
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in elderly people. Neurofibrillary tangles, composed of abnormally hyperphosphorylated tau, are key lesions of AD [1]. Abnormal hyperphosphorylation of tau converts it from a microtubule assembly-promoting to a microtubule-disrupting protein, leading to the destabilization of microtubules, the impairment of axonal transport, the dysfunction of hippocampal synaptic plasticity, and eventually the neuronal death [2]. Animal studies have consistently shown that the abnormal hyperphosphorylation of tau causes cognitive impairment [3, 4]. Therefore, proper manipulation of tau abnormal hyperphosphorylation could be promising for arresting AD neurodegeneration.
The involvement of the serotonin (5-HT) system in higher cognitive processes, such as learning and memory, has been widely described over the years and resurfaced as a new target for AD treatment. Postmortem and imaging studies demonstrated that the reduction of 5-HT and 5-HT1A receptor (5-HT1AR) in the hippocampus is correlated with cognitive decline in AD patients [5, 6]. Selective serotonin reuptake inhibitors (SSRIs), a well-known class of antidepressants, act by selectively inhibiting the reuptake of 5-HT and subsequently increase the amount of serotonin available to bind critically to 5-HT1AR. SSRIs have been proved effective in hindering the progression of the AD and improving patients’ performance [7-9]. Preclinical studies have also demonstrated a favorable cognitive-improving effect of SSRIs [10, 11]. SSRIs are also reported to increase neurotrophic factors including brain-derived neurotrophic factor (BDNF), promote neurogenesis in the hippocampus and reduce levels of toxic Amyloid-β (Aβ) [12, 13]. Interestingly, our previous study showed that escitalopram, one of the SSRIs, attenuated forskolin-induced tau hyperphosphorylation in human embryonic kidney cells that stably express human longest tau isoform tau441 (HEK293/tau441 cells) [14]. However, the mechanism has not been fully investigated.
At present, it is considered that glycogen synthase kinase-3β (GSK-3β) is a major tau kinase involved in tau hyperphosphorylation. GSK-3β activity is abnormally upregulated due to the inactivation of its upstream PI3K/Akt pathway in AD patients [15]. An activation of cortical GSK-3β has been found in 5-HT deficient mice [16, 17]. On the other hand, in vivo studies demonstrated that SSRI fluoxetine inactivated GSK-3β [18] and prevented the stress-induced inhibition of PI3K/Akt/GSK-3β pathway [19]. Furthermore, GSK-3β genetic variants play a role in the therapeutic response of SSRIs in depression [20]. A 5HT1AR agonist, 8-hydroxy-2-(din-propylamino) tetralin (8-OH-DPAT), was found to inactivate GSK-3β and the PI3K/Akt pathway was involved in this process[18, 21]. Therefore, we hypothesized that the 5-HT1AR mediated PI3K/Akt/GSK-3β pathway is responsible for reduced tau hyperphosphorylation in SSRIs-treated primary hippocampal neurons.
To this purpose, we treated the primary hippocampal neurons with Aβ1-42 to induce tau hyperphosphorylation, and then we examined whether escitalopram could attenuate tau hyperphosphorylation. Subsequently, we investigated whether the 5-HT1AR mediated PI3K/Akt/GSK-3β pathway was involved.
Results
Escitalopram attenuates Aβ1-42-induced tau hyperphosphorylation in hippocampal neurons
As revealed by coomassie blue and silver staining, the Aβ1-42 preparation was almost exclusively composed of low-molecular-weight Aβ1-42 oligomers (Figure 1A). It produced one band likely to represent Aβ monomer (molecular weight 4.5 kDa) and two evident bands probably representing Aβ trimers and tetramers (molecular weight ~17 kDa). Western blotting results indicated that Aβ1-42 at concentrations higher than 1μM significantly increased tau phosphorylation at pS396 site in primary hippocampal neuron cultures (Figure 1B). Since previous reports suggested that Aβ1-42 at higher concentrations induced neurotoxicity [22], 2 μM Aβ1-42 was considered optimum to induce tau hyperphosphorylation in our study. The MTT assay showed that escitalopram did not affect the neuronal viability at concentrations from 5 to 80 μM (Figure 1C). As shown in Figure 1D, escitalopram decreased Aβ1-42-induced tau phosphorylation in a concentration-dependent manner, while it had no effect on Tau5 that represents the total tau protein. Escitalopram (80 μM) significantly decreased tau phosphorylation at Thr231 and Ser396, while increased Tau1 that indicates the unphosphorylated tau protein. Immunofluorescence results also showed that Aβ1-42 treatment increased the tau phosphorylation, while escitalopram (80 μM) attenuated the tau phosphorylation (Figure 1E).
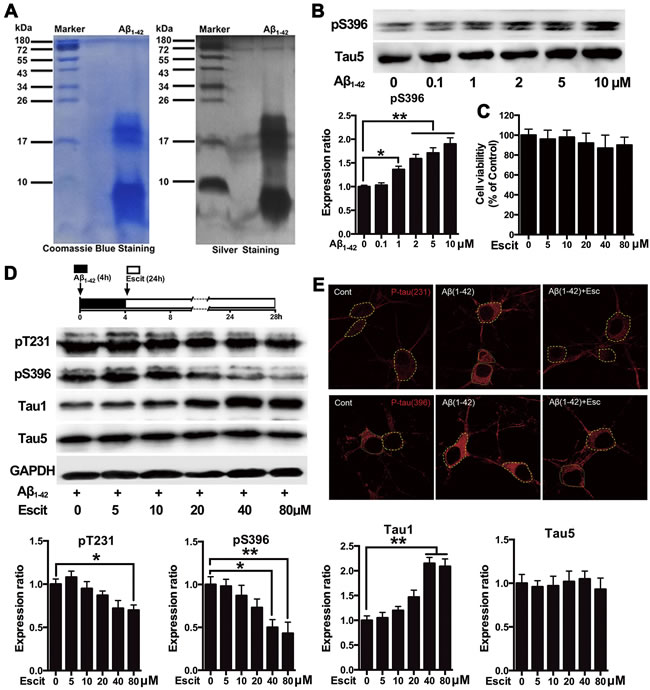
Figure 1: Escitalopram attenuates Aβ1-42-induced tau hyperphosphorylation in hippocampal neurons. A. Representative coomassie blue and silver staining of the Aβ1-42 solution used in the cultured hippocampal neurons. B. Immunoblots of tau phosphorylated at pS396 site in the cultured hippocampal neurons incubated with Aβ1-42 for 4 h at the concentrations indicated. Tau5 was used for normalization. Data were expressed as means ± SEM (n = 3; *p < 0.05, **p < 0.01). B and C. Cells were treated with Aβ1-42 (2 μM) for 4 h, and then incubated with escitalopram at the concentrations indicated for 24 h in fresh medium. Cell viability was detected by the MTT assay (C). Immunoblots of tau phosphorylated at pT231, pS396, Tau1 and Tau5 D. Tau5 or GAPDH was used for normalization. Data were means ± SEM (n = 3; *p < 0.05, **p < 0.01). E. Representative of p-Tau (Thr231) and p-Tau (Ser396) immunofluorescence from the cultured hippocampal neurons incubated with 80μM escitalopram for 24 h in the presence of pretreatment with Aβ1-42 (2 μM) for 4h. p-Tau (Thr231) and p-Tau (Ser396) were labeled with red. Similar results were observed in each of three experiments. Scale bar, 10μm. Escit, Escitalopram.
To determine whether the decreased tau phosphorylation was due to the pharmacological action of escitalopram, its enantiomer, R-citalopram, which is relatively much less active as an SSRI [23], was used. The western blotting results showed that different doses of R-citalopram had no effect on Aβ1-42-induced tau hyperphosphorylation at Thr231, Ser396 and Tau-1 epitopes (Figure 2A). To investigate whether the decreased tau phosphorylation was unique to escitalopram or for the SSRIs group, another SSRI, fluoxetine was used. As shown in Figure 2B, fluoxetine also decreased Aβ1-42-induced tau hyperphosphorylation in a concentration-dependent manner. Fluoxetine at 20 μM significantly decreased tau phosphorylation at Thr231 and Ser396, and increased Tau1, while it had no effect on Tau5.
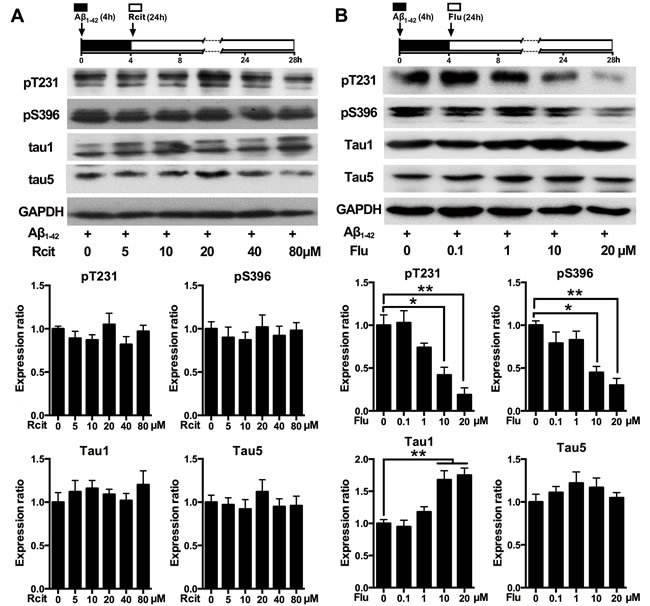
Figure 2: Effects of R-citalopram and fluoxetine on Aβ1-42-induced tau hyperphosphorylation in hippocampal neurons. Immunoblots of tau phosphorylated at pT231, pS396, Tau1 and Tau5 in the cultured hippocampal neurons. Cells were treated with Aβ1-42 (2 μM) for 4 h, and then incubated with R-citalopram A. or fluoxetine B. at the concentrations indicated for 24 h in fresh medium. Tau5 or GAPDH was used for normalization. Data were represented as means ± SEM (n = 3; *p < 0.05, **p < 0.01). Rcit, R-citalopram; Flu, fluoxetine.
Activation of PI3K/Akt/GSK-3β pathway contributes to the anti-hyperphosphorylation role of escitalopram
As GSK-3β is the crucial kinase for tau hyperphosphorylation and phosphatase 2A (PP2A) is the key phosphatase in tau dephosphorylation, the activities of GSK-3β and PP2A were measured. As shown in Figure 3A, escitalopram increased the level of pS9-GSK-3β (inactivated form) in a concentration-dependent manner, while it had no significant effect on the level of pY307-PP2AC (inactivated form). Furthermore, the phosphorylation of Akt, a critical upstream regulator of GSK-3β, was dose-dependently increased by escitalopram. In addition, LY294002, a specific inhibitor of PI3K, was found to block the phosphorylation of GSK-3β (Ser9) and Akt (Ser473 and Thr308) induced by escitalopram (80 μM) (Figure 3B). Accordingly, the attenuation of tau hyperphosphorylation at pT231, pS396 and Tau1 epitopes induced by escitalopram was reversed by LY294002 (Figure 3C). Thus, these results indicate that the PI3K/Akt/GSK-3β pathway may underlie the anti-hyperphosphorylation effect of escitalopram.
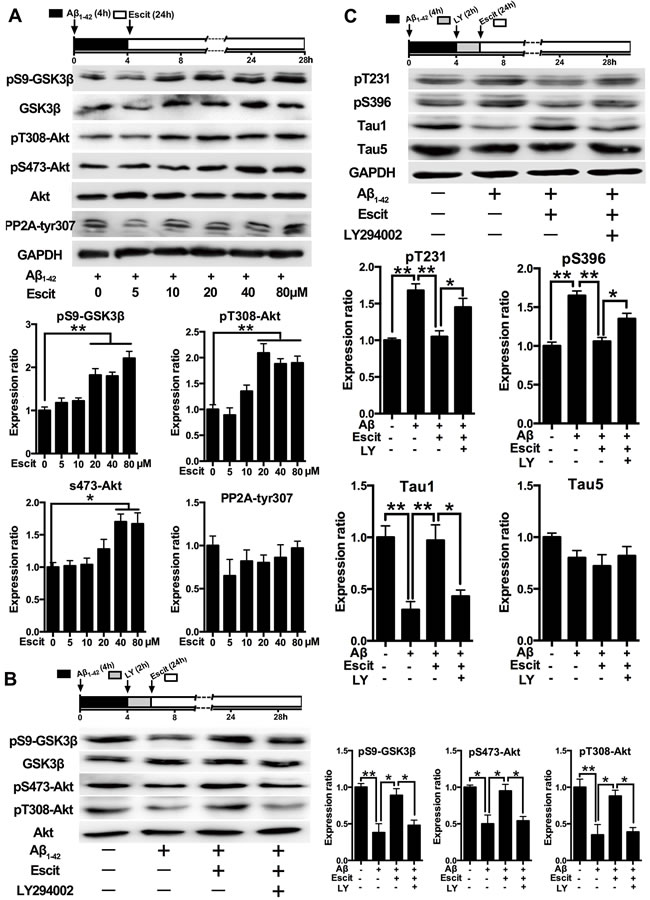
Figure 3: Activation of PI3K/Akt/GSK-3β pathway contributes to the anti-hyperphosphorylation role of escitalopram. A. Immunoblots of pS9-GSK-3β, total GSK-3β, pT308-Akt, pS473-Akt, total Akt, p-PP2Ac (tyr307) in the cultured hippocampal neurons. Cells were treated with Aβ1-42 (2 μM) for 4 h, and then incubated with escitalopram at the concentrations indicated for 24 h in fresh medium. The respective total protein or GAPDH was used for normalization. Data were expressed as means ± SEM (n = 3; *p < 0.05, **p < 0.01). B. and C. Immunoblots of pS9-GSK-3β, total GSK-3β, pT308-Akt, pS473-Akt, total Akt (B) and tau phosphorylated at pT231, pS396, Tau1 and Tau5 (C) in the cultured hippocampal neurons. Cells were treated with Aβ1-42 (2 μM) for 4 h, and then incubated with escitalopram (80 μM) for 24 h in fresh medium with or without the pretreatment with LY294002 (10 μM, 2 h). Tau5 or GAPDH was used for normalization. Data were expressed as means ± SEM (n = 3; *p < 0.05, **p < 0.01). Escit, escitalopram; LY, LY294002.
Effects of escitalopram on the PI3K/Akt/GSK-3β signaling pathway depends on 5-HT1AR
Since 5-HT1AR is a critical component in the mechanism of action of SSRIs, we next examined whether 5-HT1A is involved in the activation of PI3K/Akt/GSK-3β pathway induced by escitalopram. Similar to escitalopram, the 5-HT1AR agonist 8-OH-DPAT also decreased Aβ1-42-induced tau hyperphosphorylation at Thr231, Ser396 and Tau-1 epitopes in a concentration-dependent manner (Figure 4A). Furthermore, 8-OH-DPAT increased the level of pS9-GSK-3β, pT308-Akt and pS473-Akt in a concentration-dependent manner (Figure 4B). On the other hand, the protective effects of escitalopram on Aβ1-42-induced tau hyperphosphorylation at pT231, pS396 and Tau1 epitopes induced by escitalopram were blocked by WAY-100635, a selective antagonist of 5-HT1AR (Figure 4C). Additionally, the stimulatory effects of escitalopram on phosphorylation of both Ser9 on GSK-3β and Ser473 on Akt were significantly blocked by WAY-100635 (Figure 4D).
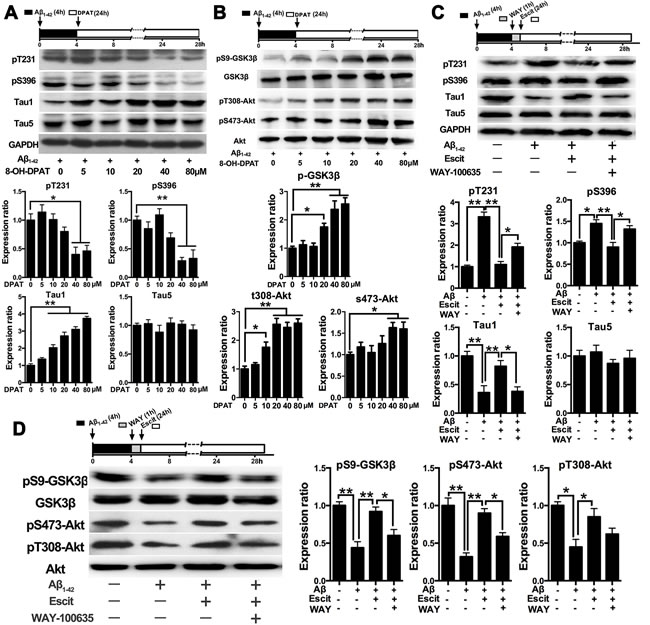
Figure 4: Effects of escitalopram on the PI3K/Akt/GSK-3β signaling pathway depends on 5-HT1AR. A. and B. Immunoblots of tau phosphorylated at pT231, pS396, Tau1 and Tau5 (A) and pS9-GSK-3β, total GSK-3β, pT308-Akt, pS473-Akt, total Akt (B) in the cultured hippocampal neurons. Cells were treated with Aβ1-42 (2 μM) for 4 h, and then incubated with 8-OH-DPAT at the concentrations indicated for 24 h in fresh medium. The respective total protein or GAPDH was used for normalization. Data were represented as means ± SEM (n = 3; *p < 0.05, **p < 0.01). C. and D. Immunoblots of tau phosphorylated at pT231, pS396, Tau1 and Tau5 (C) and pS9-GSK-3β, total GSK-3β, pT308-Akt, pS473-Akt, total Akt (D) in the cultured hippocampal neurons. Cells were treated with Aβ1-42 (2 μM) for 4 h, and then incubated with escitalopram (80 μM) for 24 h in fresh medium with or without the pretreatment with WAY-100635 (10 μM, 1 h). The respective total protein or GAPDH was used for normalization. Data were expressed as means ± SEM (n = 3; *p < 0.05, **p < 0.01). E. Immunoblots of pS9-GSK-3β, total GSK-3β, pT308-Akt, pS473-Akt, total Akt in the cultured hippocampal neurons. Cells were treated with Aβ1-42 (2 μM) for 4 h, and then incubated with escitalopram (80 μM) alone or escitalopram combined with 8-OH-DPAT (80 μM) for 24 h in fresh medium with or without the pretreatment with WAY-100635 (10 μM, 1 h). The respective total protein or GAPDH was used for normalization. Data were expressed as means ± SEM (n = 3; *p < 0.05, **p < 0.01).DPAT, 8-OH-DPAT; Escit, escitalopram; WAY, WAY-100635.
Escitalopram improves Aβ1-42 induced impairment of dendritic outgrowth
A dendritic outgrowth assay was performed to investigate whether escitalopram can regulate dendrite and spine morphology in hippocampal neurons. As shown in Figure 5A and 5B, escitalopram alone had no significant effect on dendritic outgrowth and spine density in hippocampal neurons under control conditions. Aβ1-42 treatment decreased the dendrite density and the total length of primary dendrites, while escitalopram treatment up-regulated the dendrite density and the total length of primary dendrites, neither Aβ1-42 nor escitalopram had an effect on the number of primary dendrites. Statistical analyses showed that Aβ1-42 treatment significantly decreased the density of spines, while escitalopram treatment reversed this significantly (Figure 5C). Western blotting results showed that Aβ1-42 treatment decreased levels of synaptophysin and PSD95, and again escitalopram treatment significantly reversed these effects (Figure 5D).
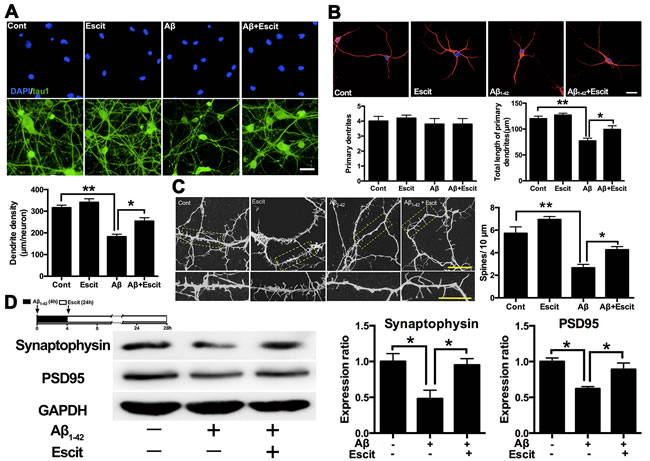
Figure 5: Escitalopram improves Aβ1-42 induced impairment of dendritic outgrowth. A. Dendrite density of the neuron was detected by immunofluorescence assay. Tau 1 was labeled as green and DAPI was blue. Data were represented as means ± SEM (n = 3; **p < 0.01). Scale bar, 10 μm. B. Number of primary dendrites and total length of primary dendrites were examined by immunofluorescence assay. Tau 5 was labeled as red and DAPI was blue. Data were represented as means ± SEM (n = 3; *p < 0.05, **p < 0.01). Scale bar, 5 μm. C. Spine density was detected by immunofluorescence assay labeled with Tau 5. Data were represented as means ± SEM (n = 3; *p < 0.05, **p < 0.01). Scale bar, 5 μm. D. Immunoblots of synaptophysin and PSD95 in the cultured hippocampal neurons. GAPDH was used for normalization. Data were represented as means ± SEM (n = 3; *p < 0.05). Escit, escitalopram.
Discussion
The present study revealed that escitalopram could protect cultured hippocampal neurons against Aβ1-42-induced tau hyperphosphorylation through the PI3K/Akt/GSK-3β pathway, with the involvement of 5-HT1AR. Furthermore, escitalopram may have a potent effect on neurite outgrowth of hippocampal neurons exposed to Aβ1-42.
SSRIs are widely used in the treatment of depression. Interestingly, recent studies have found that SSRIs reduce the risk of AD in depressed individuals [24] and have a positive role in hindering the progression of AD and improving patients’ daily performance [8, 9, 25]. In preclinical studies, a favorable cognitive-improving effect of SSRIs has been proved [10, 11, 26]. Citalopram, paroxetine and fluoxetine have been found to modulate the processing of amyloid precursor protein in vitro [27] and to lower Aβ and plaque densities in vivo [13, 26, 28]. Furthermore, it was reported that paroxetine reduced tau immunoreactive hippocampus CA1 neurons in 3xTg AD mice [26]. Our previous study has also revealed that escitalopram ameliorated forskolin-induced tau hyperphosphorylation in HEK293/tau441 cells [14]. Here we found that both escitalopram and fluoxetine attenuated Aβ1-42-induced tau hyperphosphorylation in primary hippocampal neurons, which further demonstrated that SSRIs could lessen tau pathology.
The mechanism by which SSRIs inhibit tau hyperphosphorylation is unknown. Akt/GSK-3β is the most implicated signaling pathway in regulating tau phosphorylation [29]. It was demonstrated that stimulation of GSK-3β both in vitro and in vivo induces tau hyperphosphorylation with impairments of the cognitive functions, whereas inhibition of GSK-3β improves tau pathologies and memory deficit [30]. In addition, several recent publications have implicated the Akt/GSK-3β pathway as the mechanism of action of some SSRIs antidepressants. For example, the activation effects of escitalopram, paroxetine, sertraline and fluoxetine on Akt have been previously reported in hippocampal neuron cultures, neural stem cells, and rat brain [31, 32]; the inhibition effects of fluoxetine on GSK-3β was also reported in mice brain and cultured neural precursor cells [16, 33]. Our present study showed that Akt was activated and GSK-3β was inhibited following escitalopram administration, while pharmacological inhibition of PI3K abolished the effect of escitaloram on tau phosphorylation, in agreement with previous results and suggesting that the neuroprotective effect of escitalopram on Aβ1-42-induced tau hyperphosphorylation is directly related to the activation of PI3K/Akt/GSK-3β signaling pathway.
The underlying mechanism of GSK-3β inhibition induced by escitalopram is also unknown. The primary action of SSRIs is based on the inhibition of serotonin reuptake to elevate synaptic 5-HT concentrations, thereby activating postsynaptic 5-HT receptors and triggering downstream intracellular signaling cascades. Among different serotonin receptors, the 5-HT1AR has been most implicated in mood and cognition. The density of 5-HT1AR is diminished in the brain in AD patients prior to the appearance of clinical symptoms [5]. Activation of the 5-HT1AR is a critical component in the action mechanism of SSRIs [34]. Increasing evidence indicates that the PI3K/Akt /GSK-3β pathway can be regulated by 5-HT1AR. Selective agonists for 5-HT1AR stimulated an activation of Akt [35] and an inhibition of GSK-3β [18] in vitro and in vivo. Furthermore, a follow-up study revealed that the regulation of PI3K/Akt/GSK-3β by 5-HT1AR and fluoxetine is an important signaling mechanism for serotonin-regulated behaviors [21]. Therefore, we speculate that 5-HT1AR may play an important role in escitalopram-induced activation of the Akt/GSK-3β pathway. In the present study, we found that the 5-HT1AR agonist 8-OH-DPAT activated the Akt/GSK-3β pathway, besides, the up-regulation of GSK-3β and Akt phosphorylation induced by escitalopram was blocked by the 5-HT1AR antagonist WAY-100635, providing powerful evidence to support our speculation.
Several previous studies have clearly pointed out the neuroprotective and neurotrophic potential of 5-HT1AR agonists [36, 37], however, few researches have investigated whether 5-HT1AR is directly relate to tau phosphorylation. In the present study, we found that the 5-HT1AR agonist 8-OH-DPAT significantly decreased Aβ1-42-induced tau hyperphosphorylation, while the 5-HT1AR antagonist WAY-100635 reversed the attenuation effects of escitalopram on tau hyperphosphorylation. Here we provided new evidence that a 5-HT1AR agonist decreased tau hyperphosphorylation to further support the potential of 5-HT1A receptor agonists as neuroprotectants. More importantly, these findings firmly demonstrated that 5-HT1AR is a key molecule involved in the attenuation of tau hyperphosphorylation by escitalopram.
Previous research has indicated that abnormal hyperphosphorylation of tau protein contributes to disturbance of neural plasticity in AD [38-40]. Therefore, we subsequently investigated whether escitalopram enhanced dendritic outgrowth in hippocampal neurons. We observed that escitalopram significantly enhanced dendritic outgrowth and increased dendritic spines in hippocampal neuron cultures exposed to Aβ1-42. Our previous study has reported that escitalopram rescued synaptic deficits in depressive-like rats [41]. Moreover, several studies have revealed that the disturbance of synaptic plasticity recovered when tau hyperphosphorylation was reversed pharmacologically or with genetic technology [39, 40], consistent with our results. However, whether the above-mentioned PI3K/Akt/GSK-3β pathway is also related to the enhancement of synaptic plasticity induced by escitalopram has yet to be determined. The PI3K/Akt/GSK-3β pathway is involved in long-term potentiation [42], neurite outgrowth [43], axonal outgrowth and dendritic plasticity [44] in vitro and in vivo. Moreover, a PI3K inhibitor significantly reduced the promoting effects of antidepressant drugs on dendritic outgrowth in hippocampal neurons [31]. Thus, we presume that the PI3K/Akt/GSK-3β pathway is also related to the improvement of dendritic outgrowth induced by escitalopram. However, further work is needed to fully define the mechanisms.
We acknowledge that the doses of each SSRI administered in our experiment were higher than those normally found in brain tissue, however, such high doses are routinely used in other in vitro studies [31, 45]. Moreover, we cannot draw conclusions about in vivo effects of escitalopram on tau hyperphosphorylation from our in vitro data; further studies are also needed to determine if the findings generalize to all SSRIs or even other antidepressant drugs.
In conclusion, we demonstrated that escitalopram attenuates tau hyperphosphorylation via the PI3K/Akt/GSK-3β signaling pathway that links 5-HT1AR activation. Our findings shed new light on the neuroprotective effect of escitalopram involved in tau hyperphosphorylation and support a role for 5-HT1AR mediated Akt/GSK-3β pathway in tau phosphorylation. Finally, these may provide theoretical evidence supporting the potential of escitalopram in the treatment of tau hyperphosphorylation associated disease, including AD.
Materials and methods
Drugs and reagents
Aβ protein fragment 1-42 (Aβ1-42), Fluoxetine, 8-OH-DPAT, WAY-100635, LY294002 were purchased from Sigma-Aldrich (MO, USA). Escitalopram was kindly provided by H. Lundbeck A/S. Copenhagen-Valby, Denmark. R-citalopram was purchased from Santa Cruz Biotechnology (CA, USA). Escitalopram, fluoxetine, R-citalopram, 8-OH-DPAT, WAY-100635 and LY294002 were dissolved in DMSO, then were diluted using cell culture medium without bovine serum with the final concentration of DMSO less than 0.05%.
Aβ1-42 preparation
Aβ1-42 was dissolved in DMSO, and incubated for 24 h at 37 °C to allow for fibril formation [22]. In order to examine the extent and type of Aβ1-42 fibrils formed, the Aβ1-42 preparations (20 μg) were separated by electrophoresis on a 16.5% tris-tricine gel, and then the gel was visualized by coomassie brilliant blue R-250 staining (Beyotime, Haimen, China) or silver staining using a Fast Silver Stain Kit (Beyotime).
Primary hippocampal neuron cultures
Primary cultures of hippocampal neurons were prepared from fetal brains (embryonic day 18; E18) obtained from female Sprague-Dawley rats (Experimental Animal Center of Southeast University). All studies involving animals were conducted in accordance with the National Institutes of Health guide for the care and use of Laboratory animals. Animal procedures undertaken were approved by Jiangsu Animal Care and Use Committee and every effort was made to minimize animal suffering. Briefly, the brains were exposed, and then the hippocampal tissues were dissociated in HBSS (Invitrogen, NY, USA) containing 0.125% trypsin solution (Gibco, NY, USA) for 15 min at 37°C. Subsequently, the digestion was terminated with DMEM (Gibco) containing 10% fetal bovine serum (Gibco). Finally, the dispersed tissues were centrifuged at 2000 rpm for 5 min and were resuspended in Neurobasal medium (Invitrogen) containing 2% B27 supplement (Gibco), 0.5mM L-glutamine (Gibco), 20 IU/ml penicillin and 20 IU/ml streptomycin. For the Western blotting procedure, neurons were plated onto six-well plates coated with poly-D-lysine (100μg/ml; Sigma-Aldrich) at a density of 2×106 per well. For the immunofluorescence staining procedure, neurons were plated in cover slips at a density of 2×104 cells/cm2. Cell cultures were kept in a humidified incubator containing 95% air and 5% CO2 at 37 °C. The culture medium was replaced with fresh Neurobasal/ B27 medium every 2-3 days. The purity of the neurons used in experiments was about 95%. The cultures were maintained for 14 days before being harvested for further analysis.
Cell viability assay
Cell viability was determined using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. After treatment, primary hippocampal neurons were treated with 0.5 mg/ml MTT for 4 h at 37 °C. The formazan crystals were dissolved in 100μl of DMSO and the absorbance was measured at 570 nm in a microplate reader (Multiskan GO, Thermo Scientific, NY, USA). Cell survival rates were expressed as percentages of the control group.
Western blotting
Primary hippocampal neuron cultures were collected, washed twice with ice-cold phosphate-buffered saline (PBS) and solubilized in ice-cold lysis buffer (Beyotime) containing protease inhibitor (Roche, Laval, Quebec, Canada). The cell lysates were centrifuged at 12000g for 15 min at 4 °C. The BCA kit (Pierce, Thermo Scientific, NY, USA) was used to detect the protein concentration. The samples containing equivalent amounts of protein (20 μg) were separated by SDS-PAGE and transferred to PVDF membranes (Merck Millipore, Darmstadt, Germany). The blots were blocked by 5% nonfat milk for 1h at room temperature, and then the membranes were incubated with the following primary antibodies diluted in blocking solution at 4 °C overnight: mouse monoclonal Tau5 (1:5000; BioSource, NY, USA), mouse monoclonal Tau1 (1:5000; Merck Millipore),rabbit polyclonal anti-pTau (Thr231) (1:2000; Invitrogen), rabbit polyclonal anti-pTau (Ser396) (1:2000; Invitrogen), mouse monoclonal anti-GSK-3β (1:1000; Cell Signaling, MA, USA), rabbit monoclonal anti-pGSK-3β (Ser9) (1:1000; Cell Signaling), rabbit monoclonal anti-Akt (1:1000; Cell Signaling), rabbit monoclonal anti-pAkt (Ser473) (1:1000; Cell Signaling), rabbit polyclonal anti-pAkt (Thr308) (1:500; Bioworld, MN, USA), rabbit polyclonal anti-pPP2Ac (Tyr307) (1:500; Santa Cruz), rabbit monoclonal anti-PSD95 (1:2000; Abcam, MA, USA) and rabbit monoclonal anti-synaptophysin (1:1000; Merck Millipore). Internal control was performed using GAPDH antibody (1:5000; Sigma-Aldrich). After washing with TBST buffer for three times, the membranes were incubated for 1h with horseradish peroxidase-conjugated secondary antibody (goat anti- rabbit IgG, goat anti-mouse IgG) (1:5000; Invitrogen). The membranes were then processed with ECL Western blotting reagents (Pierce), and then were detected using Image Quant LAS 4000 mini system (GE Healthcare, Japan). The sum optical density was quantitatively analyzed by Quantity One software (Bio-Rad, Richmond, CA, USA).
Immunofluorescence staining
Cells were washed three times in PBS and fixed with 4% paraformaldehyde at room temperature for 20 min. Then, cells were treated with 0.3% Triton X-100 for 5 min on ice. After washing, cells were blocked with 5% BSA for 30 min at room temperature and then incubated with rabbit polyclonal anti-pTau (Thr231) (1:400), rabbit polyclonal anti-pTau (Ser396) (1:400), mouse monoclonal Tau1 antibody (1:500) or mouse monoclonal Tau5 antibody (1:500) at 4 °C overnight. Cells were washed three times in PBST and incubated with Alexa Fluor 488 or Alexa Fluor 594 goat anti-rabbit, goat anti-mouse secondary antibody (1:2000; Invitrogen) for 1h. Finally, the cells were rinsed with PBST, stained with DAPI (Beyotime) and observed under Olympus FV 1000 Viewer (Olympus, Tokyo, Japan). For the morphological analysis of dendrites/spines, five fields were randomly selected from each sample and three independent experiments for each sample were performed. The images were captured by a person blind to their identities and were analyzed using Image J software 1.48 (NIH, Bethesda, USA).
Statistical analysis
Data were presented as mean ± standard error of the mean (SEM). One-way analysis of variance (ANOVA) followed by Tukey post hoc test were used to compare the differences between means in more than two groups by GraphPad Prism 6.01. A probability value of P < 0.05 was considered to be statistically significant.
Acknowledgments
We are grateful to H. Lundbeck A/S. (Copenhagen-Valby, Denmark) for their kindly providing of escitalopram oxalate.
Grant Support
This work was supported by grants from the National Natural Science Foundation of China (91232707 Qing-guo Ren, 81420108012 Zhi-jun Zhang, 81501173 Xiao-li Li), the National Major Science and Technology Program of China (No. 2012ZX09506-001-009 Zhi-Jun Zhang), the Key Program for Clinical Medicine and Science and Technology, Jiangsu Provincial Clinical Medical Research Center (BL2013025), National High-tech R.D Program (863 Program) (No.2015AA020508), National Basic Research Program of China (2013CB835103) and Strategic Priority Research Program of Chinese Academy of Science (XDB02020002).
Conflicts of Interest
The authors have no conflicts of interest to disclose.
References
1. Arriagada PV, Growdon JH, Hedley-Whyte ET and Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992; 42:631-639.
2. Esmaeli-Azad B, McCarty JH and Feinstein SC. Sense and antisense transfection analysis of tau function: tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J Cell Sci. 1994; 107 :869-879.
3. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005; 309:476-481.
4. Zhang Z, Song M, Liu X, Kang SS, Kwon IS, Duong DM, Seyfried NT, Hu WT, Liu Z, Wang JZ, Cheng L, Sun YE, Yu SP, Levey AI and Ye K. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med. 2014; 20:1254-1262.
5. Kepe V, Barrio JR, Huang SC, Ercoli L, Siddarth P, Shoghi-Jadid K, Cole GM, Satyamurthy N, Cummings JL, Small GW and Phelps ME. Serotonin 1A receptors in the living brain of Alzheimer’s disease patients. Proc Natl Acad Sci U S A. 2006; 103:702-707.
6. Palmer AM, Francis PT, Benton JS, Sims NR, Mann DM, Neary D, Snowden JS and Bowen DM. Presynaptic serotonergic dysfunction in patients with Alzheimer’s disease. J Neurochem. 1987; 48:8-15.
7. Mowla A, Mosavinasab M, Haghshenas H and Borhani Haghighi A. Does serotonin augmentation have any effect on cognition and activities of daily living in Alzheimer’s dementia? A double-blind, placebo-controlled clinical trial. J Clin Psychopharmacol. 2007; 27:484-487.
8. Pollock BG, Mulsant BH, Rosen J, Sweet RA, Mazumdar S, Bharucha A, Marin R, Jacob NJ, Huber KA, Kastango KB and Chew ML. Comparison of citalopram, perphenazine, and placebo for the acute treatment of psychosis and behavioral disturbances in hospitalized, demented patients. Am J Psychiatry. 2002; 159:460-465.
9. Porsteinsson AP, Drye LT, Pollock BG, Devanand DP, Frangakis C, Ismail Z, Marano C, Meinert CL, Mintzer JE, Munro CA, Pelton G, Rabins PV, Rosenberg PB, Schneider LS, Shade DM, Weintraub D, et al. Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized clinical trial. JAMA. 2014; 311:682-691.
10. Egashira N, Matsumoto Y, Mishima K, Iwasaki K, Fujioka M, Matsushita M, Shoyama Y, Nishimura R and Fujiwara M. Low dose citalopram reverses memory impairment and electroconvulsive shock-induced immobilization. Pharmacol Biochem Behav. 2006; 83:161-167.
11. Lyons L, ElBeltagy M, Bennett G and Wigmore P. Fluoxetine counteracts the cognitive and cellular effects of 5-fluorouracil in the rat hippocampus by a mechanism of prevention rather than recovery. PLoS One. 2012; 7:e30010.
12. Aboukhatwa M, Dosanjh L and Luo Y. Antidepressants are a rational complementary therapy for the treatment of Alzheimer’s disease. Mol Neurodegener. 2010; 5:10.
13. Sheline YI, West T, Yarasheski K, Swarm R, Jasielec MS, Fisher JR, Ficker WD, Yan P, Xiong C, Frederiksen C, Grzelak MV, Chott R, Bateman RJ, Morris JC, Mintun MA, Lee JM, et al. An antidepressant decreases CSF Abeta production in healthy individuals and in transgenic AD mice. Sci Transl Med. 2014; 6:236re234.
14. Ren QG, Wang YJ, Gong WG, Zhou QD, Xu L and Zhang ZJ. Escitalopram Ameliorates Forskolin-Induced Tau Hyperphosphorylation in HEK239/tau441 Cells. J Mol Neurosci. 2015; 56:500-508.
15. Pei JJ, Khatoon S, An WL, Nordlinder M, Tanaka T, Braak H, Tsujio I, Takeda M, Alafuzoff I, Winblad B, Cowburn RF, Grundke-Iqbal I and Iqbal K. Role of protein kinase B in Alzheimer’s neurofibrillary pathology. Acta Neuropathol. 2003; 105:381-392.
16. Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Cools MJ, Wetsel WC, Gainetdinov RR and Caron MG. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A. 2008; 105:1333-1338.
17. Sachs BD, Rodriguiz RM, Siesser WB, Kenan A, Royer EL, Jacobsen JP, Wetsel WC and Caron MG. The effects of brain serotonin deficiency on behavioural disinhibition and anxiety-like behaviour following mild early life stress. Int J Neuropsychopharmacol. 2013; 16:2081-2094.
18. Li XH, Zhu W, Roh MS, Friedman AB, Rosborough K and Jope RS. In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacol. 2004; 29:1426-1431.
19. Barreto RA, Walker FR, Dunkley PR, Day TA and Smith DW. Fluoxetine prevents development of an early stress-related molecular signature in the rat infralimbic medial prefrontal cortex. Implications for depression? BMC Neurosci. 2012; 13:125.
20. Tsai SJ, Liou YJ, Hong CJ, Yu YW and Chen TJ. Glycogen synthase kinase-3beta gene is associated with antidepressant treatment response in Chinese major depressive disorder. Pharmacogenomics J. 2008; 8:384-390.
21. Polter AM, Yang S, Jope RS and Li X. Functional significance of glycogen synthase kinase-3 regulation by serotonin. Cell Signal. 2012; 24:265-271.
22. Shi C, Wu F, Yew DT, Xu J and Zhu Y. Bilobalide prevents apoptosis through activation of the PI3K/Akt pathway in SH-SY5Y cells. Apoptosis. 2010; 15:715-727.
23. Sanchez C and Kreilgaard M. R-citalopram inhibits functional and 5-HTP-evoked behavioural responses to the SSRI, escitalopram. Pharmacol Biochem Behav. 2004; 77:391-398.
24. Kessing LV, Sondergard L, Forman JL and Andersen PK. Antidepressants and dementia. J Affect Disord. 2009; 117:24-29.
25. Rozzini L, Chilovi BV, Conti M, Bertoletti E, Zanetti M, Trabucchi M and Padovani A. Efficacy of SSRIs on cognition of Alzheimer’s disease patients treated with cholinesterase inhibitors. Int Psychogeriatr. 2010; 22:114-119.
26. Nelson RL, Guo Z, Halagappa VM, Pearson M, Gray AJ, Matsuoka Y, Brown M, Martin B, Iyun T, Maudsley S, Clark RF and Mattson MP. Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp Neurol. 2007; 205:166-176.
27. Pakaski M, Bjelik A, Hugyecz M, Kasa P, Janka Z and Kalman J. Imipramine and citalopram facilitate amyloid precursor protein secretion in vitro. Neurochem Int. 2005; 47:190-195.
28. Cirrito JR, Disabato BM, Restivo JL, Verges DK, Goebel WD, Sathyan A, Hayreh D, D’Angelo G, Benzinger T, Yoon H, Kim J, Morris JC, Mintun MA and Sheline YI. Serotonin signaling is associated with lower amyloid-beta levels and plaques in transgenic mice and humans. Proc Natl Acad Sci U S A. 2011; 108:14968-14973.
29. Rickle A, Bogdanovic N, Volkman I, Winblad B, Ravid R and Cowburn RF. Akt activity in Alzheimer’s disease and other neurodegenerative disorders. Neuroreport. 2004; 15:955-959.
30. Llorens-Martin M, Jurado J, Hernandez F and Avila J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014; 7:46.
31. Park SW, Lee JG, Seo MK, Lee CH, Cho HY, Lee BJ, Seol W and Kim YH. Differential effects of antidepressant drugs on mTOR signalling in rat hippocampal neurons. Int J Neuropsychopharmacol. 2014; 17:1831-1846.
32. Sutton LP and Rushlow WJ. The effects of neuropsychiatric drugs on glycogen synthase kinase-3 signaling. Neuroscience. 2011; 199:116-124.
33. Hui J, Zhang J, Kim H, Tong C, Ying Q, Li Z, Mao X, Shi G, Yan J, Zhang Z and Xi G. Fluoxetine regulates neurogenesis in vitro through modulation of GSK-3beta/beta-catenin signaling. Int J Neuropsychopharmacol. 2015; 18(5).
34. Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C and Hen R. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003; 301:805-809.
35. Cowen DS, Johnson-Farley NN and Travkina T. 5-HT receptors couple to activation of Akt, but not extracellular-regulated kinase (ERK), in cultured hippocampal neurons. J Neurochem. 2005; 93:910-917.
36. Banasr M, Hery M, Printemps R and Daszuta A. Serotonin-induced increases in adult cell proliferation and neurogenesis are mediated through different and common 5-HT receptor subtypes in the dentate gyrus and the subventricular zone. Neuropsychopharmacol. 2004; 29:450-460.
37. Mogha A, Guariglia SR, Debata PR, Wen GY and Banerjee P. Serotonin 1A receptor-mediated signaling through ERK and PKCalpha is essential for normal synaptogenesis in neonatal mouse hippocampus. Transl Psychiatry. 2012; 2:e66.
38. Scheff SW, Price DA and Sparks DL. Quantitative assessment of possible age-related change in synaptic numbers in the human frontal cortex. Neurobiol Aging. 2001; 22:355-365.
39. Ahmed T, Blum D, Burnouf S, Demeyer D, Buee-Scherrer V, D’Hooge R, Buee L and Balschun D. Rescue of impaired late-phase long-term depression in a tau transgenic mouse model. Neurobiol Aging. 2015; 36:730-739.
40. Van der Jeugd A, Hochgrafe K, Ahmed T, Decker JM, Sydow A, Hofmann A, Wu D, Messing L, Balschun D, D’Hooge R and Mandelkow EM. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012; 123:787-805.
41. Li XL, Yuan YG, Xu H, Wu D, Gong WG, Geng LY, Wu FF, Tang H, Xu L and Zhang ZJ. Changed Synaptic Plasticity in Neural Circuits of Depressive-Like and Escitalopram-Treated Rats. Int J Neuropsychopharmacol. 2015; 18:pyv046.
42. Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G and Cho K. Abeta(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3beta. Nat Neurosci. 2011; 14:545-547.
43. Rodgers EE and Theibert AB. Functions of PI 3-kinase in development of the nervous system. Int J Dev Neurosci. 2002; 20:187-197.
44. Ueno Y, Chopp M, Zhang L, Buller B, Liu Z, Lehman NL, Liu XS, Zhang Y, Roberts C and Zhang ZG. Axonal outgrowth and dendritic plasticity in the cortical peri-infarct area after experimental stroke. Stroke. 2012; 43:2221-2228.
45. Huang W, Zhao Y, Zhu X, Cai Z, Wang S, Yao S, Qi Z and Xie P. Fluoxetine upregulates phosphorylated-AKT and phosphorylated-ERK1/2 proteins in neural stem cells: evidence for a crosstalk between AKT and ERK1/2 pathways. J Mol Neurosci. 2013; 49:244-249.