
INTRODUCTION
Cancer of unknown primary site (CUP) can be referred to as ‘an orphan disease’ because it is diagnosed through metastases in various organs and the primary tumor cannot be found in spite of a defined diagnostic work-up [1, 2]. In collected autopsy data, the primary tumor was found in 73% of the cases, most frequently in the lungs (27%), pancreas (24%), liver/bile (8%), and kidney/adrenals (8%) [3]. In the Nordic countries, CUP incidence increased until about 1995-2000, followed by a sharp decline which seems to be continuing [4, 5]. In the USA, the decline in incidence started already in around 1980 [6]. The decline in CUP incidence, which has been opposite to the incidence of most cancers, has implied that the proportion of CUP cases of all cancer has dropped from about 4 to 2%; contributing factors to the decline may include new and better diagnostic methods [4-6]. In Sweden, CUP ranked as the eight most common cancer in men and women in 2012 with an incidence somewhat higher than that of pancreatic cancer [7]. Because of high fatality, CUP ranks the third to fifth among cancer deaths [6, 8, 9]; in Sweden the ranking was fifth in 2010 after lung, colorectal, prostate and breast cancers. In Sweden, the decrease in incidence has been noted for most metastatic locations but particularly for the liver [10]. Among the few known risk factors, heavy smoking conveys a risk of 3.7 and any level of smoking increased the risk for CUP with respiratory system metastases to 4.9 [11, 12]. Alcohol consumption, body mass index, waist circumference, diabetes and low educational level or socio-economic status may be other risk factors [11, 12]. Familial risk is another established risk factor with possible implications for primary sites [13, 14]. CUP was associated with many cancers in family members, including cancers of the lung, liver and kidney.
CUP can be called ‘orphan disease’ also because mechanistic research into CUP causation has been a neglected area. A review summarizing results on chromosomal aberrations, and oncogene and tumor suppressor gene mutations in CUP concluded that these appeared not to differ from those in metastatic primary cancers [15]. Recent mutational screening and genome-wide sequencing efforts have revealed frequent alterations in the receptor tyrosine kinase/Ras signaling pathways allowing possibilities for targeted therapies [16-18]. Hardly any data are available of germline variants that might predispose to CUP. In view of the familial clustering of CUP with other primary cancers, pointed out above [13], it could be speculated that some of the susceptibility genes for these primary cancers may also predispose to CUP (searchable in the GWAS Catalog, http://www.ebi.ac.uk/gwas/home).
In the present study we carried out a genome-wide association study (GWAS) on CUP using patient blood samples identified from Swedish biobanks from two centers and from German CUP clinics.
RESULTS
GWAS was successfully conducted on 515 CUP patients and 7226 healthy controls which passed all applied quality control criteria. A Manhattan plot of genotyped SNPs in CUP cases against controls is shown in Figure 1. Eight SNPs in six loci reached a significance level of p<10-6. The SNPs, including two linked SNPs on each of chromosomes 7 and 13, are listed by rs numbers in Figure 1.
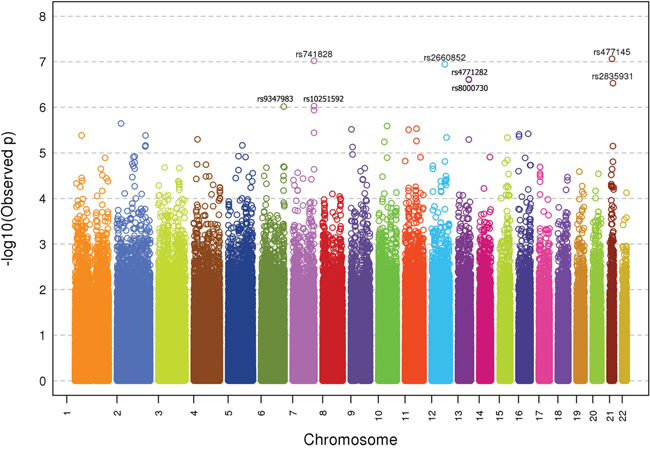
Figure 1: –Log10 p-values for association analysis of DNA from 515 CUP patients and 6227 healthy controls. The rs numbers are shown with p-values <10-6.
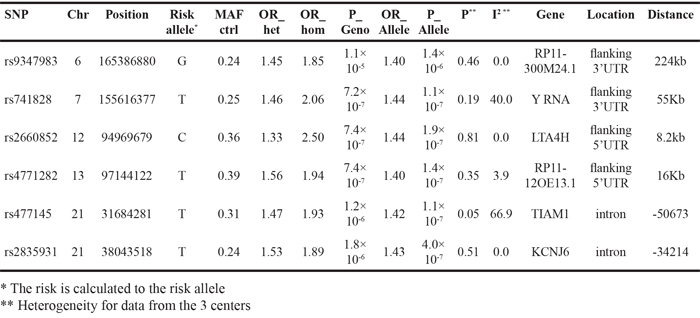
ORs and p-values of the six unlinked top SNPs (rs9347983, rs741828, rs2660852, rs4771282, rs477145 and rs2835931) are shown in Table 1. While rs9347983, rs741828, and rs4771282 were located either 3’ or 5’ to non-coding RNA genes, rs2660852 was located 5’ to LTA4H (leukotriene A4 hydrolase) on chromosome 12, rs477145 was intronic to TIAM 1 (T-cell lymphoma invasion and metastases) and rs2835931 was intronic to KCNJ6 (potassium channel, inwardly rectifying subfamily J, member 6), both on chromosome 21. Tests for heterogeneity of data from the three centers (p-value for Q > 0.05 and I2 < 75.0) showed no heterogeneity for the data on these six SNPs.
Table 1: Association data for the most significant SNPs in the case-control study of all CUP patients shown in Figure 1
A regional association plot of rs2660852 on chromosome 12 is shown in Figure 2 from 515 CUP patients and 6227 healthy controls together with functional annotation based on the ENCODE data. The SNP is flanking 5’UTR of LTA4H next to a weak or poised enhancer and histone enhancer and promoter marks, constituting the regulatory elements of LTA4H.
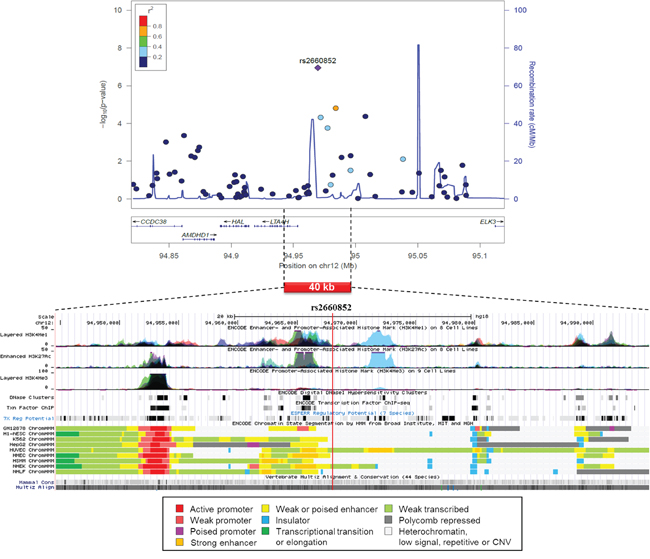
Figure 2: Regional association plot of SNP rs2660852 from 515 CUP patients and 6227 healthy controls with functional annotation based on the ENCODE data.
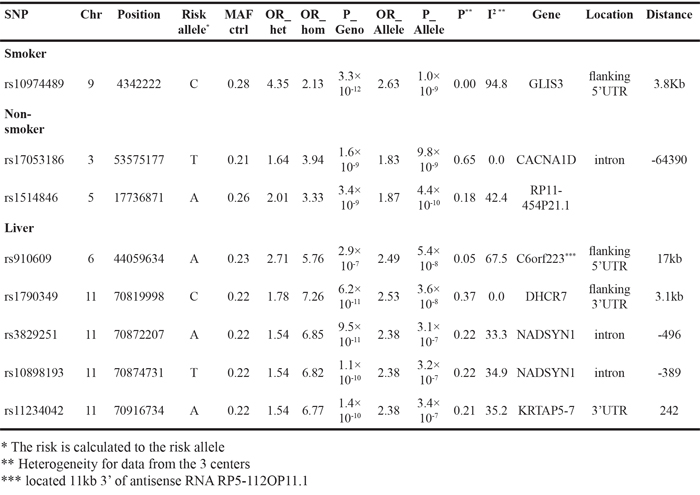
Subgroup analyses were carried out by the smoking status, histology and CUP location. Adenocarcinoma was the most common histological type and with 376 cases the data looked essentially as those for all CUP as shown in Figure 1 (data not show). Similarly, the most common location was unspecified (i.e., location unspecified) CUP which had 223 cases and with essential peaks as in Figure 1 (data not shown). However, data for smokers and non-smokers showed some novel associations reaching even genome-wide p-values (p<5x10-8) as shown in Table 2. For smokers (258 cases), rs10974489 reached a genotypic p-value of 3.3x10-12. The SNP was located 3.8 kb 5’ of the GLIS3 gene (GLIS Family Zinc Finger 3). For non-smokers (210 cases), rs17053186 reached a genotypic p-value of 1.6x10-9; this SNP was intronic to CACNA1D (calcium channel, voltage-dependent, L type, alpha 1D subunit). The p-value for rs1514846 was almost equally low 3.4x10-9; the SNP was next to an RNA coding gene. CUP with liver metastases (69 patients) showed a highly significant association for SNP rs910609 on chromosome 6 (allelic p-value 5.4x10-8) flanking 5’UTR of C6orf223. On chromosome 11 there were linked SNPs rs1790349 with genotypic p-value of 6.2x10-11, rs3829251 (9.5x10-11), rs10898193 (1.1x10-10) and rs11234042 (1.4x10-10). The adjacent genes were DHCR7 (7-dehydrocholesterol reductase), NADSYN1 (NAD synthetase 1) and KRTAP5-7 (a gene coding for a keratin associated protein). With the exception of the chromosome 9 SNP in smokers, data from the three centers were homogeneous.
Table 2: Association data for the most significant SNPs in the analysis of subgroups of CUP patients
The results on the SNPs from Table 1 and their linked SNPs (r2≥0.8) are shown in Supplementary Table S1 based on CADD and HaploReg data. The CADD score was high (18.3, over 10 is considered damaging) for rs9347983 on chromosome 6. The locus was also highly conserved (PhaseCons score 1.0) and was predicted to change binding motifs for 4 transcription factors. rs2660852 flanking 5’UTR of LTA4H was in high LD with rs2540471 located at the binding site of USF1 and NFYB, and changed the motifs of transcription factors Maf, NF-E2 and Nr2f2. The SNP, rs4771282 on chromosome 13 was linked to two conserved SNPs with CADD score over 10. The regulatory features of most other SNPs in Table 1 were changes in transcription factor binding sites.
A regional association plot and functional annotation based on the ENCODE of the smoking-related SNPs from Table 2 provided limited clues to the possible mode of action and no data are shown. However, Regulome and HaploReg data showed that rs10974489 5’ to GLIS3 had a moderate CADD score of 7.6 and it changed a motif for LBP-1 (Supplementary Table S2). rs17053186 mapped among histone marks in fetal lung tissue and changed motifs for Foxp3 and Maf. rs1514846 on chromosome 5 was linked to 4 SNPs with CADD scores of over 10, it mapped among histone marks in A549 lung carcinoma cells and changed binding motifs for SP2 and Znf143.
A regional association plot and functional annotation based on the ENCODE of rs910609 on the liver-specific association on chromosome 6 showed that the SNP was located in a poised promoter in the hepatocellular carcinoma (HepG2) cells and was mapped among enhancer and promoter histone marks in many cell lines (Figure 3). According to the Regulome and HaploReg data on rs910609, histone marks for chromatin remodeling are found for the liver, the SNP binds P300 and TCF4 and changes motif for AP-2rep (Supplementary Table S2).
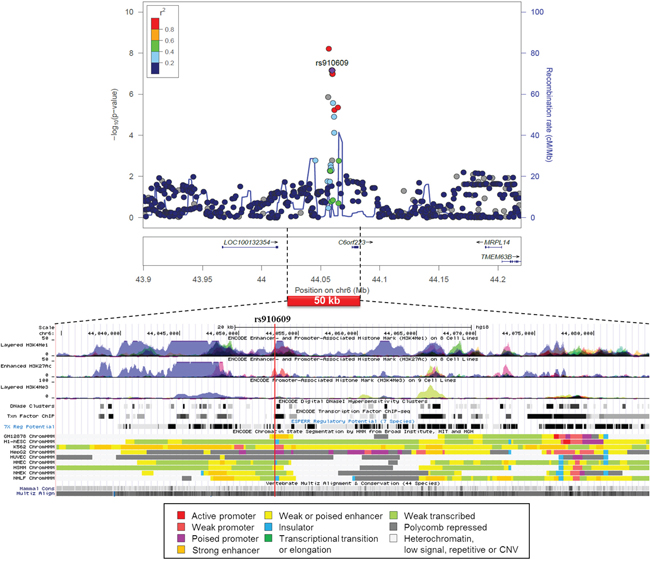
Figure 3: Regional association plot of SNP rs910609 from CUP patients with liver metastasis and controls with functional annotation based on the ENCODE data.
A similar association plot and functional annotation is shown for the 4 SNP cluster on chromosome 11 from CUP patients with liver metastases (Figure 4). The sequence between rs1790349 and rs11234042 spans less than 100 kb and covers all the 4 SNPs and numerous other SNPs in high LD. The region is actively transcribed and it contains active promoters and histone activation sites in many cell types, including adult liver and HepG2 cell line. According to the additional Regulome and HaploReg data rs1790349 is located among histone enhancer marks in adult liver and HepG2 cells and changes binding motives of several transcription factors (Supplementary Table S2). This and the other 3 SNPs are eQTLs in many tissues and each change transcription factor binding motives. rs11234042 is located in the 3’UTR and it is only 633 bp from and in high LD with a coding SNP in the KRTAP5-7 gene with a CADD value of 9.9 and a high conservation score of 1.0.
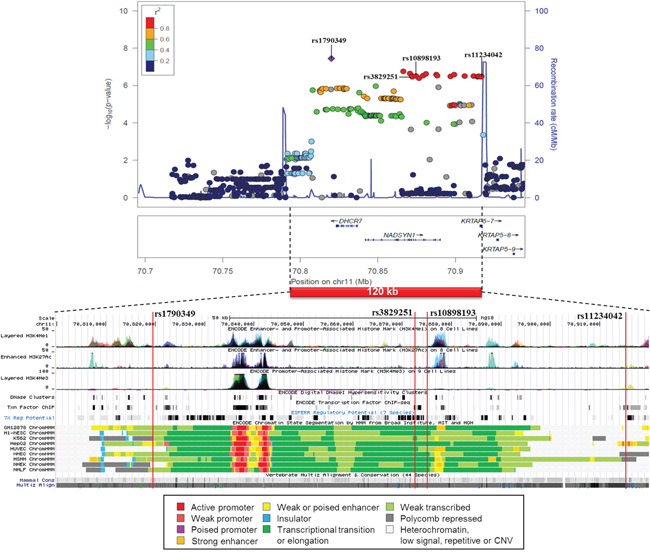
Figure 4: Regional association plot of the 4 SNP cluster on chromosome 11 from CUP patients with liver metastases and controls with functional annotation based on the ENCODE data.
DISCUSSION
CUP has been considered a heterogeneous phenotype because of its variable clinical presentation and it is speculative to ad hoc propose genetic pathways that may be related to germline risk of CUP. Yet, the association of CUP between family members and also its familial association with many primary cancers suggest that germline genetic factors contribute to this phenotype [13]. Although CUP is not a rare disease - its incidence is higher in Sweden than that of pancreatic cancer - to our knowledge the present patient series is the largest uniform collection of patients. Yet, a total of 578 patients is a not an overwhelming number for a GWAS because, alike other cancers, small relative risks are to be expected [19]. We had no separate verification populations available and thus we made sure that the data from each center was consistent with the findings.
In the association analysis of all CUP, 6 unlinked loci reached a p-value of 10-6 -10-7 but none reached the generally considered genome-wide significance level of 5x10-8, which may reflect genetic heterogeneity of CUP. Typical of the cancer-related GWASs most identified SNPs resided outside coding regions within introns or flanking coding/transcribed regions. However, as a reminder that some 75% of the human genome may be transcribed, 3 SNPs were adjacent to transcribed regions of RNA genes [20]. RP11-300M24.1 and RP11-120E13.1 (chromosomes 6 and 13) are long intergenic non-coding RNAs (lincRNA), which are expressed in many cancer cell lines (Expression Atlas, website: http://www.ebi.ac.uk/gxa/home). Y RNA (chromosome 7) is a novel miscellaneous other RNA (misc RNA) for which functional data are sparse. The proximal protein coding genes included LTA4H (leukotriene A4 hydrolase) on chromosome 12, TIAM1 (T-cell lymphoma invasion and metastases) and KCNJ6 (potassium channel, inwardly rectifying subfamily J, member 6) both on chromosome 21. For the SNPs related to LTA4H and TIAM1 a reasonable amount of functional data suggested important regulatory functions.
LTA4H is an epoxide hydrolase that catalyzes the final step in biosynthesis of the proinflammatory leukotriene B4 which is a strong chemotactic factor for mast cells and neutrophils and has been implicated in the pathogenesis of several chronic inflammatory diseases and of cancer through increasing transcription of oncogenes and interfering with apoptosis [21]. It has been shown that inflammatory markers are elevated in CUP patients [22]. Arachidonic acid is the parent compound for the synthesis of leukotriene B4 as well as of prostaglandins which are metabolized by cyclo-oxygenase (COX). This enzyme is the target of aspirin cancer prevention and it is also known to reduce the risk of CUP [23]. These data are consistent with the notion that inflammation is one of the driving forces in CUP. Protein Tiam1 modulates the activity of RHO-like proteins and it is a regulator of Rac1 mediated signaling pathways connecting extracellular signals to many types of intracellular processes, including membrane trafficking, cell migration, adhesion and invasion, and thus relating to cell growth, survival, metastasis and carcinogenesis [24]. TIAM1 is overexpressed in many tumors, including melanoma, and breast, colon, prostate and renal cancers [25]. KCNJ6 activity may be related to obesity and diabetes [26].
Among analysis of CUP subtypes the genome-wide significance was reached among smokers, non-smokers and patients with liver metastases. The GLIS3 gene that was detected among smokers is a transcription factor regulating the development of liver, kidney and pancreatic beta cells; it is associated with diabetes and also with liver cancer [27].
In patients with liver metastases SNP rs910609 on chromosome 6 flanked 5’UTR of C6orf223, a protein coding gene with unknown function but with a polymorphic variant associated with macular degeneration [28]. The <100 kb cluster of linked SNPs in the chromosome 11 encompassed 3 genes, DHCR7, NADSYN1 and KRTAP5-7. DHCR7 encodes an enzyme which catalyzes the conversion of 7-hydroxycholesterol to cholesterol. Cholesterol promotes cancer signaling, in part, due to the assembly of cholesterol-rich membrane microdomains (lipid rafts) and due to the dependency of rapidly dividing cancer cells on cholesterol and other lipids [29]. Liver is the main site of cholesterol metabolism which may explain why CUP metastases were found in this organ. Sterol metabolic pathways have been promising anticancer targets through examples such as statins and farnesyl transferase inhibitors. NAD (nicotinamide adenine dinucleotide) synthetase catalyzes the final step in the biosynthesis of NAD from nicotinic acid adenine dinucleotide. NAD has a multitude of functions, such as being a coenzyme in metabolic redox reactions, a precursor for several cell signaling molecules, and a substrate for protein posttranslational modifications. SNPs in NADSYN1 regulate serum vitamin D levels but are not related to esophageal or colon cancers or melanoma [30]. Although keratins and keratin associated proteins have numerous links with cancer the data exactly on KRTAP5-7 cannot be found. Notable in this analysis, a linked coding variant was found in KRTAP5-7 and this was predicted to be deleterious. All 4 SNPs had a fair amount of regulatory data, importantly also covering the liver, which all provide supporting evidence on the active role of this locus.
In summary, this first GWAS on CUP showed suggestive associations at the level of 10-7 in the flanking 5’UTR of LTA4H, a gene coding for an enzyme catalyzing the production of proinflammatory leukotriene B4. This is an interesting candidate involved in chronic inflammatory processes and cancer. Another gene, TIAM1 (T-cell lymphoma invasion and metastases), is not only involved in lymphoma but in many cancers where Tiam1 is proposed as a prognostic marker for breast, colon and hepatocellular cancer progression and metastasis [24]. Thus it is also an attractive candidate for CUP carcinogenicity. The associations with RNA genes remain to be explored once more functional information on these genes become available. CUP with metastases in the liver showed genome-wide associations for a chromosome 11 region clustering genes DHCR7, NADSYN1 and KRTAP5-7, the first two ones encoding key enzymes in cholesterol and NAD synthesis, and the last one encoding a keratin associated protein. All the three genes would have potential roles in carcinogenesis. These data provide a preliminary notion, in lacking direct functional evidence, for a germline architecture of CUP involving proinflammatory and metastatic gene variants in association with lipid metabolic disturbance.
PATIENTS AND METHODS
The Swedish patients were recruited from Swedish prospective biobanks (Umea and Malmö). The Umea Medical Biobank included 203 CUP patients with a median diagnostic age of 66 years. The control population was 1055 healthy persons which served as GWAS controls in the Umea Fracture and Osteoporosis (UFO) study; the mean age was 56 years at the baseline [31]. The Malmö Diet and Cancer Study and the Malmö Prevention Study allowed a joint identification of 270 CUP patients with mean diagnostic ages of 71 years. The control population was drawn from these local biobanks, including 5368 cancer-free persons. Cancers were identified up to year 2012 through the regional Oncology Centers that collect data for the Swedish Cancer Registry. The German patients were obtained from the Heidelberg clinic (52 patients with mean diagnostic age 61 years) and from a German CUP trial (53 patients with mean diagnostic age 56 years). Diagnostic years ranged from 2011 through 2013. Controls were 2205 healthy persons of German origin with a mean age of 68 years [32]. On all CUP populations data on sex, age and smoking habits (except the German trial patients) were available. Clinical data included histology and metastatic locations, classified as ‘unspecified CUP’ including spread to multiple organs, ‘liver CUP’ with liver metastasis, ‘lung CUP’ with lung involvement (including thorax and lymph nodes and organs, brain and bone, for which lung cancer is usually given as the cause of death in CUP patients [14, 33]), ‘abdominal CUP’ with abdominal metastases (including ovary) and ‘other CUP’ with other metastatic locations (any other specified site).
All samples were genotyped using Illumina Human Omni1-Quad BeadChips or OmniExpress-12 v1.0 arrays. As to the control metrics, samples were excluded if <95% of SNPs were successfully genotyped or if identity-by-state probabilities for pairs of samples were >0.20. To identify individuals of divergent ancestry we conducted a principal component analysis using smartPCA from the EIGENSTRAT package and a pruned SNP set. SNPs having a minor allele frequency <5% or a call rate <95% were excluded. Consistency of the minor allele frequency (MAF) in the control population was checked to be consistent with the the 1000 Genomes EUR population. SNPs showing departure from Hardy-Weinberg equilibrium in controls at P < 10-5 were also excluded. The analyses were conducted using PLINK (v1.07) and EIGENSTRAT software. The possibility of differential genotyping of CUP patients and controls were evaluated using quantile-quantile (Q-Q) plots of test statistics. Odds ratios (ORs) and associated 95% confidence intervals (CIs) were calculated by unconditional logistic regression. Cochran’s Q statistic was calculated to test for heterogeneity between data from the three centers and the I2 statistic was used to quantify the proportion of the total variation due to heterogeneity; I2 values ≥75% are considered a large heterogeneity. For untyped SNPs imputation was carried out to the 1000 Genomes data. However, because we had no possibility to validate imputed SNPs, these were considered only when the imputed SNPs were in linkage disequilibrium with genotyped SNPs.
Regional association plots and the epigenetic profiles of the best associated regions were used to check the chromatin state segmentation profiles (ChromHMM) in 9 cell lines, including lymphoblastoid cells (GM12878) and human liver cancer cell line HepG2, generated by the ENCODE project and available at the UCSC Genome Browser [34]. The possible functional roles of the SNPs were assessed by the ENCODE-based tool HaploReg v4.1 (www.broadinstitute.org/mammals/haploreg). To evaluate the regulatory nature and the possible functional effects of SNPs and their associated SNPs with r2 ≥ 0.8, computational predictions were performed using HaploReg v4.1 [35], CADD (combined annotation dependent depletion) [36], Regulome DB [37] and UCSC genome browser [34]. Variants with CADD scores greater than 10 are considered to be deleterious. Variants were visualized in the human genome using the Locuszoom [38] and the UCSC genome browser [34].
The study was approved by the ethics committees at Umea University and Heidelberg University.
FUNDING
This work was supported by Deutsche Krebshilfe. The UFO-study was supported by The Swedish Research Council, the Swedish Foundation for Strategic Research, the ALF/LUA research grant in Gothenburg and Umeå, the Lundberg Foundation, the Torsten and Ragnar Söderberg's Foundation, the Novo Nordisk Foundation, and the European Commission grant HEALTH-F2-2008-201865-GEFOS, BBMRI.se, the Swedish Society of Medicine, the Kempe-Foundation (JCK-1021), the Medical Faculty of Umeå University, the County Council of Västerbotten (Spjutspetsanslag VLL:159:33-2007).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Pavlidis N, Fizazi K. Carcinoma of unknown primary (CUP). Crit Rev Oncol Hematol. 2009; 69:271-278.
2. Greco FA, Oien K, Erlander M, Osborne R, Varadhachary G, Bridgewater J, Cohen D, Wasan H. Cancer of unknown primary: progress in the search for improved and rapid diagnosis leading toward superior patient outcomes. Ann Oncol. 2012; 23:298-304.
3. Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet. 2012; 379:1428-1435.
4. Brustugun OT, Helland A. Rapid reduction in the incidence of cancer of unknown primary. A population-based study. Acta Oncol. 2014; 53:134-137.
5. Shu X, Sundquist K, Sundquist J, Hemminki K. Time trends in incidence, causes of death, and survival of cancer of unknown primary in Sweden. Eur J Cancer Prev. 2012; 21:281-288.
6. Urban D, Rao A, Bressel M, Lawrence YR, Mileshkin L. Cancer of unknown primary: a population-based analysis of temporal change and socioeconomic disparities. Br J Cancer. 2013; 109:1318-1324.
7. Centre for Epidemiology. (2013). Cancer incidence in Sweden 2012. (Stockholm: The National Board of Health and Welfare).
8. Hemminki K, Bevier M, Hemminki A, Sundquist J. Survival in cancer of unknown primary site: population-based analysis by site and histology. Ann Oncol. 2012; 23:1854-1863.
9. Brewster DH, Lang J, Bhatti LA, Thomson CS, Oien KA. Descriptive epidemiology of cancer of unknown primary site in Scotland, 1961-2010. Cancer epidemiology. 2014; 38:227-234.
10. Bevier M, Sundquist J, Hemminki K. Incidence of cancer of unknown primary in Sweden: analysis by location of metastasis. Eur J Cancer Prev. 2012; 21:596-601.
11. Kaaks R, Sookthai D, Hemminki K, Krämer A, Boeing H, Wirfält E, Weiderpass E, Overvad K, Tjønneland A, Olsen A, Peeters PH, Bueno-de-Mesquita HB, Panico S, Pala V, Vineis P, Quirós JR, et al. Risk factors for cancers of unknown primary site (CUP) – results from the prospective EPIC cohort. Int J Cancer. 2014; 135:2475-2481.
12. Hemminki K, Chen B, Melander O, Manjer J, Hallmans G, Hemminki A. Smoking and Body-Mass-Index as Risk Factors for Subtypes of Cancer of Unknown Primary. Int J Cancer. 2015; 136:246-247.
13. Hemminki K, Ji J, Sundquist J, Shu X. Familial risks in cancer of unknown primary: tracking the primary sites. J Clin Oncol. 2011; 29:435-440.
14. Hemminki K, Bevier M, Sundquist J, Hemminki A. Cancer of unknown primary (CUP): does cause of death and family history implicate hidden phenotypically changed primaries? Ann Oncol. 2012; 23:2720-2724.
15. Kamposioras K, Pentheroudakis G, Pavlidis N. Exploring the biology of cancer of unknown primary: breakthroughs and drawbacks. European Journal of Clinical Investigation. 2013; 43:491-500.
16. Tothill RW, Li J, Mileshkin L, Doig K, Siganakis T, Cowin P, Fellowes A, Semple T, Fox S, Byron K, Kowalczyk A, Thomas D, Schofield P, Bowtell DD. Massively-parallel sequencing assists the diagnosis and guided treatment of cancers of unknown primary. The Journal of Pathology. 2013; 231:413-423.
17. Pentheroudakis G, Kotteas EA, Kotoula V, Papadopoulou K, Charalambous E, Cervantes A, Ciuleanu T, Fountzilas G, Pavlidis N. Mutational profiling of the RAS, PI3K, MET and b-catenin pathways in cancer of unknown primary: a retrospective study of the Hellenic Cooperative Oncology Group. Clinical & Experimental Metastasis. 2014; 31:761-769.
18. Ross J, Wang K, Gay L, Otto G, White E, Iwabik K, Palmer G, Yelensky R, Lipson D, Chmielecki J, Erlich R, Rankin A, Ali S, Elvin J, Morosini D, Miller V, et al. Comprehensive genomic profiling of carcinoma of unknown primary site. JAMA Oncol. 2015; 1:40-49.
19. Fletcher O, Houlston RS. Architecture of inherited susceptibility to common cancer. Nat Rev Cancer. 2010; 10:353-361.
20. Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, Greger L, van Iterson M, Almlof J, Ribeca P, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013; 501:506-511.
21. Amirian ES, Ittmann MM, Scheurer ME. Associations between arachidonic acid metabolism gene polymorphisms and prostate cancer risk. Prostate. 2011; 71:1382-1389.
22. Mohamed Z, Pinato DJ, Mauri FA, Chen KW, Chang PM, Sharma R. Inflammation as a validated prognostic determinant in carcinoma of unknown primary site. Br J Cancer. 2014; 110:208-213.
23. Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011; 377:31-41.
24. Boissier P, Huynh-Do U. The guanine nucleotide exchange factor Tiam1: a Janus-faced molecule in cellular signaling. Cellular Signalling. 2014; 26:483-491.
25. Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene. 2014; 33:4021-4035.
26. Anderson D, Cordell HJ, Fakiola M, Francis RW, Syn G, Scaman ES, Davis E, Miles SJ, McLeay T, Jamieson SE, Blackwell JM. First genome-wide association study in an Australian aboriginal population provides insights into genetic risk factors for body mass index and type 2 diabetes. PLoS One. 2015; 10:e0119333.
27. Xu L, Hazard FK, Zmoos AF, Jahchan N, Chaib H, Garfin PM, Rangaswami A, Snyder MP, Sage J. Genomic analysis of fibrolamellar hepatocellular carcinoma. Hum Mol Genet. 2015; 24:50-63.
28. Cheng CY, Yamashiro K, Chen LJ, Ahn J, Huang L, Huang L, Cheung CM, Miyake M, Cackett PD, Yeo IY, Laude A, Mathur R, Pang J, Sim KS, Koh AH, Chen P, et al. New loci and coding variants confer risk for age-related macular degeneration in East Asians. Nature Communications. 2015; 6:6063.
29. Gabitova L, Gorin A, Astsaturov I. Molecular pathways: sterols and receptor signaling in cancer. Clin Cancer Res. 2014; 20:28-34.
30. Wang JB, Dawsey SM, Fan JH, Freedman ND, Tang ZZ, Ding T, Hu N, Wang LM, Wang CY, Su H, Qiao YL, Goldstein AM, Taylor PR, Abnet CC. Common genetic variants related to vitamin D status are not associated with esophageal squamous cell carcinoma risk in China. Cancer Epidemiology. 2015; 39:157-159.
31. Englund U, Nordström P, Nilsson J, Bucht G, Björnstig U, Hallmans G, Svensson O, Pettersson U. Physical activity in middle-aged women and hip fracture risk: the UFO study. Osteoporos Int. 2011; 22:499-505.
32. Weinhold N, Johnson DC, Rawstron AC, Forsti A, Doughty C, Vijayakrishnan J, Broderick P, Dahir NB, Begum DB, Hosking FJ, Yong K, Walker BA, Hoffmann P, Muhleisen TW, Langer C, Dorner E, et al. Inherited genetic susceptibility to monoclonal gammopathy of unknown significance. Blood. 2014; 123:2513-2517.
33. Hemminki K, Bevier M, Sundquist J, Hemminki A. Site-specific cancer deaths in cancer of unknown primary diagnosed with lymph node metastasis may reveal hidden primaries. Int J Cancer. 2013; 132:944-950.
34. Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, Harte RA, Heitner S, Hickey G, Hinrichs AS, Hubley R, Karolchik D, et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Research. 2015; 43:D670-681.
35. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research. 2012; 40:D930-934.
36. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genet. 2014; 46:310-315.
37. Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome Research. 2012; 22:1790-1797.
38. Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010; 26:2336-2337.