]. Population-based case-control association studies using single nucleotide variants (SNVs) powerfully fine mapped susceptible variants for common diseases [14, 15]. To fine map and identify risk loci of PCa in the population, over 700 candidate gene association studies and 19 GWAS have been published in the past 25 years (between 1990 and 2015). However, the statistical association results claimed by these studies are not consistent. Meta-analysis is a helpful procedure to increase statistical power and to estimate whether the genetic association of a disease is true or not through combining data from the initial reports and subsequent replication studies [16].
To better assess and explain the association of genetic variants with PCa, we have collected and catalogued all population-based case-control genetic association studies, which were published in the field of PCa between August 1, 1990 and August 1, 2015. Subsequently, we systematically meta-analyzed gene-specific SNVs using genotyping data available from at least three independent case-control studies.
RESULTS
Publication searches
After screening over 40,000 titles and abstracts, we obtained 728 publications reporting on 217 genetic SNVs in 136 different genes (Supplementary Table S1). Based on our inclusion criteria: 1) at least three independent case-control studies for a gene-specific SNV; 2) each SNV with not more than two alleles distribution, 560 papers containing 19 GWAS publications which reported 66 SNVs in 51 different genes were finally included for the meta-analyses. Next, we searched publications in EMBASE and Cochrane database with the keywords “prostate cancer AND 7 random chosen SNVs or 7 matched genes”, respectively. This process obtained 453 publications from the EMBASE and 3 publications from the Cochrane. 13 of 453 and 2 of 3 publications fulfilled our inclusion criteria. In parallel, PubMed yielded 37 fulfilled publications using the same keywords above. No additional eligible publications from the EMBASE and the Cochrane database were included compared with that from PubMed.
Meta-analyses
Among the 217 SNVs, 66 variants in 51 different genes with sufficient data were finally included for our meta-analysis (Supplementary Table S1 and Supplementary Figure S1). Combining studies of all ethnic populations, we calculated the summary OR and 95% c.i. values across the 66 included variants using an allelic contrast model. Of these 66 SNVs, 20 in 19 different genes (KLK3, IGFBP3, ESR1, SOD2, CAT, CYP1B1, VDR, RFX6, HNF1B, SRD5A2, FGFR4, LEP, HOXB13, FAS, FOXP4, SLC22A3, LMTK2, EHBP1 and MSMB) had significant summary ORs (referred to as “positive” association) (Tables 1 and 2; Supplementary Figures S2 and S3). These results suggested an increased or a decreased risk for prostate cancer. The total of sample size involved in these positive meta-analyses was 584,100. The average sample size combining cases with controls across these 20 positive meta-analyses was 29,205 subjects (ranging from 2,581 to 84,391). On average, ~8 independent studies were included for each SNV (ranging from 3 to 18) (Tables 1 and 2). Fourteen of twenty positive SNVs (summary ORs >1, ranging from 1.039–3.788) increased the risk for prostate cancer by an average of 1.34-fold. Six SNVs (in VDR, FAS, KLK3, RFX6 and HNF1B) with an average protective summary OR of 0.838 (ranging from 0.757–0.896) decreased the risk for PCa by approximately 14%. Apart from 17 SNVs that have been previously meta-analyzed using published data (Tables 1 and 2), 3 additional positive associations were identified by this studies (including FOXP4 rs1983891, LMTK2 rs6465657 and RFX6 rs339331).
Table 1: Fixed-effects meta-analyses using allelic contrasts for SNVs showing significant summary ORs (as of August 1, 2015)
Gene |
SNV |
Putative function |
Model |
OR (95% c.i.) a P-value |
Q-value b |
Heterogeneity c P-value |
Cases versus controls (Number of independent samples) |
|---|---|---|---|---|---|---|---|
SRD5A2 |
rs9282858 |
Exon (p.Ala49Thr) |
A vs. G, all ethnicities |
1.323 (1.111–1.575) P = 0.002 |
12.337 |
0.419 |
4241 vs. 4015 (13) |
A vs. G, all excl. initial study |
1.315 (1.104–1.566) P = 0.002 |
11.021 |
0.442 |
4135 vs. 3903 (12) |
|||
A vs. G, all excl. HWE study |
1.191 (0.969–1.464) P = 0.097 |
7.594 |
0.474 |
3785 vs. 3298 (9) |
|||
FGFR4 |
rs351855 |
Exon (p.Gly388Arg) |
T vs. C, all ethnicities |
1.156 (1.057–1.263) P = 0.002 |
5.358 |
0.374 |
2618 vs.2157 (6) |
T vs. C, all excl. initial study |
1.129 (1.029–1.238) P = 0.010 |
1.329 |
0.722 |
2289 vs. 1966 (4) |
|||
VDR |
rs731236 |
Promoter |
C vs. T, all ethnicities |
0.757 (0.645–0.889) P = 0.001 |
7.005 |
0.637 |
1198 vs. 1753 (10) |
C vs. T, all excl. initial study |
0.743 (0.628–0.879) P = 0.001 |
6.468 |
0.595 |
1098 vs. 1551 (9) |
|||
C vs. T, all excl. HWE study |
0.760 (0.636–0.908) P = 0.003 |
5.941 |
0.547 |
988 vs. 1378 (8) |
|||
LEP |
rs2167270 |
5’UTR |
A vs. G, all ethnicities |
1.163 (1.042–1.299) P = 0.007 |
3.723 |
0.155 |
1343 vs. 1238 (3) |
A vs. G, all excl. initial study |
1.124 (1.001–1.262) P = 0.049 |
0.189 |
0.663 |
1200 vs. 1120 (2) |
|||
HOXB13 |
rs138213197 |
Exon (p.Gly84Glu) |
A vs. G, all ethnicities |
3.788 (2.450–5.855) P = 0.000 |
8.599 |
0.197 |
11524 vs. 63753 (7) |
A vs. G, all excl. initial study |
3.649 (2.316–5.749) P = 0.000 |
8.284 |
0.141 |
9989 vs. 61994 (6) |
|||
A vs. G, all excl. HWE study |
3.743 (2.412–5.810) P = 0.000 |
8.450 |
0.133 |
10807 vs. 62061 (6) |
|||
FAS |
rs1800682 |
Promoter |
G vs. A, all ethnicities |
0.866 (0.773–0.971) P = 0.014 |
3.625 |
0.163 |
1451 vs. 1174 (3) |
G vs. A, all excl. initial study |
0.928 (0.692–1.245) P = 0.619 |
3.496 |
0.062 |
794 vs. 927 (2) |
|||
FOXP4 |
rs1983891 |
Intron |
T vs. C, all ethnicities |
1.107 (1.065–1.150) P = 0.000 |
1.380 |
0.502 |
11128 vs. 13738 (3) |
T vs. C, all excl. initial study |
1.104 (1.052–1.157) P = 0.000 |
1.343 |
0.246 |
8127 vs. 8323 (2) |
|||
SLC22A3 |
rs9364554 |
Intron |
T vs. C, all ethnicities |
1.039 (1.005–1.076) P = 0.026 |
3.840 |
0.428 |
15763 vs. 50197 (5) |
T vs. C, all excl. initial study |
1.033 (0.994–1.074) P = 0.097 |
3.412 |
0.332 |
14038 vs. 14798 (4) |
|||
LMTK2 |
rs6465657 |
Intron |
T vs. C, all ethnicities |
1.060 (1.027–1.094) P = 0.000 |
7.623 |
0.106 |
16376 vs. 50128 (5) |
T vs. C, all excl. initial study |
1.016 (0.922–1.118) P = 0.755 |
7.457 |
0.059 |
14652 vs. 14770 (4) |
|||
EHBP1 |
rs721048 |
Intron |
A vs. G, all ethnicities |
1.100 (1.057–1.146) P = 0.000 |
6.405 |
0.269 |
16385 vs. 50290 (6) |
A vs. G, all excl. initial study |
1.098 (1.054–1.144) P = 0.000 |
5.665 |
0.226 |
15517 vs. 49412 (5) |
|||
MSMB |
rs10993994 |
Promoter |
T vs. C, all ethnicities |
1.210 (1.170–1.250) P = 0.000 |
5.648 |
0.342 |
14714 vs. 14463 (6) |
T vs. C, all excl. initial study |
1.220 (1.177–1.265) P = 0.000 |
4.130 |
0.389 |
11851 vs. 12762 (5) |
Note that ‘All ethnicities’ represented that all studies across all ethnic groups were meta-analyzed; ‘all excl. initial study’ represented that the first publication reporting on statistic association for any variant was excluded; ‘all excl. HWE study’ represented that the studies deviating from HWE were excluded. a, the summary OR and 95% c.i. values. b, Q statistic across crude ORs was calculated for each independent study. c, P < 0.1 was usually considered as a significant evidence for between-study heterogeneity.
Table 2: Random-effects meta-analyses using allelic contrasts for SNVs showing significant summary ORs (as of August 1, 2015)
Gene |
SNV |
Putative function |
Model |
OR (95% c.i.) a, P-value |
Q-value b |
Heterogeneity c P-value |
Cases versus controls (Number of independent samples) |
|---|---|---|---|---|---|---|---|
KLK3 |
rs2735839 |
Intron |
A vs. G, all ethnicities |
0.795 (0.694–0.911) P = 0.001 |
71.124 |
0.000 |
17964 vs. 19099 (9) |
A vs. G, excl. initial study |
0.845 (0.731–0.975) P = 0.021 |
27.005 |
0.000 |
12842 vs. 13839 (6) |
|||
IGFBP3 |
rs2854744 |
Promoter |
C vs. A, all ethnicities |
1.169 (1.047–1.304) P = 0.005 |
13.721 |
0.089 |
2788 vs. 3020 (9) |
C vs. A, excl. initial study |
1.172 (1.035–1.327) P = 0.012 |
13.717 |
0.056 |
2481 vs. 2748 (8) |
|||
ESR1 |
rs9340799 |
Intron |
G vs. A, all ethnicities |
1.151 (1.028–1.288) P = 0.014 |
39.394 |
0.001 |
3666 vs. 5066 (16) |
G vs. A, excl. initial study |
1.150 (1.022–1.294) P = 0.020 |
39.125 |
0.000 |
3584 vs. 4829 (15) |
|||
G vs. A, excl. HWE study |
1.150 (1.015–1.303) P = 0.028 |
33.101 |
0.001 |
3208 vs. 4257 (13) |
|||
SOD2 |
rs4880 |
Exon (p.Val16Ala) |
C vs. T, all ethnicities |
1.121 (1.024–1.227) P = 0.013 |
27.234 |
0.018 |
4210 vs. 6907 (15) |
C vs. T, excl. initial study |
1.111 (1.012–1.221) P = 0.027 |
25.796 |
0.018 |
4011 vs. 6716 (14) |
|||
C vs. T, excl. HWE study |
1.108 (1.013–1.213) P = 0.026 |
25.138 |
0.022 |
4159 vs. 6752 (14) |
|||
CAT |
rs1001179 |
Promoter |
T vs. C, all ethnicities |
1.211(1.045–1.404) P = 0.011 |
10.505 |
0.033 |
3867 vs.28224 (5) |
T vs. C, excl. initial study |
1.270 (1.052–1.534) P = 0.013 |
8.285 |
0.040 |
3359 vs. 26821 (4) |
|||
T vs. C, excl. HWE study |
1.234 (0.971–1.570) P = 0.086 |
10.454 |
0.015 |
2338 vs.3040 (4) |
|||
CYP1B1 |
rs1056836 |
Exon (p.Leu432Val) |
G vs. C, all ethnicities |
1.129 (1.004–1.270) P = 0.042 |
36.379 |
0.000 |
5999 vs. 5438 (11) |
G vs. C, excl. initial study |
1.091 (0.986–1.207) P = 0.091 |
25.300 |
0.003 |
5949 vs. 5388 (10) |
|||
G vs. C, excl. HWE study |
1.132 (0.958–1.337) P = 0.144 |
20.424 |
0.001 |
3341 vs. 3220 (6) |
|||
VDR |
rs1544410 |
Promoter |
A vs. G, all ethnicities |
0.896 (0.823–0.975) P = 0.011 |
41.674 |
0.001 |
7270 vs. 8009 (18) |
A vs. G, excl. initial study |
0.888 (0.815–0.969) P = 0.007 |
40.454 |
0.001 |
7119 vs. 7835 (17) |
|||
A vs. G, excl. HWE study |
0.909 (0.827–0.999) P = 0.048 |
35.691 |
0.001 |
6273 vs. 6840 (14) |
|||
RFX6 |
rs339331 |
Intron |
T vs. C, all ethnicities |
0.854 (0.787–0.927) P = 0.000 |
10.733 |
0.013 |
12638 vs. 15897 (4) |
T vs. C, excl. initial study |
0.846 (0.743–0.963) P = 0.011 |
8.758 |
0.013 |
9637 vs. 10482 (3) |
|||
HNF1B |
rs4430796 |
Intron |
G vs. A, all ethnicities |
0.859 (0.793–0.930) P = 0.000 |
55.853 |
0.000 |
26822 vs. 57569 (11) |
G vs. A, excl. initial study |
0.866 (0.784–0.956) P = 0.004 |
48.460 |
0.000 |
17488 vs. 50505 (10) |
Note that ‘All ethnicities’ represented that all studies across all ethnic groups were meta-analyzed; ‘all excl. initial study’ represented that the first publication reporting on statistic association for any variant was excluded; ‘all excl. HWE study’ represented that the studies deviating from HWE were excluded. a, the summary OR and 95% c.i. values. b, Q statistic across crude ORs was calculated for each independent study. c, P < 0.1 was usually considered as a significant evidence for between-study heterogeneity.
After the initial publications were removed, twelve (60%) of these twenty positive meta-analyses showed notable changes in statistical significance. The P values of eleven variants clearly decreased, and three positive variants (rs1800682 in FAS, rs9364554 in SLC22A3 and rs6465657 in LMTK2) became insignificant (P >0.05) (Tables 1 and 2), due to a substantial decrease in sample size (range: 34-56%). By contrast, one variant rs1544410 in VDR became more significant (P value changed from 0.011 to 0.007). Of twenty positive SNVs, eleven variants showed study homogeneity (P >0.1 in Q statistic) in the meta-analyses combining all ethnic groups, and their summary ORs and 95% c.i. values were calculated using the fixed-effect model (Table 1). Nine other positive associations showed large between-study heterogeneity (P < 0.1 in Q statistic), and the summary ORs of them were calculated using the random-effect model (Table 2).
Forty-six SNVs in thirty-five different genes did not show significant summary ORs (referred to as negative SNVs) utilizing random-effect models, after all published population-based case-control studies were allelic-specifically meta-analyzed in all ethnic groups (Supplementary Table S2). The combined sample sizes of these negative meta-analyses ranged from 1,070-32,738 (mean of 9,096 individuals), and the average case-control studies were 15 (range from 3 to 33). Thirty-eight of these 46 variants were close to statistical significance. The average of OR was 1.05 (ranging from 0.921-1.249).
Eight of the twenty positive variants in the control subjects showed deviation from Hardy-Weinberg equilibrium (HWE). While four variants (rs9282858 in SRD5A2, rs1001179 in CAT, rs1056836 in CYP1B1 and rs1544410 in VDR) became insignificant after the exclusion of HWE-deviation studies (Tables 1 and 2). For positive variants that still showed between-study heterogeneity after excluding the HWE-deviation studies, we then stepwise removed outlier studies until homogeneity was obtained. Eight (~90%) of nine variants with heterogeneity were corrected (Heterogeneity P-value >0.1), except RFX6 rs339331. Although the overwhelming majority of significant results only showed slight changes after the process, one positive variant (rs9340799 in ESR1) lost significant effect size (Supplementary Table S4).
Apart from two positive variants (HOXB13 rs138213197 and LEP rs2167270) for which all published case-control studies were of Caucasian-ancestry, we stratified other eighteen positive variants into racial subgroups and re-meta-analyzed summary ORs and 95% c.i. values. Two variants (in EHBP1 and HNF1B) consistently showed significant association (P values: 0.036 to 0.000) with PCa across Asian-ancestry, Caucasian-ancestry and African-ancestry; Five variants (RFX6 rs339331, FOXP4 rs1983891, CYP1B1 rs1056836, MSMB rs10993994 and FGFR4 rs351855) showed positive results in both Asian-ancestry and Caucasian-ancestry; Six variants in KLK3, SRD5A2, SOD2, CAT, SLC22A3 and LMTK2 only maintained statistical significance in Caucasian-ancestry; Two variants in the VDR gene showed positive results only in Asian ancestry; While the variant rs9340799 in ESR1 was significantly associated with PCa only in African-ancestry patients. Of note, no positive results were seen in IGFBP3 rs2854744 and in FAS rs1800682 in all ethnic subgroups (Table 3, Figure 1 and Supplementary Figure S5).
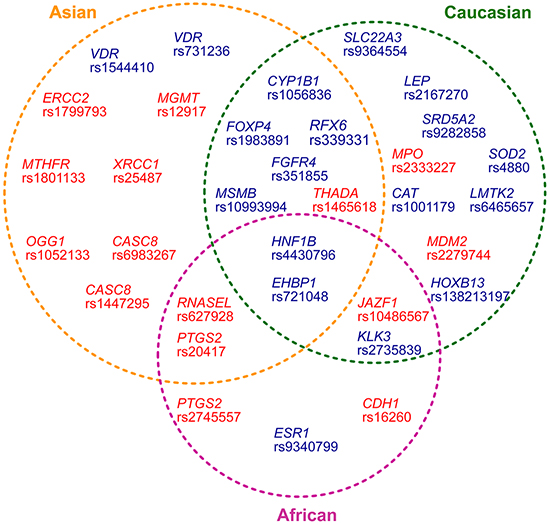
Figure 1: Distribution of thirty-three prostate cancer-associated variants in different ethnic ancestries. The variants showing statistical significance in both the initial meta-analyses combining all ethnic ancestries and in the analyses based on classified racial groups were indicated in dark blue text. The positive variants that only showed statistic significance in the analyses based on classified racial groups were indicated in red.
Table 3: Meta-analyses based on ethnic subgroups using allelic contrasts for20 positive SNVs (as of August 1, 2015)
Gene |
SNV |
Model |
OR (95% c.i.) P-value b |
Q-value c |
Heterogeneity d P-value |
Cases versus controls (Number of independent samples) |
|---|---|---|---|---|---|---|
KLK3 a |
rs2735839 |
African |
0.783 (0.625–0.981) P = 0.033 |
0.000 |
1.000 |
454 vs. 301 (1) |
Asian |
0.895 (0.649–1.234) P = 0.498 |
13.238 |
0.001 |
1501 vs. 2047 (3) |
||
Caucasian |
0.736 (0.571–0.947) P = 0.017 |
38.416 |
0.000 |
13676 vs. 14364 (3) |
||
Mixed |
0.767 (0.678–0.867) P = 0.000 |
1.094 |
0.296 |
2333 vs.2387 (2) |
||
SRD5A2 |
rs9282858 |
African |
0.778 (0.042–14.236) P = 0.865 |
0.000 |
1.000 |
30 vs. 261 (1) |
Caucasian |
1.325 (1.113–1.578) P = 0.002 |
12.209 |
0.348 |
4211 vs. 3754 (12) |
||
ESR1 a |
rs9340799 |
African |
1.531 (1.132–2.071) P = 0.006 |
0.064 |
0.800 |
129 vs. 422 (2) |
Asian |
1.157 (0.928–1.442) P = 0.194 |
4.574 |
0.206 |
537 vs. 850 (4) |
||
Caucasian |
1.113 (0.974–1.271) P = 0.116 |
27.623 |
0.001 |
3000 vs. 3794 (10) |
||
SOD2 a |
rs4880 |
African |
1.100 (0.803–1.509) P = 0.552 |
3.231 |
0.199 |
182 vs. 672 (3) |
Caucasian |
1.153 (1.030–1.291) P = 0.013 |
20.386 |
0.016 |
3372 vs. 4781 (10) |
||
Mixed |
1.055 (0.825–1.351) P = 0.668 |
2.398 |
0.122 |
656 vs.1454 (2) |
||
CAT a |
rs1001179 |
Asian |
1.064 (0.780–1.452) P = 0.694 |
0.000 |
1.000 |
1417 vs. 1008 (1) |
Caucasian |
1.338 (1.053–1.700) P = 0.017 |
7.504 |
0.023 |
1942 vs. 25813 (3) |
||
Mixed |
1.054 (0.884–1.256) P = 0.559 |
0.000 |
1.000 |
508 vs.1403 (1) |
||
FGFR4 |
rs351855 |
African |
1.154 (0.731–1.823) P = 0.538 |
0.004 |
0.948 |
191 vs. 174 (2) |
Asian |
1.244 (1.022–1.514) P = 0.030 |
0.000 |
1.000 |
492 vs. 344 (1) |
||
Caucasian |
1.133 (1.022–1.255) P = 0.018 |
4.666 |
0.097 |
1935 vs. 1639 (3) |
||
VDR |
rs731236 |
Asian |
0.767 (0.640–0.920) P = 0.004 |
6.912 |
0.546 |
1065 vs.1596 (9) |
Caucasian |
0.722 (0.513–1.017) P = 0.062 |
0.000 |
1.000 |
133 vs. 157 (1) |
||
CYP1B1 a |
rs1056836 |
Asian |
1.621 (1.206–2.179) P = 0.001 |
0.991 |
0.320 |
236 vs.355 (2) |
Caucasian |
1.080 (1.022–1.141) P = 0.006 |
28.384 |
0.000 |
5763 vs. 5083 (9) |
||
VDR a |
rs1544410 |
African |
0.886 (0.590–1.330) P = 0.559 |
5.187 |
0.075 |
375 vs. 361 (3) |
Asian |
0.533 (0.326–0.871) P = 0.012 |
9.268 |
0.026 |
665 vs. 760 (4) |
||
Caucasian |
0.959 (0.910–1.009) P = 0.108 |
7.876 |
0.547 |
5791 vs. 6411 (10) |
||
Mixed |
0.873 (0.723–1.053) P = 0.155 |
0.000 |
1.000 |
439 vs.477 (1) |
||
IGFBP3 a |
rs2854744 |
African |
1.166 (0.965–1.410) P = 0.111 |
0.036 |
0.849 |
451 vs. 414 (2) |
Asian |
1.123 (0.969–1.302) P = 0.124 |
2.276 |
0.320 |
1196 vs. 1199 (3) |
||
Caucasian |
1.294 (0.755–2.217) P = 0.349 |
2.987 |
0.084 |
218 vs. 380 (2) |
||
Mixed |
1.107 (0.861–1.424) P = 0.427 |
3.983 |
0.046 |
923 vs.1027 (2) |
||
LEP |
rs2167270 |
Caucasian only e |
1.163 (1.042–1.299) P = 0.007 |
3.723 |
0.155 |
1343 vs. 1238 (3) |
HOXB13 |
rs138213197 |
Caucasian only e |
3.788 (2.450–5.855) P = 0.000 |
8.599 |
0.197 |
11524 vs. 63753 (7) |
FAS |
rs1800682 |
Asian |
0.878 (0.766–1.007) P = 0.063 |
3.496 |
0.062 |
794 vs. 927 (2) |
Caucasian |
0.839 (0.682–1.003) P = 0.099 |
0.000 |
1.000 |
657 vs. 247 (1) |
||
RFX6 a |
rs339331 |
Asian |
0.831 (0.787–0.877) P = 0.000 |
1.666 |
0.435 |
4814 vs. 7867 (3) |
Caucasian |
0.929 (0.885–0.976) P = 0.003 |
0.000 |
1.000 |
7824 vs. 8030 (1) |
||
FOXP4 |
rs1983891 |
Asian |
1.122 (1.054–1.193) P = 0.000 |
1.084 |
0.298 |
3290 vs. 5702 (2) |
Caucasian |
1.097 (1.045–1.152) P = 0.000 |
0.000 |
1.000 |
7838 vs. 8036 (1) |
||
SLC22A3 |
rs9364554 |
African |
1.000 (0.861–1.161) P = 0.999 |
0.000 |
1.000 |
2652 vs. 2659 (1) |
Asian |
0.945 (0.847–1.054) P = 0.312 |
0.109 |
0.741 |
1392 vs. 1675 (2) |
||
Caucasian |
1.053 (1.015–1.093) P = 0.006 |
0.066 |
0.797 |
11719 vs. 45863 (2) |
||
LMTK2 |
rs6465657 |
African |
1.000 (0.898–1.114) P = 0.998 |
0.000 |
1.000 |
3266 vs. 2631 (1) |
Asian |
0.911 (0.785–1.057) P = 0.221 |
1.177 |
0.278 |
1392 vs. 1674 (2) |
||
Caucasian |
1.075 (1.039–1.112) P = 0.000 |
0.736 |
0.391 |
11718 vs. 45823 (2) |
||
EHBP1 |
rs721048 |
African |
1.264 (1.062–1.503) P = 0.008 |
0.000 |
0.998 |
2951 vs. 2994 (2) |
Asian |
1.338 (1.020–1.757) P = 0.036 |
0.113 |
0.736 |
1392 vs. 1677 (2) |
||
Caucasian |
1.087 (1.042–1.133) P = 0.000 |
1.522 |
0.217 |
12042 vs. 45619 (2) |
||
HNF1B a |
rs4430796 |
African |
1.093 (1.014–1.179) P = 0.021 |
0.000 |
1.000 |
3112 vs. 2911 (1) |
Asian |
0.849 (0.776–0.929) P = 0.000 |
3.013 |
0.698 |
2500 vs. 2379 (6) |
||
Caucasian |
0.816 (0.794–0.838) P = 0.000 |
1.580 |
0.664 |
21210 vs. 52279 (4) |
||
MSMB |
rs10993994 |
Asian |
1.155 (1.052–1.267) P = 0.002 |
1.951 |
0.377 |
1627 vs. 2005 (3) |
Caucasian |
1.218 (1.175–1.262) P = 0.000 |
2.598 |
0.273 |
13087 vs. 12458 (3) |
Note that these 20 variants showing positive association in all ‘ethnic groups’ were re-meta-analyzed based on different ethnic ancestries. a, positive variants with between-study heterogeneity calculated in all ethnic groups; b, the summary OR and 95% c.i. values. c, Q statistic across crude ORs was calculated for each included study. d, P >0.1 is usually considered as an evidence for no between-study heterogeneity; P <0.1 as an evidence for significant between-study heterogeneity. e, HOXB13 rs138213197 and LEP rs2167270 only were reported in Caucasian-ancestry. ‘Q statistic=0 and P=1’ indicated that a SNV was only investigated once in one ethnic group.
Fifteen of forty-six negative variants (calculation using random-effects model combining all subjects in all ethnic populations) became positive results (P value range: 0.04-0.000) in one or two ethnic groups, when re-analyzed them based on different ethnic ancestries. Of these fifteen positive variants, seven (~47%) were seen only in Asian-ancestry population. Except for one protective variant (MTHFR rs1801133) for prostate cancer (OR = 0.684, 95% c.i.=0.565–0.828), six of the seven variant (in MGMT, XRCC1, OGG1, ERCC2 and CASC8) showed risks for prostate cancer. The average risk OR was 1.417 (range: 1.148-1.911), and the mean sample size combining case-control individuals in all Asian-ancestry studies was 2091 (range: 407-3430). Six of the fifteen significant variants were observed in African and Caucasian ancestries, respectively (that is, RNASEL rs627928, CDH1 rs16260 and PTGS2 rs2745557 in African-ancestry; MPO rs2333227, MDM2 rs2279744 and THADA rs1465618 in Caucasian-ancestry). Among the fifteen variants, only one (rs10486567 in JAZF1) shared association with prostate cancer in 5,747 African (OR=0.855, 95% c.i.=0.787–0.929) and in 19,461 Caucasian (OR=0.847, 95% c.i.=0.808–0.889) (Figure 1, Supplementary Table S3 and Supplementary Figure S6).
We estimated the potential publication bias of all twenty positive variants in combining all sample sizes across all ethnic groups using Egger’s linear regression test. The results suggested that five (SOD2 rs4880, ESR1 rs9340799, VDR rs1544410, FOXP4 rs1983891 and EHBP1 rs721048) showed evidence of significant publication bias (P value from 0.048 to 0.01) (Supplementary Figure S7). The results produced by trim and fill algorithm showed that sixteen of the twenty positive variants had a adjusted effect size. The imputed missing studies ranged from 1 to 5 (Supplementary Figure S7).
The statistical power for detection of these twenty significant summary ORs (meta-analyzed in all ethnic groups) ranged from 0.28 to 1, and the average power was 0.77. The power of nine meta-analyses with OR<1.2 was below 80%. The genetic power of five variants (VDR rs731236 and KLK3 rs2735839 with OR <0.8; SRD5A2 rs9282858, HOXB13 rs138213197 and MSMB rs10993994 with OR > 1.2) approached 80% (Supplementary Table S5).
To estimate the sufficiency and stability of the positive meta-analyses, we performed one-study-removed tests and cumulative meta-analyses (see methods). The results from the one-study-removed test revealed that most of positive meta-analyses (85%) had a slight change in the summary ORs and 95% c.i. values in one direction, but the summary ORs of three variants (LEP rs2167270, LMTK2 rs6465657 and FAS rs1800682) became negative when the studies with a large sample size were removed (Supplementary Figure S8). Cumulatively meta-analyzing all twenty positive variants, we found that all meta-analyses achieved consistent and significant effect sizes when an average of two-thirds of the independent studies were accumulated (mean of cumulative OR = 0.83 for protective variants, range:0.72-0.91; mean of cumulative OR=1.29 for risk variants, range:1.04-3.03) (Supplementary Figure S9 and Supplementary Table S6). Four variants (ESR1 rs9340799, CYP1B1 rs1056836, SLC22A3 rs9364554 and LMTK2 rs6465657) reached significant cumulative ORs until their all samples were included.
DISCUSSION
As of 1 August, 2015, we collected and extracted data from 728 publications reporting on 217 genetic SNVs in 136 different genes. Based on our inclusion criteria,we systematically assessed the summary ORs of 66 SNVs across 51 different genes using meta-analysis. 35 meta-analyzed SNVs in 32 genes showed statistical significance. 20 of these positive results were derived from the meta-analyses combining all ethnic groups. The other 15 significant SNVs resulted from the analysis procedure in different ethnic ancestries. The average allelic risk summary OR was 1.338, and the average protective summary OR was 0.791.
~70% (46/66) of SNVs in 371 independent studies involving 418,393 subjects showed no significant association with PCa when combining all samples across all ancestral groups. As we detected the sample size using the medium genotype relative risk of 1.16 that was observed in the present study, α defined as 0.05 and a disease prevalence of 0.1%, the sample size of case subjects ranged from ~3,500 to 14,500 with minor allelic frequencies between 0.1 and 0.5, when a statistical power approached 80% (estimated using genetic power calculator, see method). According to this estimation, 29 and 44 of 46 negative results could not meet these sample size requirements, respectively. Therefore, the small effect sizes across the current negative meta-analyses might account for the insufficient power.
Meta-analysis is a powerful way not only to measure heterogeneity of associations across independent studies, but also to quantify the extent of between-study heterogeneity [17]. In total, 43 (~65%) of 66 current meta-analyses showed evidence of significant heterogeneity (P<0.1 in Q statistic) across 560 published studies (range of heterogeneity P-value: 0.093-0.000). Of them, nine were positive associations and thirty-four were negative associations.
The initial positive study (the first association study) often exhibits an underlying statistical inflation [18]. In our current analyses, we observed the same phenomenon. After excluding the initial study from each of the twenty positive meta-analyses, we found that over half (11/20) of positive meta-analyses showed a reduction in statistical significance. The P value decreased, on average, by 9.6-fold (range: 1.429 to 44.214) (Tables 1 and 2, Supplementary Table S2). Four meta-analyses with weak significance became negative results, supported by the evidence from cumulative meta-analysis plots (Supplementary Figure S9). These results suggested that the actual genetic effect of these positive association might be overestimated by the “winner’s curse” bias [19].
After removing 37 HWE-deviated studies (including 9 positive associations and 26 negative associations), the heterogeneity values of only nine negative analyses were corrected (heterogeneity P-value >0.1). We further excluded the outlier studies from nine positive meta-analyses with significant between-study heterogeneity until reaching homogeneity. Eight of nine could be reduced to display no between-study heterogeneity. We also stratified all studies into the same racial groups and re-performed the meta-analyses. Thirty-three of sixty-six meta-analyses showed positive associations in one or more racial groups. Fifteen negative SNVs identified in the initial meta-analyses became positive (Supplementary Table S3). Eight of forty-three meta-analyses with significant heterogeneity across all ethnic ancestries showed no between-study heterogeneity in ethnic sub-groups (Table 3 and Supplementary Table S3). Notably, thirty-six of sixty-six meta-analyses existed significant between-study heterogeneity (heterogeneity P-value <0.1) within a single-ancestry population, suggesting that an evidence of bias for positive results might partly be attributed to undetected population stratification in Asian-, African- and Caucasian-ancestry.Finally, ten initially “negative variants” (in CASC8, CYP1A1, CYP3A4, GPX1, IL18, JAZF1, MDM2, MPO, THADA and TP53) under the random-effects model became significant when analyzed using the fixed-effects model (Supplementary Table S7). However, Cochran’s Q test suggested that all ten variants have a large between-study heterogeneity (the average P value=0.021).
Publication bias tends to occur when small studies with insignificant results are not published. We observed the presence of significant publication bias in five of the current 20 positive meta-analyses using Egger’s regression. The bias was further shown by the trim and fill procedure (an average of imputed missing studies was three) (Supplementary Figure S7). These bias evidence suggested that small studies led to larger effects than larger studies for these five positive variants (FOXP4 rs1983891, EHBP1 rs721048, SOD2 rs4880, ESR1 rs9340799 and VDR rs1544410). Although the trim and fill analyses also showed that thirteen positive variants need to impute average missing studies of 2, only small shift of adjusted ORs was observed (shift range: 0.1%-0.7%) (Supplementary Figure S7). Statistical methods used to test publication bias are generally underpowered [20, 21], thus the conclusion of the genetic association of these variants with PCa should be treated cautiously.
Of the twenty positive associations identified here, three significant SNVs (two in FOXP4 and LMTK2 with risk effects and one in RFX6 with protective effects), to our current knowledge, were not reported in previous publications. The vast majority of differences between the results of previously published meta-analyses and the results reported here arose from the different inclusion- and exclusion criteria. For example, five previous meta-analyses included family-based publications. In addition, one difference likely arose from miscalculation in one previous meta-analysis (Supplementary Table S8).
In this meta-analyses, five protein-coding variants (SRD5A2 p.Ala49Thr, FGFR4 p.Gly388Arg, HOXB13 p.Gly84Glu, CYP1B1 p.Leu432Val and SOD2 p.Val16Ala) were significantly associated with PCa risk, but the last two variants with small summary ORs (an average of 1.12) had significant between-study heterogeneity (heterogeneity P-value <0.1). The rs351855 C>T resides in exon 9 of the FGFR4 gene leads to a glycine to arginine substitution at codon 388 (p.Gly388Arg). The risk allele (T) of the variant was significantly associated with a poor cancer prognosis [22, 23]. The rs138213197 C>T in HOXB13, yielding a rare G84E missense variant, significantly increases the risk of prostate cancer, and the biological function of this 84E remains unclear.
In summary, we have performed a comprehensive estimation of the genetic association between population-based gene-specific SNVs and PCa using currently available data. Our meta-analyses yielded twenty significant associations, but the bulk of their genetic effects were small or modest. Thus, the interpretation of these positive results should be cautious. Further analyses with sufficient power and investigations of the potential biological roles of these genetic variants are needed.
MATERIALS AND METHODS
Inclusion criteria
To be considered for inclusion in this meta-analysis, a study must be satisfy four criteria. (i) It must be an association study between a SNV and PCa. Note that in this study, as we exclusively focused on population-based case-control designs to investigate the genetic risk loci of disease, studies based on family designs or quantitative trait analyses were not included in any of the statistical analyses. Studies on microsatellite variants or on genetic markers with complex allelic architecture (for example, SNVs with more than two alleles for which it is difficult to obtain a consistent distribution of alleles and genotypes across different studies) were also not included. As SNVs occurring in known gene sequences can exert severe effects on biological functions, we thus exclusively meta-analyzed gene-specific SNVs here. (ii) The study must be a research published in a peer reviewed journal. (iii) Only studies published in English were included. Because non-English-publishing genetic association papers in PubMed and EMBASE represent 5.6% of all population-based case-control association studies of PCa, the exclusion of these papers is not expected to produce a drastic effect on any of the overall conclusions. (iv) The SNVs were reported on at least three independent case-control studies. For studies with underlying duplicate publications, we contacted the corresponding authors by e-mail to ask for clarification. Those publications that could not be clarified by authors after twice e-mails were considered as duplication publications. We only included the initial publication in our study.
We first performed a search for all publications included in the PubMed database using the keywords “prostate cancer AND (polymorphism OR association OR variation OR variant OR risk OR susceptible OR susceptibility OR sequencing OR case-control OR gene)” between August 1, 1990 and August 1, 2015. A total of 41,955 articles were obtained. The titles and/or abstracts of these papers were scanned for the inclusion criteria above. This step yielded 560 studies containing 66 SNVs in 51 different genes that were eligible for the inclusion criteria in this study. Next, to test the completeness of our search strategies, we searched the EMBASE and Cochrane database using 7 random chosen SNVs or 7 matched genes combined with the keyword “prostate cancer”, respectively.
Data extraction
First, full-text versions of all papers used for meta-analyses were obtained. Next, the first author, year of publication, ethnic group, genotype or allele data and PubMed ID were extracted from each paper. In this study, ethnic groups encompassed four general populations (African, Asian, Caucasian and Mixed population). All 66 SNVs for subsequent meta-analyses were represented using dbSNP identifiers (“rs” numbers). For the papers in which genotype or allele distributions were not provided, we requested genotype information by directly e-mailing the first or corresponding authors. For SNVs for which we could not obtain the genotype data after two e-mail, genotype distributions were deduced from their allelic data. All information extracted from the publications was summarized in Supplementary Table S1.
Statistical analyses
SNVs with genotype and/or allele data in three or more independent studies were included for meta-analyses. We performed meta-analyses with i) all studies including general populations (all ethnic groups); ii) studies excluding the initial publication (because the initial publication often shows inflated evidence of association); iii) studies after the exclusion of samples deviating from HWE; iv) studies sorted by subgroups which have same ancestries. We calculated the crude ORs and 95% c.i. values using allele contrasts. Summary ORs and 95% c.i. values were calculated using the Mantel-Haenszel fixed-effects model [24] and the DerSimonian and Laird random-effects model [25]. Statistical heterogeneity across studies was assessed using Cochran’s Q test, for which a P value< 0.1 showed the presence of significant heterogeneity. For meta-analyses that still showed evidence of between-study heterogeneity (Q statistic, P value< 0.1) after excluding deviations from HWE, we performed a heterogeneity correction by the stepwise removal of outlier studies until homogeneity was achieved. We performed one-study-removed analyses to show the effect of each study on the summary OR. In the one-study-removed analysis, a single study was removed from the meta-analysis and the summary ORs and 95% c.i. values of the remaining studies were re-calculated. We estimated publication bias for all statistically significant SNVs. Egger’s regression procedure was used to test the extent of publication bias, with a funnel plot to show bias. Duval and Tweedie’s trim and fill procedure to impute a shift of ORs and to impute the number of missing studies when a apparent bias was to be removed [26]. We also estimated sufficiency and stability of the positive meta-analyses using meta-cumulative method [27]. In this approach, studies are sorted by the time of publication and summary ORs and 95% c.i. values are calculated iteratively when each study was added. In this study, statistical analyses were conducted using the comprehensive meta-analysis version 2.0 software [Biostat, Englewood, NJ, USA]. Hardy-Weinberg equilibrium was estimated in the control samples and deviation from HWE was defined as a P value < 0.05. The statistical power was detected using the Genetic Power Calculator online tool (http://pngu.mgh.harvard.edu/purcell/gpc/cc2.html). The prevalence of PCa was 0.1%. The minor allele frequencies of each SNV in combined cases were used as high-risk allele frequencies, and the genotype relative risks were calculated utilizing the genotype distribution from the combined case-control genotype data and sample size. The type I error rate was 0.05.
ACKNOWLEDGMENTS
We thank the twelfth 5-year National Program from the Ministry of Scientific Technology (2012BAI10B01) and the Natural Science Foundation of China (81061120527, 81370445, 81472408 and 81400790) for supporting this study.
CONFLICTS OF INTEREST
The authors declare no competing financial interests relevant to this article.
GRANT SUPPORT
This study was funded by the twelfth 5-year National Program from the Ministry of Scientific Technology (2012BAI10B01) and the Natural Science Foundation of China (81061120527,81370445,81472408, and 81400790).
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69-90.
2. Steinberg GD, Carter BS, Beaty TH, Childs B, Walsh PC. Family history and the risk of prostate cancer. Prostate. 1990; 17:337-47.
3. Carter BS, Beaty TH, Steinberg GD, Childs B, Walsh PC. Mendelian inheritance of familial prostate cancer. Proc Natl Acad Sci U S A. 1992; 89:3367-71.
4. Stanford JL, Ostrander EA. Familial prostate cancer. Epidemiol Rev. 2001; 23:19-23.
5. Kiciński M, Vangronsveld J, Nawrot TS. An epidemiological reappraisal of the familial aggregation of prostate cancer: a meta-analysis. PLoS One. 2011; 6:e27130.
6. Page WF, Braun MM, Partin AW, Caporaso N, Walsh P. Heredity and prostate cancer: a study of World War II veteran twins. Prostate. 1997; 33:240-5.
7. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000; 343:78-85.
8. Gayther SA, de Foy KA, Harrington P, Pharoah P, Dunsmuir WD, Edwards SM, Gillett C, Ardern-Jones A, Dearnaley DP, Easton DF, Ford D, Shearer RJ, Kirby RS, et al. The frequency of germ-line mutations in the breast cancer predisposition genes BRCA1 and BRCA2 in familial prostate cancer. The Cancer Research Campaign/British Prostate Group United Kingdom Familial Prostate Cancer Study Collaborators. Cancer Res. 2000; 60:4513-8.
9. Gayther SA, Russell P, Harrington P, Antoniou AC, Easton DF, Ponder BA. The contribution of germline BRCA1 and BRCA2 mutations to familial ovarian cancer: no evidence for other ovarian cancer-susceptibility genes. Am J Hum Genet. 1999; 65:1021-9.
10. Shang Z, Zhu S, Zhang H, Li L, Niu Y. Germline homeobox B13 (HOXB13) G84E mutation and prostate cancer risk in European descendants: a meta-analysis of 24,213 cases and 73, 631 controls. Eur Urol. 2013; 64:173-6.
11. Xu J. Combined analysis of hereditary prostate cancer linkage to 1q24-25: results from 772 hereditary prostate cancer families from the International Consortium for Prostate Cancer Genetics. Am J Hum Genet. 2000; 66:945-57.
12. Lange EM, Gillanders EM, Davis CC, Brown WM, Campbell JK, Jones M, Gildea D, Riedesel E, Albertus J, Freas-Lutz D, Markey C, Giri V, Dimmer JB, et al. Genome-wide scan for prostate cancer susceptibility genes using families from the University of Michigan prostate cancer genetics project finds evidence for linkage on chromosome 17 near BRCA1. Prostate. 2003; 57:326-34.
13. Edwards S, Meitz J, Eles R, Evans C, Easton D, Hopper J, Giles G, Foulkes WD, Narod S, Simard J, Badzioch M, Mahle L; International ACTANE Consortium. Results of a genome-wide linkage analysis in prostate cancer families ascertained through the ACTANE consortium. Prostate. 2003; 57:270-9.
14. Doris PA. Hypertension genetics, single nucleotide polymorphisms, and the common disease:common variant hypothesis. Hypertension. 2002; 39:323-31.
15. Watanabe Y, Metoki H, Ohkubo T, Katsuya T, Tabara Y, Kikuya M, Hirose T, Sugimoto K, Asayama K, Inoue R, Hara A, Obara T, Nakura J, et al. Accumulation of common polymorphisms is associated with development of hypertension: a 12-year follow-up from the Ohasama study. Hypertens Res. 2010; 33:129-34.
16. Munafò MR, Flint J. Meta-analysis of genetic association studies. Trends Genet. 2004; 20:439-44.
17. Lau J, Ioannidis JP, Schmid CH. Summing up evidence: one answer is not always enough. Lancet. 1998; 351:123-7.
18. Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007; 39:17-23.
19. Zollner S, Pritchard JK. Overcoming the winner’s curse: estimating penetrance parameters from case-control data. Am J Hum Genet. 2007; 80:605-15.
20. Sterne JA, Gavaghan D, Egger M. Publication and related bias in meta-analysis: power of statistical tests and prevalence in the literature. J Clin Epidemiol. 2000; 53:1119-29.
21. Kicinski M. How does under-reporting of negative and inconclusive results affect the false-positive rate in meta-analysis? A simulation study. BMJ Open. 2014; 4:e004831.
22. Falvella FS, Frullanti E, Galvan A, Spinola M, Noci S, De Cecco L, Nosotti M, Santambrogio L, Incarbone M, Alloisio M, Calabrò E, Pastorino U, Skaug V, et al. FGFR4 Gly388Arg polymorphism may affect the clinical stage of patients with lung cancer by modulating the transcriptional profile of normal lung. Int J Cancer. 2009; 124:2880-5.
23. Frullanti E, Berking C, Harbeck N, Jézéquel P, Haugen A, Mawrin C, Parise O Jr, Sasaki H, Tsuchiya N, Dragani TA. Meta and pooled analyses of FGFR4 Gly388Arg polymorphism as a cancer prognostic factor. Eur J Cancer Prev. 2011; 20:340-7.
24. MANTEL N, HAENSZEL W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959; 22:719-48.
25. DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986; 7:177-88.
26. Duval S, Tweedie R. Trim and fill: A simple funnel-plot-based method of testing and adjusting for publication bias in meta-analysis. Biometrics. 2000; 56:455-63.
27. Lau J, Antman EM, Jimenez-Silva J, Kupelnick B, Mosteller F, Chalmers TC. Cumulative meta-analysis of therapeutic trials for myocardial infarction. N Engl J Med. 1992; 327:248-54.