
INTRODUCTION
Gastric cancer was once one of the most common cancer types and the second leading cause of cancer-related death in the world [1]. Whereas gastric cancer incidence has declined dramatically in Western countries, it still remains high in Eastern Asian countries [2]. Many genes have been analyzed in an attempt to understand gastric cancer pathogenesis and to improve clinical outcomes. Despite these efforts, only a few genetic changes, including the amplification and/or overexpression of MET and ERBB2 [3], as well as the mutations and downregulation of APC [4] and RUNX3 [5], have been associated with gastric cancer and the underlying molecular mechanisms remain largely undefined. Therefore, the identification of additional genes critical for the development and maintenance of gastric cancer is required.
The ERBB family plays an important role in normal embryonic development, angiogenesis [6], metastasis [7], cell proliferation [8] and apoptosis resistance [9]. Their aberrant signaling pathways are often found in human cancers [10, 11]. Among the ERBB family members ERBB2 receptor tyrosine kinase (RTK) is a pivotal driver of tumorigenesis [12, 13], and an important prognostic factor for patients with gastric cancer [14–16]. ERBB3 is unique as its intracellular tyrosine kinase domain is considered inactive or weak [17, 18], mainly gaining elevated activity through hetero-dimerization with other members of the ERBB family [17, 19]. Upon ligand-binding, these receptors homo-dimerize or hetero-dimerize, leading to their autophosphorylation and subsequent activation of the downstream phosphatidylinositol 3-kinase (PI3K). These mechanisms lead to mitogen-activated protein kinase (MAPK) signaling cascades [12]. The complex formed by orphan ERBB2 receptor and functionally weak ERBB3 has been recognized as being one of the most mitogenic signaling complexes in cancer development and maintenance [13, 20]. Therefore, better understanding the molecular mechanism underlying their activation through dimerization is key.
APIP has been associated with various types of cell death processes. The direct binding of APIP to APAF-1 highly inhibits mitochondria-mediated apoptosis [21]. During hypoxia, APIP reduces significantly hypoxic cell death by inducing the sustained activation of AKT and ERK1/2 in muscle cells [22]. It was recently shown that APIP inhibits caspase-1-mediated pyroptosis in response to Salmonella infection through its role within the methionine salvage pathway [23]. However, even though there are indications that the level of APIP is elevated in gastric tumor compared with normal tissues (www.proteinatlas.org) [24, 25], its role in tumorigesisis is unknown. APIP (located in chromosome region 11p13) amplification has also been observed in gastric cancer cell lines [26] and gastric cancers [27–29]. Finally, APIP is altered in non-small cell lung carcinoma (NSCLC) tumor [30]. In this study, we reveal a novel oncogenic function of APIP, which stimulates gastric cell proliferation and tumorigenesis through its interaction with ERBB3.
RESULTS
APIP is upregulated in human gastric cancers and cell lines
To characterize the role of APIP in gastric tumorigenesis, we examined APIP expression in human gastric cancer tissues. A total of 110 pairs of human gastric cancer tissues and adjacent gastric mucosa were examined in this study. Western blot analysis revealed an increased expression of APIP in 29 (26.4%) samples out of all gastric cancer tissues (Table 1). Among these 29 samples, the results from western blotting of 7 representative samples are shown in Figure 1A. Moderately and poorly differentiated tumors were associated with APIP expression (P = 0.039). However, there were no statistically significant correlations between APIP expression and histology, TNM stage or lymphatic invasion (Table 1). When we further assessed the clinicopathological and prognostic roles of APIP expression in human gastric tissues using immunohistochemistry (IHC), we observed a strong staining of APIP in gastric adenocarcinoma specimens, compared to normal samples (data not shown). We also evaluated APIP mRNA and protein levels in a panel of human gastric cancer cell lines (SNU-1, -5, -16, -216, -484, -601, -620, -638, -668 and -719) [31]. Most human gastric cancer cell lines expressed APIP but highly metastatic SNU-16 cells showed the highest expression of them all (Figure 1B).
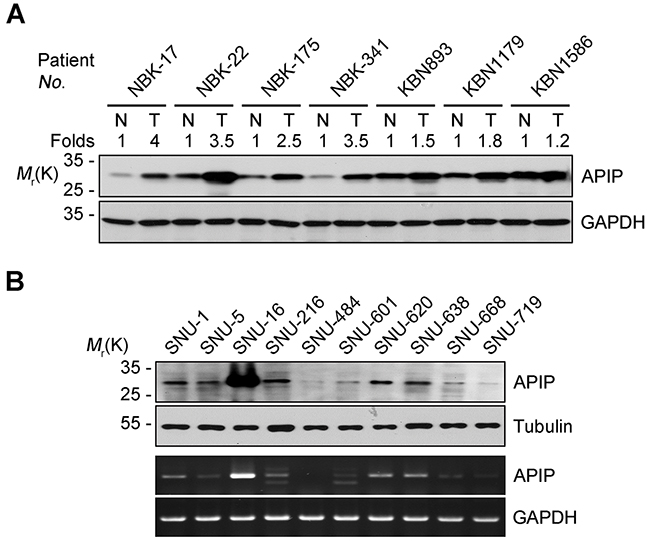
Figure 1: APIP is overexpressed in human gastric cancer cells and tissues. A. Upregulation of APIP expression in human gastric cancer tissues. Cancer tissues and paired non-cancerous normal tissues of gastric cancer patients were analyzed by Western blotting. N, normal tissue; T, tumor tissue. B. Expression levels of APIP in human gastric cancer cell lines. APIP protein (upper) and mRNA (lower) levels were evaluated by Western blotting and RT-PCR analysis, respectively.
Table 1: Correlation between APIP expression and clinicopathological characteristics of 110 gastric tumors cases
Tumor tissue parameters |
n* |
APIP expression |
P value |
|
|---|---|---|---|---|
High (%) |
Low (%) |
|||
All |
110 |
29 (26.4) |
81 (73.6) |
|
Gender |
ns |
|||
Male |
79 |
20 (18.2) |
59 (53.6) |
|
Female |
31 |
9 (8.2) |
22 (20) |
|
Age (years) |
ns |
|||
Average, Range |
63.4,32-85 |
|||
<55 |
25 |
6 (5.5) |
19 (17.3) |
|
≥55 |
85 |
23 (20.9) |
62 (56.3) |
|
WHO classification |
0.039 |
|||
W/D |
13 |
3 (2.7) |
10 (9.1) |
|
M/D |
44 |
14 (12.8) |
30 (27.3) |
|
P/D |
45 |
11 (10) |
34 (30.9) |
|
SRC |
8 |
1 (0.9) |
7 (6.3) |
|
Lauren classification |
ns |
|||
Intestinal |
102 |
26 (23.6) |
76 (69.1) |
|
Diffuse |
8 |
3 (2.8) |
5 (4.5) |
|
TNM stage |
ns |
|||
I |
30 |
3 (2.7) |
27 (24.5) |
|
II |
37 |
9 (8.1) |
28 (25.5) |
|
III |
33 |
10 (9.1) |
23 (20.9) |
|
IV |
10 |
7 (6.5) |
3 (2.7) |
|
Abbreviations: W/D, well differentiated tubular adenocarcinoma; M/D, moderately differentiated tubular adenocarcinoma; P/D, poorly differentiated tubular adenocarcinoma; SRC; signet ring cell carcinoma; ns, not significant. * Numbers of cases in each group.
APIP stimulates cell proliferation and gastric tumor growth
We then assessed whether APIP expression influences growth characteristics of gastric cancer cells. Cell proliferation was significantly enhanced by APIP overexpression in SNU-620 gastric cancer cells (SNU-620/APIP) (Figure 2A). Furthermore, xenograft assays in nude mice revealed a dramatic increase in tumor growth for APIP-overexpressing SNU-620 cells compared with control cells (Figure 2A). On the contrary, APIP knockdown suppressed the proliferation in SNU-16 gastric cancer cells (SNU-16/shAPIP #2 and #3) (Figure 2B; Supplementary Figure S1A to S1C). In agreement with cell line results, tumors derived from SNU-16 APIP knockdown cells grew substantially slower compared to those derived from SNU-16 control cells (Figure 2B). APIP knockdown itself did not trigger any cell death in SNU-16 cells (Supplementary Figure S1D). We confirmed that the observed shAPIP-mediated effect was specifically dependent on APIP knockdown by generating a shRNA-resistant APIP cDNA (APIP*); ectopic expression of APIP* cDNA in APIP knockdown cells restored the inhibitory effect of APIP shRNA on cell proliferation (Figure 2C). These results show that the regulation of APIP expression is crucial for cell proliferation and gastric tumor growth in vivo.
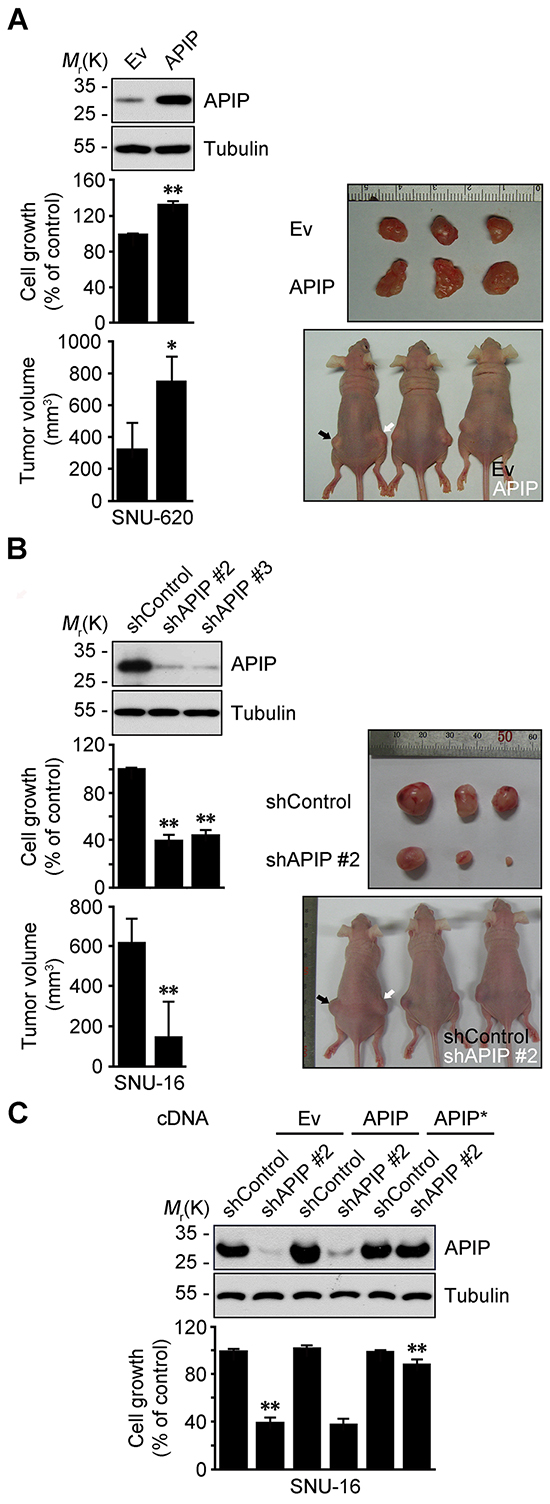
Figure 2: APIP positively regulates cell growth and tumorigenic potential in gastric cancer. A. Overexpression of APIP in SNU-620 gastric cancer cells enhances cell proliferation (middle, n = 3) and promotes tumor growth (bottom, n = 5). Ev, pcDNA3 empty; APIP, pAPIP. Representative xenograft tumors of sacrificed mice (right). B. Downregulation of APIP expression in SNU-16 gastric cancer cells suppresses cell proliferation (middle, n = 3) and in vivo tumor growth (bottom, n = 5). shControl, pSUPER.neo; shAPIP #2 and #3, APIP shRNAs. C. Expression of shRNA-resistant APIP* rescues cell growth-inhibitory phenotype in SNU-16 APIP knockdown cells. SNU-16 control and APIP knockdown cells were transfected with pcDNA, pAPIP or pAPIP* (shRNA-resistant APIP) for 72 h. Cell growth rates (lower) and APIP protein levels (upper) were assessed. All data are represented as mean ± S.D. (n ≥ 3). Statistical significance is indicated as follows: *, P < 0.05; **, P < 0.01.
APIP activates the AKT and ERK1/2 pathways for cell proliferation
We previously demonstrated that APIP sustains AKT and ERK1/2 activation under hypoxic condition in C2C12 mouse myoblast cells [21]. Therefore, we tested whether or not APIP stimulates cell proliferation via AKT and ERK1/2. In SNU-16 gastric cancer cells, APIP knockdown decreased the phosphorylation of AKT (Ser473 and Thr308) and ERK1/2 (Figure 3A). Inversely, APIP overexpression increased the activation of those pathways in SNU-620 cells (Figure 3B). As expected, overexpression of APIP* restored the activities of AKT and ERK1/2 in SNU-16 APIP knockdown cells (Figure 3C). In addition, APIP knockdown in SNU-16 cells reduced the reporter activity of c-Fos and Elk-1, downstream targets of ERK1/2 (Supplementary Figure S2A). These results indicate that APIP increases the activity of AKT and ERK1/2 in gastric cancer cells.
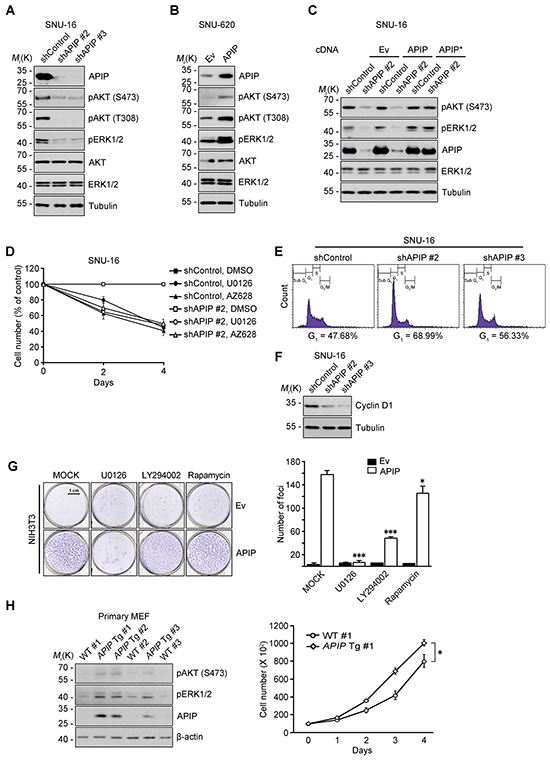
Figure 3: APIP affects both AKT and ERK1/2 pathways for cell proliferation. A. and B. Knockdown or overexpression of APIP regulates AKT and ERK1/2 phosphorylation. Whole-cell extracts of SNU-16 control and APIP knockdown cells (A) or of SNU-620 control and APIP overexpression cells (B) were analyzed by western blotting. C. Reconstitution of APIP* rescues AKT and ERK1/2 phosphorylation in APIP knockdown SNU-16 cells. SNU-16 control and APIP knockdown cells were transfected with pcDNA, pAPIP or pAPIP* for 36 h. D. Effects of RAF1-MEK inhibition on cell proliferation in SNU-16 APIP knockdown cells. SNU-16 control and APIP knockdown cells were treated with 2 μM U0126 or 1 μM AZ628 for 4 days. E. Altered cell cycle distributions by APIP knockdown in asynchronous culture. SNU-16 control and APIP knockdown cells were stained with propidium iodide and analyzed by flow cytometry. Numbers indicates cell percentages at G1 phase. F. APIP knockdown decreases the expression level of cyclin D1. Asynchronous SNU-16 control and APIP knockdown cells were analyzed by western blotting. G. Overexpressed APIP exhibits focus-forming activity through MEK1/2 and PI3K. NIH3T3 control and APIP-overexpressing cells were assayed for focus formation with 2 μM U0126, 10μM LY294002 or 5 ng/ml rapamycin for 14 days, as visualized by crystal violet. Representative plates (left) and quantification of foci (right) are shown. Scale bar, 1 cm. H. Enhanced phosphorylation of AKT and ERK1/2 and cell growth in the primary APIP transgenic MEFs. Primary MEFs from wild-type (#1) and APIP transgenic (#1) were analyzed by western blotting (left) or maintained for 4 days to measure cell growth rate (right). All data are represented as mean ± S.D. (n = 3). Statistical significance is indicated as follows: **, P < 0.01. ***, P < 0.005.
We then determined whether the AKT or ERK1/2 activation by APIP is responsible for the enhanced cell proliferation using kinase inhibitors. Treatment with PLX4032, a potent inhibitor of B-RafV600E in V600E-positive cells, or LY294002, a PI3K inhibitor, did not affect cell death or proliferation in SNU-16 cells. On the other hand, U0126, a MEK1/2 inhibitor, or AZ628, a RAF1 inhibitor, effectively suppressed cell proliferation without detectible induction of cell death (Figure 3D and Supplementary Figure S2B). This observation was similar to the suppression obtained through APIP knockdown in SNU-16 cells. There was no further additive effect of either U0126 or AZ628 on the inhibition of cell growth in APIP knockdown SNU-16 cells (Figure 3D), suggesting that APIP activates the RAF1 signaling pathway and ERK1/2 activation by APIP stimulates cell proliferation in SNU-16 cells. In addition, APIP knockdown increased the proportion of cells in the G1 phase by approximately 20% (Figure 3E) and resulted in the loss of cyclin D1 (Figure 3F), a key regulator involved in the G0/G1 phase transition. Furthermore, focus-formation assays revealed a dramatic increase in the number of foci in APIP-overexpressing cells (NIH3T3/APIP) compared with control cells (Figure 3G). However, the focus-forming activity of APIP was totally or partially disabled by U0126 and LY294002 respectively, while unaffected by rapamycin (Figure 3G).
To examine the potential role of APIP in cell proliferation in vivo, we generated APIP knockout mice and APIP transgenic mice which ubiquitously overexpress human APIP. An analysis of mouse embryonic fibroblasts (MEFs) showed a significant increase of cell proliferation rates as well as ERK1/2 and AKT activation in APIP transgenic MEFs compared with wild type MEFs (Figure 3H). On the other hand, APIP knockout had no obvious effect on cell proliferation in primary MEF cultures (Supplementary Figure S2C and S2D).
Oncogenic function of APIP is independent of its enzymatic activity in methionine metabolism
We examined whether APIP’s methylthioribulose-1-phosphate dehydratase (MtnB) enzymatic activity is associated with its oncogenic function. As APIP is part of the zinc-dependent class II aldolase family and forms a tetrameric active enzyme in the protein crystal structure [32], we employed histidine mutants-H115A, H117A, H195A and their triple mutants with disabled enzymatic activity due to their incapacity to coordinates zinc ions. Like APIP, all of those APIP mutants stimulated cell growth with ERK1/2 and AKT activation (Figure 4A) and formed APIP/APIP multimers (Figure 4B).
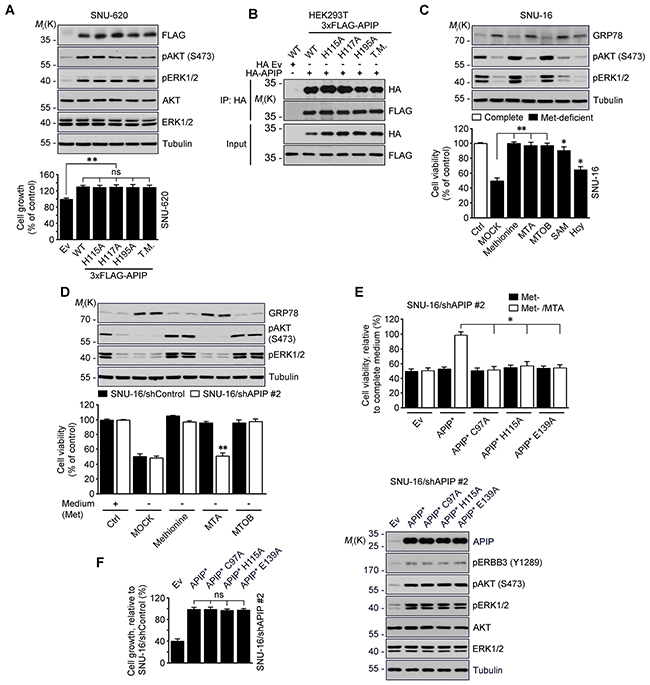
Figure 4: Evaluation of APIP/MtnB enzymatic activity in cell proliferation. A. APIP/MtnB enzymatic activity is not associated with oncogenic function of APIP. SNU-620 cells expressing APIP (WT), single (H115A, H117A and H195A) or triple (T.M., H115/H117/H195A) mutants were measured for growth rates for 4 days. B. APIP mutants also form APIP/APIP multimers. HEK293T cells were co-transfected with 3xFLAG-tagged APIP (WT), single (H115A, H117A and H195A) or triple (T.M., H115/H117/H195A) mutant for 18 h. Whole-cell lysates were then subjected to immunoprecipitation (IP) assay. C. and D. Evaluation of APIP/MtnB enzymatic activity in SNU-16 cells. SNU-16 cells (C) or SNU-16 APIP knockdown cells (D) were maintained for 24 h in methionine-deficient media with the supplementations indicated. E. Reconstitution assay with APIP/MtnB mutants in methionine salvage pathway. SNU-16 APIP knockdown cells were transfected with the indicated APIP and incubated for 48 h in methionine-deficient media with 300 μM MTA. F. APIP/MtnB mutants display oncogenic activities in SNU-16 APIP knockdown cells. Cell growth rates (left) and APIP expression levels (right) were assessed. All data are represented as mean ± S.D. (n = 3). Statistical significance is indicated as follows: *, P < 0.05; **, P < 0.01; ns, not significant.
Next, we checked the function of the methionine (Met) salvage pathway in SNU-16 cells. As previously reported for different cells [23, 33], deficiency of methionine in culture media induced endoplasmic reticulum (ER) stress and reduced cell viability in SNU-16 cells. However, when reintroducing precursors or intermediates of methionine salvage pathway, including 5’-methylthioadenosine (MTA), 4-Methylthio-2-oxobutanoic acid (MTOB), S-adenosyl methionine (SAM) and homocysteine (Hcy), cell viability was restored (Figure 4C). Unlike methionine and MTOB, MTA did not reduce the level of GRP78. Thus, this ER stress response was used as a control of methionine salvage pathway in the following step of the analysis. When we assessed the effects of methionine, MTA and MTOB on the survival of SNU-16 cells and SNU-16/APIP knockdown cells, we found that APIP deletion in SNU-16 cells impaired cell growth in the medium in which methionine was replaced by MTA (Figure 4D). Furthermore, reconstitution of SNU-16/APIP knockdown cells with APIP C97, H115 or E139 mutants, all of which are essential for MtnB activity in APIP, did not restore cell proliferation (Figure 4E). An assessment of the oncogenic activity of these mutants revealed that all of them stimulated cell proliferation as well as AKT and ERK1/2 activation, in a similar way with APIP in SNU-16/APIP* cells (Figure 4F). These findings suggest that the oncogenic function of APIP is dissociated from its enzymatic activity in the methionine salvage pathway.
APIP enables HRG-β1-mediated ERBB3 activation
To explore how APIP regulates AKT and ERK1/2 signaling in gastric cancer cells, we employed a pull-down assay using 3xFLAG-tagged APIP followed by a liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Interestingly, we found that ERBB3 is an APIP-binding partner (Figure 5A). We investigated the effects of several growth factors on the activation of RTKs and their downstream AKT and ERK1/2. Whereas insulin, IGF-1, EGF, HRG-β1 and FGF2 all activated ERK1/2 and/or AKT in SNU-16 cells, HRG-β1 was also a very potent stimulator of ERBB3 phosphorylation (Y1289), an activated form of ERBB3 [13] (Figure 5B). Assessment of ERBB3 expression in gastric cancer cell lines revealed high levels of phosphorylated ERBB3 (Y1289) in growing SNU-5 and SNU-16 cells but not in SNU-620 cells (Figure 5C). There was no difference in the levels of ERBB3 mRNA in these cells (Supplementary Figure S3A). Importantly, the activation of ERBB3, ERK1/2, and AKT by HRG-β1 was disabled by APIP knockdown in SNU-16 cells without affecting levels of ERBB3, EGFR or ERBB2 (Figure 5B and 5D; Supplementary Figure S3B). In addition, the HRG-1 β1-induced increase of pAKT, pERK and the reporter activity of c-Fos and Elk-1 were also impaired by APIP knockdown (Supplementary Figure S3C~SE). On the other hand, APIP overexpression enabled HRG-β1-induced activation of AKT and ERK1/2 in SNU-620 cells (Figure 5E and Supplementary Figure S3F). These results suggest that APIP is essential for the HRG-β1-induced activation of ERBB3 as well as for the downstream AKT and ERK1/2 pathways.
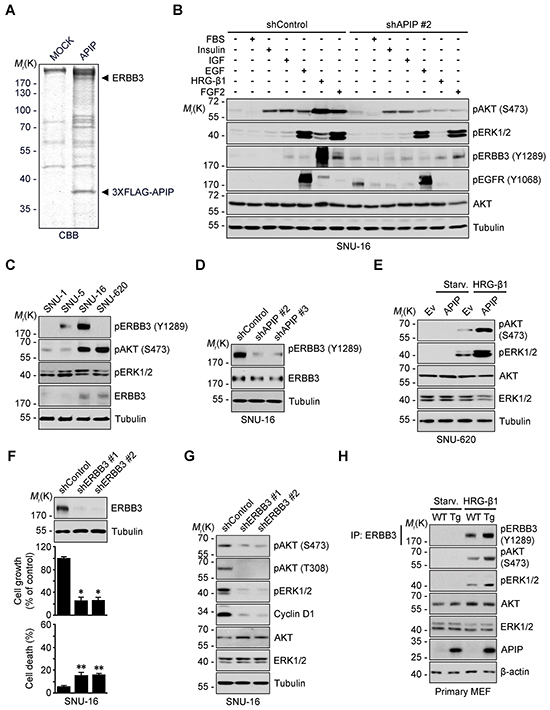
Figure 5: APIP is an essential activator of HRG-β1/ERBB3 in gastric cancer cells. A. APIP-interacting proteins were purified from SNU-16 cells expressing 3xFLAG-tagged APIP by co-immunoprecipitation assay using FLAG M2 affinity gel. The bound proteins were resolved by SDS-PAGE and prepared for LC-MS/MS analysis. CBB, coomassie brilliant blue. B. Inhibition of HRG-β1-dependent ERBB3 phosphorylation and its downstream signals by APIP knockdown. Serum-starved SNU-16 control and APIP knockdown cells were treated with 10% FBS, 2 μM Insulin, 50 ng/ml IGF, 100 ng/ml EGF, 50 ng/ml HRG-β1 or 50 ng/ml FGF2 for 10 min and subjected to western blotting. C. Analysis of phospho-ERBB3 (Y1289) in a panel of gastric cancer cell lines by western blotting.D. Inhibition of ERBB3 activity by APIP knockdown in complete medium. Whole-cell extracts of SNU-16 control and APIP knockdown cells were analyzed with western blotting. E. APIP overexpression sensitizes SNU-620 cells to HRG-β1. SNU-620 control and APIP-overexpressing cells were stimulated with 2 ng/ml HRG-β1 and subjected to western blotting. Starv., serum starved. F. and G. ERBB3 knockdown inhibits cell growth and suppresses AKT and ERK1/2 activity. Cell growth (middle) and death rates (bottom) of SNU-16 control cells (shControl) or ERBB3 knockdown cells (shERBB3 #1 and #2) were assessed and analyzed by western blotting (top). The results represent mean ± S.D. (n = 3). (F). Whole cell lysates were examined by western blotting (G). H. Enhanced HRG-β1 signaling in APIP transgenic MEFs. WT and APIP transgenic MEFs were treated with 10 ng/ml HRG-β1 and harvested for immunoprecipitation (IP) assay. Statistical significance is indicated as follows: *, P < 0.05; **, P < 0.01.
ERBB3 knockdown by shRNA inhibited cell growth while having a low incidence of cell death (Figure 5F), and generated a decrease in AKT and ERK1/2 activities (Figure 5G). Correspondingly, ERBB3 knockdown led to a significant decrease of cyclin D1 levels (Figure 5G). Overall, ERBB3 knockdown showed a very similar effect to APIP knockdown on both the delay in cell growth and inhibition of downstream signaling (Figure 5F and 5G). We further validated the potential role of APIP in the HRG-β1/ERBB3 pathway in APIP Tg and Apip knockout MEFs. In comparison with wild-type MEFs, APIP Tg MEFs showed an enhanced activation of ERBB3, AKT and ERK1/2 following HRG-β1 stimulation (Figure 5H). On the other hand, such differences in the activation of AKT and ERK1/2 between wild-type and Apip knockout MEFs in response to HRG-β1 could not be observed (Supplementary Figure S3G). These observations indicate that the gain-of-function of APIP in ERBB3 signaling is also present for the stimulation of cell proliferation in mice.
APIP binds to the C-terminal tail of ERBB3
To gain insight into the molecular mechanism by which APIP regulates HRG-β1/ERBB3 signaling, we focused on the binding process between APIP and ERBB3. In proliferating SNU-16 cells, we detected ERBB3 in APIP protein complexes isolated by immunoprecipitation assay using an anti-APIP antibody and reciprocally (Figure 6A). We also confirmed the interaction between APIP and ERBB3 in vitro (Figure 6B). We evaluated the effect of HRG-β1 on this binding, showing that HRG-β1 treatment increased the binding of APIP to ERBB3 in SNU-16 cells, particularly to phosphorylated ERBB3 (Y1289) (Figure 6C and Supplementary Figure S4A). Alternatively, an investigation of the same binding process in a living cell using a split-Venus-based bimolecular fluorescence complementation (BiFC) assay enabled us to visualize the formation of ERBB3-VN/APIP-VC complexes in the plasma membrane and cytosol (Supplementary Figure S4B). The reverse combination of complexes (APIP-VN and ERBB3-VC) generated the same fluorescence pattern. Under this condition, co-expression of unlabeled full-length ERBB3 reduced the fluorescence in a dose-dependent manner (Supplementary Figure S4C), indicating that APIP specifically binds to ERBB3 in these cells.
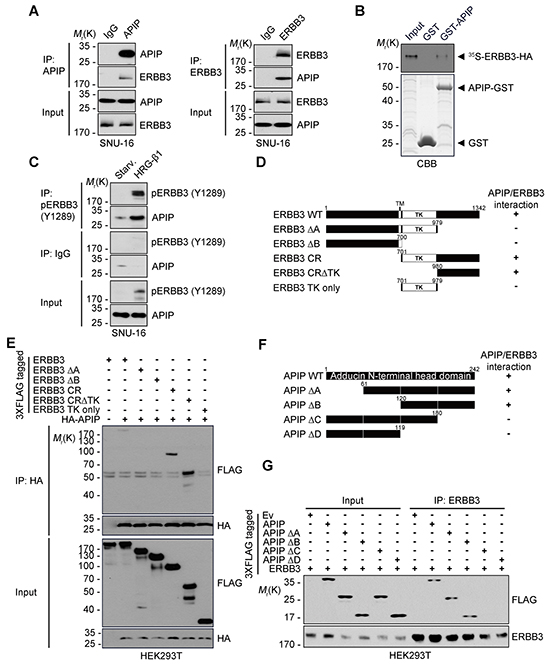
Figure 6: APIP interacts with ERBB3 via its C-terminus. A. Co-immunoprecipitation of APIP and ERBB3 in SNU-16 cells. Whole cell extracts were subjected to immunoprecipitation (IP) assay with anti-APIP (left) or anti-ERBB3 (right) antibody. B. In vitro binding assay. In vitro translated and 35S-methionine-labeled ERBB3 protein was incubated with the purified GST or GST-APIP protein immobilized on glutathione-Sepharose 4B beads. The bound proteins were separated by SDS-PAGE and detected by autoradiography (upper) and CBB staining (lower). C. APIP binds to ERBB3 in a HRG-β1-dependent manner. SNU-16 cells were treated with 50 ng/ml HRG-β1 for 10 min and subjected to immunoprecipitation (IP) assay. D. Schematic representation of ERBB3 and its deletion mutants (Δ). The binding activities of ERBB3 WT and mutants to APIP are summarized based on the results in (E). E. Mapping of APIP-binding domain within ERBB3. HEK293T cells were co-transfected with pHA-APIP and either pERBB3-3xFLAG or deletion mutant for 24 h and whole-cell lysates were subjected to immunoprecipitation (IP) assay. F. Schematic representation of the conserved domains of APIP and its deletion mutants (Δ). The binding activities of APIP WT and mutants to ERBB3 are summarized based on the results in (G). G. The C-terminal region of APIP is essential for the binding to ERBB3. The immunoprecipitates were analyzed as in (E).
To explore the binding regions in APIP and ERBB3 in greater detail, we generated serial deletion mutants of APIP and ERBB3 based on their secondary structures and reported domains (Figure 6D and 6F). The results of the immunoprecipitation assay revealed that APIP binds to the ERBB3-cytosolic region (CR), harboring a tyrosine kinase domain (TK, residues 701-979) and a C-terminal region (residues 980-1342), and the ERBB3-CRΔTK (residues 980-1342). However, APIP does not attach to the ERBB3-TK only or the ERBB3-ΔA lacking the C-terminal tail region (Figure 6D and 6E). These results suggest that APIP binds to the C-terminal tail region (residues 980-1342) of ERBB3. Similar assays revealed that APIP-ΔA and APIP-ΔB mutants lacking the N-terminal regions (residues 1-60 and 1-119, respectively) bind to ERBB3, whereas APIP-ΔC mutant lacking the C-terminal region (residues 181-242) did not interact with ERBB3 (Figure 6F and 6G), confirming the necessity of the C-terminal region of APIP for its binding to ERBB3.
APIP enhances the binding of ERBB3 and ERBB2 in response to HRG-β1
ERBB3 requires hetero-dimerization with a suitable kinase-competent ERBB family member to activate HRG-β1/ERBB3 signaling. We first assessed the phosphorylation status of EGFR, ERBB2 and ERBB3 in serum-starved SNU-16 cells pretreated with EGF or HRG-β1 which differentially induces hetero-dimerization of ERBB family proteins [12, 34]. HRG-β1 mainly increased the phosphorylation of ERBB2 (Y1121/1122) and ERBB3 (Y1289) as well as AKT (S473) and ERK1/2 (Figure 7A), whereas EGF induced the phosphorylation of EGFR (Y1068), ERBB2 (Y1121/1122) and ERK1/2. We next examined whether APIP could stimulate the binding of ERBB3 with ERBB2, and thereby affect the activities of AKT and ERK1/2. A three-way immunoprecipitation assay showed that APIP formed a ternary protein complex with ERBB3 and ERBB2 (APIP/ERBB3/ERBB2) in growing SNU-16 cells (Figure 7B). On the other hand, there was no binding between ERBB3 and ERBB2 in serum-starved SNU-16 cells (Figure 7C). Furthermore, APIP knockdown in SNU-16 cells remarkably reduced the binding of ERBB3 with ERBB2, even in the presence of HRG-β1 (Figure 7D) or serum (Supplementary Figure S5). These results demonstrate that APIP increases the binding of ERBB3 to ERBB2 in response to HRG-β1. An evaluation of the contribution of ERBB2 to cell proliferation revealed that, as seen in ERBB3 knockdown, the loss of ERBB2 expression also suppressed cell proliferation in gastric cells (Figure 7E). However, this effect was lower than with ERBB3 knockdowns (Figure 5E). Overall, these results suggest that APIP stimulates cell proliferation via ERBB3, acting in conjunction with ERBB2, in gastric cancer cells (Figure 7F).
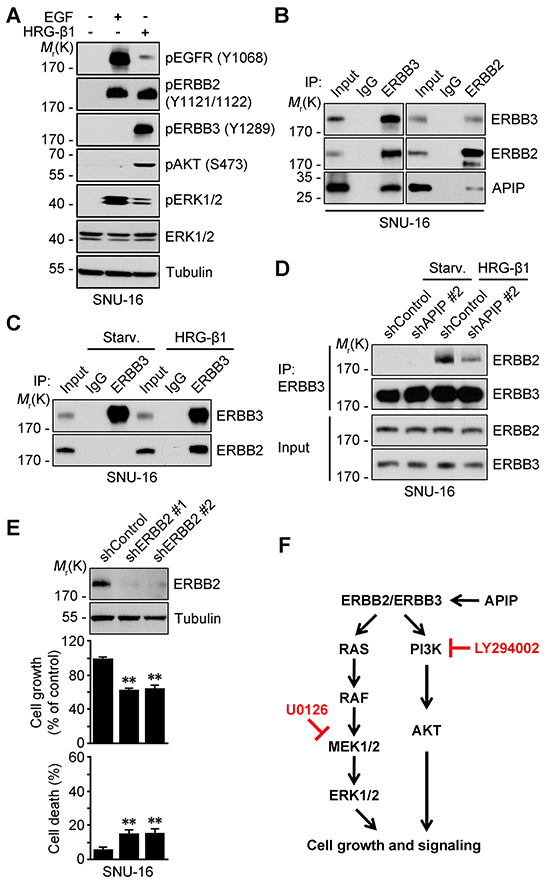
Figure 7: APIP stimulates the formation of ERBB3/ERBB2 heterodimer. A. HRG-β1 induces the transactivation of ERBB2 in SNU-16 cells. SNU-16 cells were treated with 50 ng/ml HRG-β1 or 100 ng/ml EGF for 10 min and subjected to western blotting. B. APIP binds to ERBB3/ERBB2 complex in growing SNU-16 cells. SNU-16 cells were analyzed by immunoprecipitation (IP) assay with anti-ERBB3 (left) or anti-ERBB2 (right) antibody. C. HRG-β1 induces ERBB3/ERBB2 hetero-dimerization. SNU-16 cells were treated with 50 ng/ml HRG-β1 for 10 min and analyzed by immunoprecipitation (IP) assay. D. HRG-β1 induces ERBB3/ERBB2 interaction through APIP. SNU-16 control and APIP knockdown cells were treated with 50 ng/ml HRG-β1 for 10 min and analyzed as in (C). E. ERBB2 knockdown has a modest inhibitory effect on cell growth in SNU-16 cells. Cell growth (middle) and cell death (bottom) rates of SNU-16 control (shControl) and ERBB2 knockdown (shERBB2 #1 and #2) cells were assessed and whole-cell extracts were analyzed by western blotting (top). The results represent mean ± S.D. (n = 3). Statistical significance is indicated as follows: *, P < 0.05; **, P < 0.01. F. Proposed model of APIP/ERBB3/ERBB2 protein complex in cell growth signaling in gastric cancer cells.
DISCUSSION
In the present study, we report an important function of APIP in ERBB2/ERBB3 signaling in gastric cancer. Our findings provide APIP with an oncogenic function which had previously been suggested by reports showing increased expression or gene amplification of APIP in a variety of human cancers (early and advanced), including gastric cancer [27, 29]. The increased expression of APIP in gastric cancer cells enhances the HRG-β1/ERBB3-induced activation of AKT and ERK1/2 which stimulate cell proliferation and positively increase tumorigenesis by elevating ELK-1, C-FOS or cyclin D1 [35]. This signaling pathway is also conserved in other tumor cells, such as HeLa cervical carcinoma cells, as well as in MEFs. Therefore, the function of APIP in stimulating cell proliferation potentially operates in many cell types, including tumor cells and normal cells.
Then, how does APIP stimulate cell proliferation? It has been reported that ERBB3 alone is not sufficient to activate downstream signaling pathways because it is considered to be inactive and is classified as a pseudokinase [10]. It is thus conceivable to employ other ERBB receptors, such as ERBB2 or ERBB4, to activate ERBB3-mediated signaling pathway [13, 20]. ERBB4 was not detected in SNU-16 cells (Supplementary Figure S3H) and consequently not required for the amplification of ERBB3 signaling in SNU-16 cells. On the contrary, ERBB2 hetero-dimerized with ERBB3. As previously shown in EGFR [36], APIP binds to the cytoplasmic domain of ERBB3 to possibly stimulate dimer formation. Moreover, this formation of ERBB2/ERBB3 heterodimer was stimulated by APIP. As a model describing the role of APIP in stimulating the formation of this heterodimer, we suggest that APIP induces conformational change of ERBB3 after binding to it, resulting in an increase of the binding of APIP/ERBB3 to ERBB2 to stimulate heterodimer formation (Figure 7F). We also hypothesize that the binding of APIP and ERBB3 enhances the susceptibility of ERBB3 to HRG-β1 [37–39].
Another possible role of APIP in ERBB3 activation is receptor clustering, as shown in ligand-dependent receptor clustering [40]. A recent report showed that APIP exists as a tetrameric complex which is essential for the methionine salvage pathway [32]. In gastric cancer, APIP also forms a tetrameric complex and the inhibition of APIP/APIP interactions blocks its oncogenic activity. Thus, tetramer forms are important for the APIP oncogenic activity and might play a role in clustering its binding protein ERBB3. Because active forms of ERBB receptors are usually dimeric complexes and there is no direct interaction between ERBB2 and APIP, APIP can function as an enhancer of ERBB3 homo-dimerization or oligomerization [41]. In that sense, the presence of significant kinase activity of ERBB3 was recently emphasized. Although this activity of ERBB3 is weaker than the EGFR’s, it is sufficient to phosphorylate its intracellular region and enable ERBB3-mediatedtrans-autophosphorylation [18].
There are several known non-ERBB regulators of ERBB3, such as MET [42], BRK [43], EBP-1 [44] and CDK5 [45], in gastric cancer. All these non-ERBB regulators of ERBB3 directly phosphorylate ERBB3 [46] or recruit other protein(s) into the receptor complex [47]. APIP exhibits a similar activity, stimulating auto-phosphorylation of ERBB2 and ERBB3 as well as their activities. However, APIP is discriminated from other ERBB3-binding partners, such as c-Src [48, 49] and p85 subunit of PI3K [50] which also bind to the C-terminal regulatory (CR) domain of ERBB3, enabling its phosphorylation and activation in cancer cells. While these binding partners bind to ERBB3 through either their PTB or SH-2 domains [48, 49], there is no such domain in APIP. Indeed, APIP binds to ERBB3 through its uncharacterized domain. The possibility that APIP might form a protein complex with ERBB3 through binding to those ERBB3-binding partners or that it creates a signaling pathway along with non-ERBB regulators of ERBB3 cannot be excluded and needs to be further investigated.
In summary, this is the first report showing that APIP displays a pivotal oncogenic activity by binding to ERBB3 to stimulate the formation of ERBB3/ERBB2 heterodimer, leading to amplification of HRG-mediated signaling involved in cell proliferation and tumorigenesis in gastric cancer. Therefore, APIP can serve as a potential therapeutic target in gastric cancer, specifically targeting ERBB3 signaling in APIP-overexpressing cancers.
MATERIALS AND METHODS
Human gastric cancer tissues
Tissue samples from 110 patients with gastric cancer were studied after surgical resection at Chonbuk National University Hospital in Korea. The frozen fresh human tissue specimens and paraffin sections were supplied according to the regulation of the institutional review board (IRB). This study was approved by the IRB of Seoul National University (IRB No. 1009/001-002).
FACS analysis
Cells were washed with ice-cold PBS and kept in 70% ethanol for one day for fixation. Fixed cells were washed with ice-cold PBS twice and incubated with propidium iodide (PI) (Sigma-Aldrich) solution (0.1% Nonidet P-40, 100 μg/ml RNase A and 200 μg/ml PI) for 10 min. Stained cells were analyzed by FACSCalibur™ flow cytometer (BD Biosciences), and the number of the cells in each stage was calculated with the ModFit LT™ cell cycle analysis program (Verity Software House) according to the manufacturer’s instructions.
Luciferase reporter assay
Cells cultured on 6-well plates were transfected with pFR-luciferase reporter plasmid and either pFA2-Elk-1-GAL4 or pFA2-C-Fos-GAL4 for 18 h. Whole-cell lysates were lysed in chemiluminescence lysis buffer and analyzed for luciferase activity with a luciferase assay kit (Promega) as recommended by the manufacturer. All luciferase activities were normalized by β-galactosidase activity.
Bimolecular fluorescence complementation (BiFC) assay
Cells were transfected with VN or VC-fusion construct alone or in combination and incubated at 37°C for 18 h. After staining cell nuclei with Hoechst 33342, fluorophore formation in the living cells was imaged using a fluorescence microscope (Olympus, Hamburg, Germany) [51].
Immunoprecipitation assay
For endogenous immunoprecipitation assay, 5 × 106 SNU-16 cells were lysed in 600 μl CHAPS buffer containing 30 mM Tris-Cl pH 7.5, 150 mM NaCl, 1% CHAPS, 1 mM PMSF and 1 mg/ml each of aprotinin, leupeptin and pepstatin A. Whole-cell lysates were incubated for 12 h at 4°C with anti-APIP antibody or IgG after preclearance with 30 μl protein A/G–Bead (Santa Cruz Biotechnology). For co-immunoprecipitation assay, 1 × 107 HEK293T cells were transfected with 3xFLAG-tagged APIP and HA-tagged APIP, and lysed in RIPA buffer (50 mM Tris-Cl pH 8.0, 15 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF and 1 mg/ml each of aprotinin, leupeptin and pepstatin A). Whole-cell lysates were incubated for 2 h at 4°C with monoclonal HA or FLAG antibody. After adding another 30 μl protein A/G–Bead, the mixtures were incubated for an additional 2 h at 4°C. The immunocomplexes were collected by centrifugation at 5,000 rpm for 3 min, washed 3 times with lysis buffer and detected by western blotting, as described above.
Reverse transcription-PCR
Total RNA was purified using TRIZOL® reagent (Invitrogen) according to the manufacturer’s protocol. Reverse transcription was performed with a SuperScript II First-Strand Synthesis Kit (Invitrogen). PCR was performed using the following synthetic oligonucleotides sets: GAPDH (5’-GAA GGT GAA GGT CGG AGT CAA C-3’, 5’’-CCT GGA AGA TGG TGA TGG GAT T-3’); APIP (5’-CTG GGT CAC TGG GAC TGG AGG AGG-3’, 5’-GAA TAA AGA AAT GTA CTT CCG GAG GG-3’); EGFR (5’- ATG CGA CCC TCC GGG ACG GCC-3’, 5’-GAT AAG ACT GCT AAG GCA TAG G-3’; ERBB2 (5’-CAC AGA CAC GTT TGA GTC CA-3’, 5’-AAA GCT CTC CGG CAG AAA TG-3’); ERBB3 (5’-GGC AGC ACA CAG AGT TGC CC-3’, 5’-TCT CTG GGC ATT AGC CTT GGG G-3’); and ERBB4 (5’-GGA AGA TAT GAT GGA TGC TGA GGA G-3’, 5’-TTT GGG TTT GTC TCG CAT AGG AG-3’).
Plasmid construction
APIP cDNA was subcloned into the Xho1 site of pcDNA3-HA (pHA-APIP) or p3x-FLAG-CMV14 (3xFLAG-APIP). Empty vectors were used as control (Ev). The shRNAs were constructed using forward and reverse synthetic 19-nucleotides, which were synthesized, annealed, and sub-cloned into BglII and HindIII sites of pSUPER.neo. Sequences were as follows: shAPIP (#2, 5’-GGA CGG GAG TTT AAA ATT A-3’; #3, 5’-CCA TGT GTG AGT GTT ATG A-3’); shERBB2 (#1, 5’-TGG AAG AGA TCA CAG GTT A-3’; #2, 5’-GCC AGG TGG TGC AGG GAA A-3’); and shERBB3 (#1, 5’- GAG CGA CTA GAC ATC AAG C-3’;- AAG AGG ATG TCA ACG GTT A-3’).
Complementation assay using shRNA-resistant APIP
APIP* that carries multiple silent mutations at the binding site of shAPIP #2 was constructed by site-directed mutagenesis using pfu DNA polymerase (Neurotics) and the oligonucleotide primer 5’-GGT CGC GAA TTC AAG ATA A-3’ (mutated residues are underlined).
Cell culture
Human gastric cancer cell lines, SNU-1, -5, -16, -216, -484,-601, -620, -638, -668 and -719, were provided by Cancer Research Institute of Seoul National Hospital, Seoul National University [31]. Each cell line was maintained in a medium as follows: SNU cell lines in RPMI-1640 supplemented with 10% FBS (HyClone) and 2 mM glutamine; and MEFs, NIH3T3, HeLa and HEK293T cells in DMEM supplemented with 10% FBS. All cell lines were incubated in a humidified atmosphere of 5% CO2 at 37°C.
Generation of APIP transgenic and Apip knockout mouse
A 729 bp fragment encoding human APIP cDNA (NM_015957.2) was subcloned into XhoI site of the mammalian expression vector pCAGGS [52]. The pCAGGS-APIP was digested with SalI and HindIII to isolate the transgenic cassette consisting of CMV enhancer, chicken β-actin promoter, APIP cDNA and rabbit β-globin poly(A) sequence. The transgenic cassette was injected into mouse embryos with a C57BL/6 genetic background and four independent founders (F0) were identified by PCR analysis using synthetic oligonucleotides (forward, pCAGGS-for; reverse, APIP Tg-rev) (Macrogen, Seoul, Korea).
Apip/Mmrp19 gene-trapped embryonic stem cells (XK742) were provided by BayGenomics of the International Gene Trap Consortium. Mice heterozygotes for Apip were generated by following the protocol provided by BayGenomics. Insertion of the gene-trap vector was confirmed by genomic PCR with a β-gal probe recommended by BayGenomics. The expression of APIP was examined using RT-PCR with synthetic oligonucleotides derived from Apip (Apip-5’ and Apip-3’) and β-gal. The absence of APIP expression in homozygous Apip KO (Apip−/−) mice was confirmed by Western analysis of the proteins extracted from the various tissues of Apip KO mice (4 weeks old). All animal experiments were performed with the approval of the Institutional Animal Care and use Committee of Seoul National University (IACUC No. SNU-120928-3). The animal treatments were carried out in accordance with the guidelines of the International Association for the Study of Pain (IASP). All mice were housed in an animal facility with a specific pathogen-free (SPF) barrier under a 12 h light/dark cycle.
Primary culture of mouse embryonic fibroblasts
APIP Tg or Apip KO heterozygous mice were interbred to generate homozygous, heterozygous and wild-type embryos. Primary MEFs were derived from day 13.5 littermate embryo. The embryos were washed once with DMEM and 3 times with PBS. The tissue was then minced with a scalpel blade and digested with 0.01% Trypsin-EDTA (Invitrogen) for 30 min at 37 °C. The tissue was shaken vigorously every 5 min during the incubation, and the mixture was then passed 3 times through an 18G needle to further dissociate any remaining clumps. Trypsin-EDTA was inactivated by addition of a 15-fold excess of DMEM containing 10% FBS. Cells from each embryo were put into 100 mm dish and cultured in DMEM with 10% FBS. Primary MEF from each embryo were plated separately and their genotypes were examined by Western blotting and genomic PCR. For standard culture condition, primary MEFs [passage (P) 2 or P3] were cultured in DMEM with 10% FBS.
DNA transfection and reagents
Transfection was carried out using Polyfect® reagent (Qiagen, Valencia, CA), Lipofectamine® (Invitrogen) or PEI (Sigma-Aldrich) according to the manufacturers’ instructions. Recombinant human HRG-β1 was purchased from R&D systems (Minneapolis, MN). EGF, IGF, FGF2 and insulin were obtained from Sigma-Aldrich.
Generation of stable cell lines
SNU-620 and NIH3T3 cells were transfected with pcDNA3-HA or pHA-APIP for 24 h and then cultivated in selection medium containing 1 mg/ml G418 (Invitrogen) for 2 weeks. SNU-16 cells were transfected with pSUPER.neo (OligoEngine) or pAPIP shRNAs for 24 h and selected as above followed by western blot analysis.
Cell death and cell proliferation assays
To determine cell death, cells were analyzed with trypan blue assay. To assess cell proliferation using DNA contents, 2.5 × 103 cells were fluorometrically analyzed for their DNA contents with CyQUANT® Cell Proliferation Assay kit (Molecular Probes) according to the manufacturer’s instructions. Fluorescence was measured for excitation at 480 nm and emission detection at 520 nm on EnVision® Multilabel Plate Reader (PerkinElmer, Wellesley, MA).
Xenograft assay
Female athymic nude mice (4–5 weeks old) were obtained from the Central lab. Animal Inc. (Seoul, Korea) and injected subcutaneously with 2× 106 cells in 0.1 ml of PBS containing 50 μl of Matrigel™ (BD Bioscience) into the flanks. Tumor volumes were measured weekly and calculated using the formula V=ab2/2 in millimeters, where ‘a’ is the length and ‘b’ is the width. Statistical analysis was performed by Student’s t-test applied to the final time point.
Focus-formation assay
Triplicate 6 well plates of confluent NIH3T3 control and APIP overexpression cells were left untreated or treated with a variety of inhibitors and cultured for additional 2 weeks. Media was replaced every 24 h thereafter until termination of the experiment. To inhibit focus formation, the inhibitors were added at every media change at the concentrations indicated. Cells were then washed with PBS, fixed with 10% formaldehyde and stained with 0.05% Crystal violet and the transformed foci were scored by manual counting.
Western blotting and antibodies
Cells were lysed in a lysis buffer (50 mM Tris–Cl pH 7.4, 30 mM NaCl, 1% Triton X-100, 0.1% SDS, 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF and 1 mg/ml each of aprotinin, leupeptin and pepstatin A) and sonicated briefly. The lysates were clarified by centrifugation, separated by SDS–PAGE and blotted onto nitrocellulose membrane (Millipore, Billerica, MA, USA). The membrane was blocked with 3% BSA in TBST (10 mM Tris-Cl pH 8.0, 150 mM NaCl, 0.5% Tween-20) and incubated overnight at 4°C with primary antibodies. The following monoclonal (m) and polyclonal (p) antibodies (Ab) were used: anti-APIP pAb (Imgenix); anti-PCNA mAb, anti-GAPDH mAb, anti-Tubulin mAb, anti-GFP pAb and Cyclin D1 mAb (Santa Cruz Biotechnology); anti-AKT pAb, anti-ERK1/2 pAb, anti-EGFR mAb, anti-ERBB2 mAb, anti-ERBB3 mAb, anti-AKT phospho (pS473) pAb, anti-AKT phospho (pT308) mAb, anti-ERK1/2 phospho (pT202 /pY204) mAb, anti-EGFR phospho (pY1068) mAb, anti-ERBB2 phospho (pY1221/pY1222) mAb and anti-ERBB3 phospho (pY1289) mAb (Cell signaling); and anti-FLAG mAb (Sigma-Aldrich).
Statistical analysis
Data are expressed as mean ± S.D. Statistical comparisons between groups were performed using one-way analysis of variance (ANOVA) followed by Bonferroni’s test or by the Student’s t-test as appropriate. Probabilities of *P < 0.05, **P < 0.01 and ***P < 0.001 were considered statistically significant.
ACKNOWLEDGMENTS
Ms. B. Lim was in part supported by the Brain Korea 21 program. The biospecimens for this study were provided by the Biobank of Chonbuk National University Hospital, a member of the National Biobank of Korea, which is supported by the Ministry of Health, Welfare and Family Affairs. All samples were obtained with informed consent under institutional review board-approved protocols.
FUNDING
This work was supported by Global Research Laboratory (NRF-2010-00341) and a CRI grant (NRF-2013R1A2A1A01016896) funded by the Ministry of Education, Science and Technology.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
Author contributions
S.-H.H., W.L, D.-H.C. and Y.-K.J. designed research; S.-H.H., Y.-J.J., Y.K., H.K., W.L. and B.L. performed experiments; H.-K.Y., W.D.H., J.K.Y., D.-H.Y. and C.-Y.K. contributed new reagents/analytic tools; S.-H.H., W.L. and Y.-K.J. analyzed all experiments and wrote the manuscript.
REFERENCES
1. Parkin DM, Bray F, Ferlay J and Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005; 55:74-108.
2. Jemal A, Bray F, Center MM, Ferlay J, Ward E and Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69-90.
3. Nessling M, Solinas-Toldo S, Wilgenbus KK, Borchard F and Lichter P. Mapping of chromosomal imbalances in gastric adenocarcinoma revealed amplified protooncogenes MYCN, MET, WNT2, and ERBB2. Genes, chromosomes & cancer. 1998; 23:307-316.
4. Cho JH, Noguchi M, Ochiai A and Hirohashi S. Loss of heterozygosity of multiple tumor suppressor genes in human gastric cancers by polymerase chain reaction. Lab Invest. 1996; 74:835-841.
5. Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, Kim HM, Kim WJ, Yamamoto H, Yamashita N, Yano T, Ikeda T, et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002; 109:113-124.
6. Ciardiello F, Caputo R, Bianco R, Damiano V, Fontanini G, Cuccato S, De Placido S, Bianco AR and Tortora G. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res. 2001; 7:1459-1465.
7. Xue C, Liang F, Mahmood R, Vuolo M, Wyckoff J, Qian H, Tsai KL, Kim M, Locker J, Zhang ZY and Segall JE. ErbB3-dependent motility and intravasation in breast cancer metastasis. Cancer research. 2006; 66:1418-1426.
8. Yamaguchi T, Yanagisawa K, Sugiyama R, Hosono Y, Shimada Y, Arima C, Kato S, Tomida S, Suzuki M, Osada H and Takahashi T. NKX2-1/TITF1/TTF-1-Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell. 2012; 21:348-361.
9. Zhu SM, Belkhiri A and El-Rifai W. DARPP-32 Increases Interactions Between Epidermal Growth Factor Receptor and ERBB3 to Promote Tumor Resistance to Gefitinib. Gastroenterology. 2011; 141:1738-U1297.
10. Hynes NE and MacDonald G. ErbB receptors and signaling pathways in cancer. Current opinion in cell biology. 2009; 21:177-184.
11. Sithanandam G and Anderson LM. The ERBB3 receptor in cancer and cancer gene therapy. Cancer gene therapy. 2008; 15:413-448.
12. Baselga J and Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nature reviews Cancer. 2009; 9:463-475.
13. Hynes NE and Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nature reviews Cancer. 2005; 5:341-354.
14. Garcia I, Vizoso F, Martin A, Sanz L, Abdel-Lah O, Raigoso P and Garcia-Muniz JL. Clinical significance of the epidermal growth factor receptor and HER2 receptor in resectable gastric cancer. Annals of surgical oncology. 2003; 10:234-241.
15. Allgayer H, Babic R, Gruetzner KU, Tarabichi A, Schildberg FW and Heiss MM. c-erbB-2 is of independent prognostic relevance in gastric cancer and is associated with the expression of tumor-associated protease systems. J Clin Oncol. 2000; 18:2201-2209.
16. Ross JS and McKenna BJ. The HER-2/neu oncogene in tumors of the gastrointestinal tract. Cancer investigation. 2001; 19:554-568.
17. Steinkamp MP, Low-Nam ST, Yang S, Lidke KA, Lidke DS and Wilson BS. erbB3 is an active tyrosine kinase capable of homo- and heterointeractions. Molecular and cellular biology. 2014; 34:965-977.
18. Shi F, Telesco SE, Liu Y, Radhakrishnan R and Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:7692-7697.
19. Yarden Y and Sliwkowski MX. Untangling the ErbB signalling network. Nature reviews Molecular cell biology. 2001; 2:127-137.
20. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF and Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. P Natl Acad Sci USA. 2003; 100:8933-8938.
21. Cho DH, Hong YM, Lee HJ, Woo HN, Pyo JO, Mak TW and Jung YK. Induced inhibition of ischemic/hypoxic injury by APIP, a novel apaf-1-interacting protein. J Biol Chem. 2004; 279:39942-39950.
22. Cho DH, Lee HJ, Kim HJ, Hong SH, Pyo JO, Cho C and Jung YK. Suppression of hypoxic cell death by APIP-induced sustained activation of AKT and ERK1/2. Oncogene. 2007; 26:2809-2814.
23. Ko DC, Gamazon ER, Shukla KP, Pfuetzner RA, Whittington D, Holden TD, Brittnacher MJ, Fong C, Radey M, Ogohara C, Stark AL, Akey JM, Dolan ME, Wurfel MM and Miller SI. Functional genetic screen of human diversity reveals that a methionine salvage enzyme regulates inflammatory cell death. P Natl Acad Sci USA. 2012; 109:E2343-E2352.
24. Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Bjorling L and Ponten F. Towards a knowledge-based Human Protein Atlas. Nature biotechnology. 2010; 28:1248-1250.
25. Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E, Andersson AC, Angelidou P, Asplund A, Asplund C, Berglund L, Bergstrom K, Brumer H, Cerjan D, Ekstrom M, Elobeid A, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Molecular & cellular proteomics. 2005; 4:1920-1932.
26. Fukuda Y, Kurihara N, Imoto I, Yasui K, Yoshida M, Yanagihara K, Park JG, Nakamura Y and Inazawa J. CD44 is a potential target of amplification within the 11p13 amplicon detected in gastric cancer cell lines. Genes Chromosomes & Cancer. 2000; 29:315-324.
27. Liu YY, Chen HY, Zhang ML, Tian D, Li S and Lee JY. Loss of fragile histidine triad and amplification of 1p36.22 and 11p15.5 in primary gastric adenocarcinomas. World journal of gastroenterology : WJG. 2012; 18:4522-4532.
28. Seruca R, Castedo S, Correia C, Gomes P, Carneiro F, Soares P, de Jong B and Sobrinho-Simoes M. Cytogenetic findings in eleven gastric carcinomas. Cancer genetics and cytogenetics. 1993; 68:42-48.
29. Zhang D, Wang ZN, Luo Y, Xu Y, Liu YS, Yang W and Zhang X. Analysis of DNA copy Number Aberrations by Multiple Ligation-Dependent Probe Amplification on 50 Intestinal Type Gastric Cancers. J Surg Oncol. 2011; 103:124-132.
30. Moravcikova E, Krepela E, Prochazka J, Rousalova I, Cermak J and Benkova K. Down-regulated expression of apoptosis-associated genes APIP and UACA in non-small cell lung carcinoma. International journal of oncology. 2012; 40:2111-2121.
31. Park JG, Frucht H, LaRocca RV, Bliss DP, Jr., Kurita Y, Chen TR, Henslee JG, Trepel JB, Jensen RT, Johnson BE and et al. Characteristics of cell lines established from human gastric carcinoma. Cancer research. 1990; 50:2773-2780.
32. Kang W, Hong SH, Lee HM, Kim NY, Lim YC, Le le TM, Lim B, Kim HC, Kim TY, Ashida H, Yokota A, Hah SS, Chun KH, Jung YK and Yang JK. Structural and biochemical basis for the inhibition of cell death by APIP, a methionine salvage enzyme. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:E54-61.
33. Henkel AS, Dewey AM, Anderson KA, Olivares S and Green RM. Reducing endoplasmic reticulum stress does not improve steatohepatitis in mice fed a methionine- and choline-deficient diet. American journal of physiology Gastrointestinal and liver physiology. 2012; 303:G54-59.
34. Normanno N, Bianco C, Strizzi L, Mancino M, Maiello MR, De Luca A, Caponigro F and Salomon DS. The ErbB receptors and their ligands in cancer: An overview. Curr Drug Targets. 2005; 6:243-257.
35. Sithanandam G, Fornwald LW, Fields J and Anderson LM. Inactivation of ErbB3 by siRNA promotes apoptosis and attenuates growth and invasiveness of human lung adenocarcinoma cell line A549. Oncogene. 2005; 24:1847-1859.
36. Yu X, Sharma KD, Takahashi T, Iwamoto R and Mekada E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Molecular biology of the cell. 2002; 13:2547-2557.
37. Yen L, Benlimame N, Nie ZR, Xiao D, Wang T, Al Moustafa AE, Esumi H, Milanini J, Hynes NE, Pages G and Alaoui-Jamali MA. Differential regulation of tumor angiogenesis by distinct ErbB homo- and heterodimers. Molecular biology of the cell. 2002; 13:4029-4044.
38. Kim HH, Vijapurkar U, Hellyer NJ, Bravo D and Koland JG. Signal transduction by epidermal growth factor and heregulin via the kinase-deficient ErbB3 protein. The Biochemical journal. 1998; 334 ( Pt 1):189-195.
39. Sliwkowski MX, Schaefer G, Akita RW, Lofgren JA, Fitzpatrick VD, Nuijens A, Fendly BM, Cerione RA, Vandlen RL and Carraway KL, 3rd. Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. The Journal of biological chemistry. 1994; 269:14661-14665.
40. Bray D, Levin MD and Morton-Firth CJ. Receptor clustering as a cellular mechanism to control sensitivity. Nature. 1998; 393:85-88.
41. Kani K, Warren CM, Kaddis CS, Loo JA and Landgraf R. Oligomers of ERBB3 have two distinct interfaces that differ in their sensitivity to disruption by heregulin. The Journal of biological chemistry. 2005; 280:8238-8247.
42. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039-1043.
43. Kamalati T, Jolin HE, Fry MJ and Crompton MR. Expression of the BRK tyrosine kinase in mammary epithelial cells enhances the coupling of EGF signalling to PI 3-kinase and Akt, via erbB3 phosphorylation. Oncogene. 2000; 19:5471-5476.
44. Zhang Y and Hamburger AW. Specificity and heregulin regulation of Ebp1 (ErbB3 binding protein 1) mediated repression of androgen receptor signalling. British journal of cancer. 2005; 92:140-146.
45. Li BS, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, Kulkarni AB and Pant HC. Cyclin-dependent kinase-5 is involved in neuregulin-dependent activation of phosphatidylinositol 3-kinase and Akt activity mediating neuronal survival. The Journal of biological chemistry. 2003; 278:35702-35709.
46. Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, Scher HI and Sliwkowski MX. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer cell. 2002; 2:127-137.
47. Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM and Moasser MM. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007; 445:437-441.
48. Ishizawar RC, Miyake T and Parsons SJ. c-Src modulates ErbB2 and ErbB3 heterocomplex formation and function. Oncogene. 2007; 26:3503-3510.
49. Contessa JN, Abell A, Mikkelsen RB, Valerie K and Schmidt-Ullrich RK. Compensatory ErbB3/c-Src signaling enhances carcinoma cell survival to ionizing radiation. Breast cancer research and treatment. 2006; 95:17-27.
50. Schulze WX, Deng L and Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Molecular systems biology. 2005; 1:2005.0008.
51. Hu CD, Chinenov Y and Kerppola TK. Visualization of interactions among bZip and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002; 9:789-798.
52. Galbiati F, Volonte D, Liu J, Capozza F, Frank PG, Zhu L, Pestell RG and Lisanti MP. Caveolin-1 expression negatively regulates cell cycle progression by inducing G(0)/G(1) arrest via a p53/p21(WAF1/Cip1)-dependent mechanism. Mol Biol Cell. 2001; 12:2229-2244.