
INTRODUCTION
Monocytoid-lineage leukaemias encompass the monocytoid subtypes of acute myeloid leukaemia (AML) (acute myelomonocytic leukaemia [previously FAB type M4] and acute monoblastic/monocytic leukaemia [M5]), as well as chronic myelomonocytic leukaemia (CMML) and juvenile myelomonocytic leukaemia (JMML) [1, 2]. Although the overall prognosis of AML has improved gradually over the last 40 years, the majority of patients continue to succumb to the disease with clinical prospects remaining particularly bleak for older patients; there has been little change to standard chemotherapeutic treatment approaches over this time [3]. CMML is a neoplasm that is classified within the WHO sub-categorisation of ‘myelodysplastic / myeloproliferative neoplasms’ [2] that has a high median age of presentation (70–75 years), a median survival of only 11–17 months and currently very limited treatment options [4, 5]. There is a pressing need to exploit advances made in the understanding of the pathogenetic mechanisms that underpin monocytoid malignancies by developing novel therapeutic agents, applicable ideally to the treatment of patients of all ages, that are able to effectively deliver targeted effects to malignant cell populations while avoiding significant systemic toxicity.
Histone acetylation is an important molecular modification used to regulate gene transcription that affects many cellular processes including cell proliferation, differentiation, DNA repair, cell survival and angiogenesis [6–8]. Histone deacetylases (HDACs) are a family of epigenetic modifiers that alter chromatin structure by removing lysine acetylation from histones, resulting in a transcriptionally-closed state and subsequent gene silencing or loss of expression. Alterations in epigenetic programming have been commonly reported in the initiation, progression and maintenance of cancer; and aberrant localization of HDACs and resultant promoter silencing has been implicated in several malignancies [7], [9] including AML and the myelodysplastic syndromes (MDS) [10–12] and has been reported in connection with common oncogenic fusion proteins such as AML1ETO (t8:21), and Inv16 (core binding protein) abnormalities [13]. Myelomonocytoid and monocytoid / monoblastic AML (FAB M4 and M5) accounts for approximately 25% of total AML cases [44]; these cases have a distinct clinical profile with frequent extramedullary manifestations and leucocytosis along with emerging associations with abnormalities in epigenetic regulation in the form of DNMT3A mutations [45, 46].
Given the reversible nature of acetylation modifications, therapeutic targeting of HDACs has been an active area of drug development with the promise of correcting the effects of aberrant gene expression [14]. HDAC inhibitors may exert their activity by multiple mechanisms of action including: cell differentiation, DNA repair inhibition [15], induction of reactive oxygen species [16], and replication stalling [17]. Clinical trials of several HDAC inhibitors including valproic acid, vorinostat, romidepsin, belinostat and panabinostat have been conducted in both solid tumours and haematological malignancies including AML, MDS and CMML patients [18–22]. In general, reported clinical responses to single-agent HDAC inhibitory therapy have been modest with dose escalation of HDAC inhibitors being limited by a relatively restricted therapeutic window. Off-target effects of HDAC inhibition have been associated with significant systemic toxicities including gastrointestinal disturbances, thrombocytopenia, fatigue and insomnia which have limited the wider clinical uptake of these agents. It is highly desirable to develop mechanisms through which HDAC inhibitory activity can be more-selectively concentrated within tumour cells while sparing non-disease cell populations.
Tefinostat (CHR-2845) is a novel pan HDAC inhibitor which is cleaved to an active acid, CHR-2847, by the intracellular esterase human carboxylesterase-1 (hCE-1), the expression of which is limited to cells of monocytoid lineage and some hepatocytes, allowing selective accumulation of active drug within monocytoid cells. [23]. A phase I dose escalation study of tefinostat in patients with relapsed/refractory haematological malignancies demonstrated early signs of clinical efficacy without any dose limiting toxicity. [23].
We examined the pre-clinical activity of tefinostat in a large cohort of primary AML and CMML patient samples in order to assess lineage specific activity, potential therapeutic window and combination studies with Cytarabine to build a rationale for future therapeutic evaluation in monocytoid leukaemias.
RESULTS
Monocytoid leukaemias show selective high sensitivity to tefinostat
The in vitro efficacy of tefinostat was first assessed by MTS cell viability assay in AML cell lines HL60 (M2 FAB type), MV411 (M4, FLT3-ITD), OCIAML3 (M4 NPM1mut) and THP1 (M5) (EC50 = 2300 nM +/−226 vs. 57 nM +/−6.2 vs. 110 nM +/− vs. 560 nM +/−17.12 respectively, Figure 1A). Annexin V/PI incorporation showed strong apoptotic induction in myelo-monocytic cell lines THP1, MV411 (FLT3-ITD) and OCIAML3 within 24 hours of tefinostat treatment that was only reached in non-monocytic HL60 cells at much higher drug concentrations (Figure 1B–1C). Dose response to tefinostat was assessed in a cohort of 66 primary AML and 7 primary CMML samples (Ave EC50 2.7 μM +/− 3.1). Significant growth inhibitory effects were seen in M4 (myelomonocytic)/M5 (monocytic / monoblastic) AMLs and CMML samples with lower EC50s in comparison to non-M4/M5 AML FAB types (mean EC50 M4/M5 = 1.1 μM +/−1.8, CMML = 1.9 +/−1.6 vs. M0/M1 = 5.1 μM +/−4.7 respectively, *p = 0.009 spearman’s correlation, Figure 1D). This selectivity between M0/M1 and M4/M5 FAB groups was abolished when the t-butyl tefinostat analogue CHR-8185 (which is not cleaved by hCE-1) was substituted as an alternative HDACi, further supporting the monocytoid selectivity of tefinostat. M2 FAB type AMLs displayed a wide range of sensitivity of response to tefinostat; overall responses of M2 samples were not significantly different from the M4/M5 sub-groups. Importantly, there was no differential response between tefinostat and CHR8185 in the M2 subgroup, suggesting responses to be non hCE-1 mediated in this group (Figure 1D).
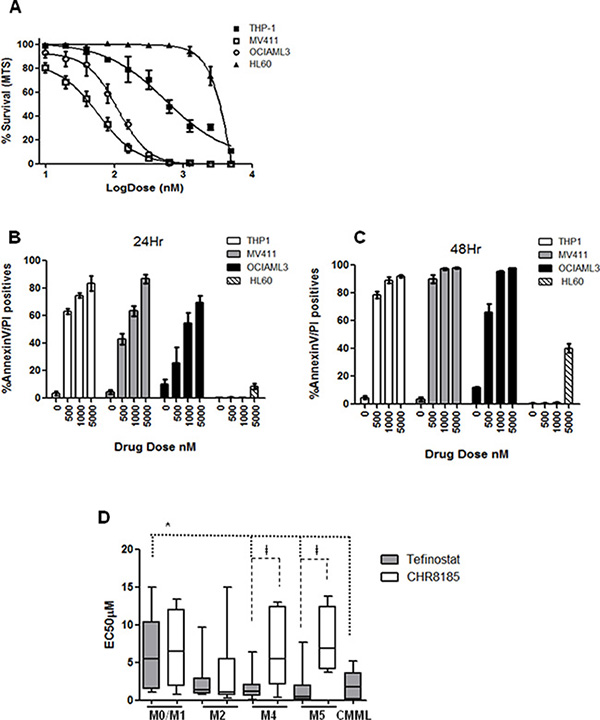
Figure 1: Monocytoid leukaemias show selective high sensitivity to Tefinostat. (A) Dose response plot for AML cell lines HL60 (M2), MV411 (M4), OCIAML3 (M4) and THP-1 (M5) treated with serial dilutions of Tefinostat. Tefinostat-induced Annexin V/PI incorporation measured by flow cytometry in THP1, MV411, OCIAML3 and HL60 at (B) 24 Hrs and (C) 48 Hrs post treatment. (D) Box and whiskers plots showing range of FAB type-specific MTS-derived EC50s in response to Tefinostat and the comparative non hCE-1-dependent t-butyl analogue CHR8185 in a cohort of 66 primary AML and 7 primary CMML samples. (Tefinostat EC50 M4/M5/CMML vs. M0/M1 *p = 0.009 spearman’s correlation. Tefinostat vs. t-butyl analogue CHR8185, only significant in M4/M5 groups ǂp < 0.007).
Further analysis of the relationship between in vitro tefinostat sensitivity (log10(EC50) and patient characteristics revealed no significant differences in drug efficacy according to other disease parameters including clinical outcome, presenting cytogenetics and FLT3/NPM1 mutational status.(Supplementary Table S1).
Monocytic targeting of HDACi therapy spares normal bone marrow progenitor cells
Analysis of selective response to tefinostat in sub-populations of primary cells was carried out using flow cytometric 7AAD exclusion assays. CD14+ AML blasts were highly sensitive to tefinostat treatment in contrast to CD34+/CD14− AML blasts and CD45high lymphocyte populations (Figure 2A). This sensitivity was significantly reduced in when equivalent concentrations of the non-hCE-1-dependent analogue CHR8185 were used, demonstrating the myelo-monocytic specificity of Tefinostat (Supplementary Figure S1). Importantly, no growth inhibitory effects were seen in normal bone marrow (NBM) CD34+ cells exposed to AML-toxic doses of tefinostat while, in comparison, equivalent concentrations of CHR-8185 caused considerable cytotoxicity (Figure 2B). Exposure of NBM progenitor cells to increasing concentrations of tefinostat in longer-term colony assays failed to reduce colony forming units in comparison to vehicle-treated controls and AML treated samples (pre-selected for high CD14+ percentage), which showed significant reduction in colony formation (Figure 2C, p < 0.02).
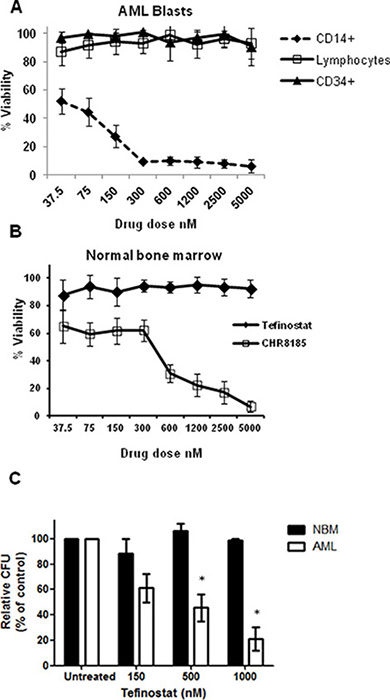
Figure 2: Monocytic targeting of HDACi therapy spares normal bone marrow progenitor cells. (A) Flow cytometric analysis of viability of cell subpopulations in primary AML samples in response to Tefinostat dosing measured by 7AAD exclusion (n = 8). (B) Dose response effects of Tefinostat and CHR8185 in NBM cells (n = 4) measured by 7AAD exclusion. (C) Tefinostat effects on Colony forming units (CFUs) in NBM CD34+ cells (no significant difference, n = 4) and primary AML blasts (*p < 0.02 MWU, n = 6) following 14 days culture on methocult.
hCE-1 expression levels dictate efficacy of tefinostat in monocytic leukaemias
hCE-1 expression in primary samples was quantified by intracellular flow cytometry in combination with cell surface markers CD14, CD64 and CD45 to allow identification of hCE-1 levels in different cellular sub-populations (Supplementary Figure S2). Significant correlation was seen between levels of the mature monocytic marker CD14+ and hCE-1 expression in AML samples (Figure 3A). Further analysis of a larger cohort of 40 primary AMLs and 7 CMMLs by western blot confirmed highest hCE-1 levels in myelomonocytic and monocytic FAB types, with M5 AMLs displaying significant overexpression in comparison to NBM levels (Figure 3B–3C, p = 0.01). This observation was validated by microarray analysis of hCE-1 mRNA in a further 130 AML samples, with M4/M5 AMLs showing significant overexpression compared to NBM CD34+ cells (Supplementary Figure S3). High hCE-1 levels was associated with low EC50 values to tefinostat across the cohort as measured by western blotting (Figure 3D p < 0.001). To supplement western blot expression levels, intracellular flow analysis of hCE-1 in a smaller cohort of M2, M4 and M5 monocytoid AML samples was divided into tefinostat-sensitive primary AMLs (EC50 < 1 μM Ave) and tefinostat-resistant samples (EC50 > 2.5 μM). Significantly higher intracellular flow levels of hCE-1 were observed in tefinostat-sensitive AMLs compared to resistant samples (Figure 3E p < 0.001).
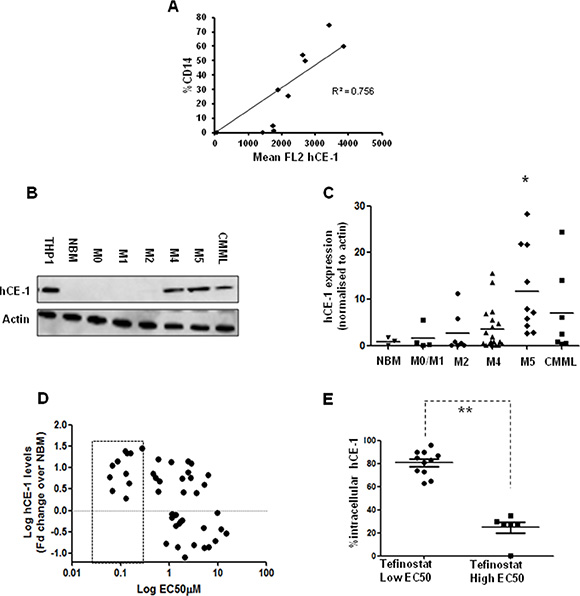
Figure 3: hCE-1 expression levels dictate efficacy of Tefinostat in monocytic leukaemias. (A) Correlation plot of CD14+ expression vs. hCE-1 mean fluorescence measured by intracellular flow in primary AML samples (n = 10) (B) Representative western blot of hCE-1 expression in NBM, AML and CMML patient samples. (C) Comparison of hCE-1 protein levels across FAB types (NBM vs. M5 *p = 0.01 Wilcoxon rank sum). (D) Correlation plot of hCE-1 protein levels measured by western blot (fold change relative to NBM levels, n = 3) compared to tefinostat sensitivity (EC50) in a cohort of 40 primary AML and 7 CMML samples, p = 0.001 Spearman’s correlation per 10 fold increase in EC50). Dotted box represents low EC50 samples. (E) Monocytoid intracellular hCE-1 levels in tefinostat-sensitive and tefinostat-resistant primary samples gated by CD14+/CD64+ (EC50 high (n = 6) and low patients (n = 11), **p < 0.001 MWU).
Increases in intracellular acetylation and DNA damage induction are biomarkers of tefinostat efficacy
In order to identify biomarkers of tefinostat sensitivity in AML blasts we undertook sub-population analysis by flow cytometry to look for differential effects on intracellular acetylation. CD14-expressing cells showed a maximum induction of intracellular protein acetylation at nanomolar tefinostat concentrations after 6 hours of drug exposure (Figure 4A). This analysis was extended to compare tefinostat sensitive (n = 8, EC50 < 1 μM) and resistant AML samples (n = 5, EC50 > 2.5 μM); significant acetylation was induced at low nanomolar doses in low EC50 samples, but was absent in those patients with resistance to tefinostat (Figure 4B). Strong acetylation induction was also observed in several CMML patient samples (Figure 4C), although this appeared to be less variable with Tefinostat sensitivity. Tefinostat-sensitive samples also showed strong phosphorylation induction of the cell cycle arrest and DNA double strand break damage sensor protein γ-H2A.X, which is associated with DNA cleavage during apoptosis. Induction was seen within 24 hours of drug treatment (Figure 4D–4E); suggesting γ-H2A.X may be a potential future biomarker of patient responsiveness to this drug.
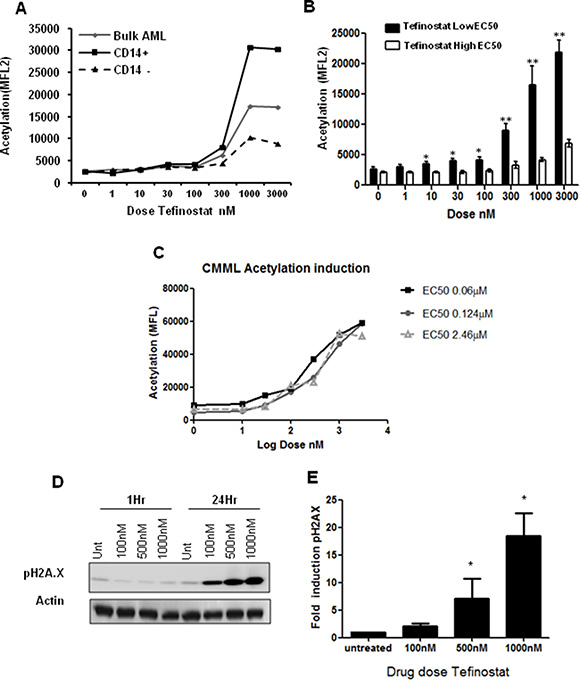
Figure 4: Increases in intracellular acetylation and DNA damage induction are biomarkers of Tefinostat efficacy. (A) Tefinostat dose-dependent intracellular acetylation staining in a representative primary AML sample using acetylated lysine monoclonal antibody and sub-population analysis by flow cytometry. (B) Acetylation induction in Tefinostat sensitive (low EC50/CD14+ black bars, n = 8) compared to insensitive (high EC50, white bars, n = 5) primary AML samples, *p < 0.05, **p < 0.005. (C) Tefinostat dose-dependent intracellular acetylation induction in primary CMML samples (n = 3). All samples exhibit > 80% CD14+ and > 70% hCE-1 expression. (D) Representative western blot of phospho-H2A.X induction by tefinostat at 1 and 24 hours post-treatment. (E) Western blot quantification of dose-dependent tefinostat-induced γ-H2A.X induction at 24 hrs compared to vehicle treated control (n = 9 AML samples, *p < 0.01 Kruskal Wallis).
Tefinostat is synergistic with cytarabine
To further inform clinical development of tefinostat we investigated the synergistic potential between tefinostat and the conventional cytotoxic agent cytosine arabinoside (AraC) at a fixed ratio of 1:10 (Tefinostat:AraC). In vitro synergy was demonstrated in combination experiments with clinically-relevant concentrations of tefinostat and cytarabine (AraC) firstly in MV411 cells (Figure 5A) and then in primary AML blasts (Figure 5B, Mean AML combination index (CI) = 0.51, n = 31 patient samples, Mean CMML CI = 0.53, n = 6 samples, CI at 50% fraction affected; values < 0.9 = synergistic, 0.9–1.2 additive, > 1.2antagonistic)). This synergism was observed across a range of drug effects (Figure 5C and Supplementary Table 3). Sequential administration of AraC followed by tefinostat did not increase synergistic effects beyond those observed with simultaneous application of the two agents (Figure 5D–5E); pre-treatment with tefinostat, however, was found to antagonise the combination response. Interestingly, tefinostat-responsive AMLs frequently stimulated NFκB p65 levels, commonly associated with drug resistance mechanisms, although these samples showed good synergy in combination with AraC (Supplementary Figure S4). Clonogenic assessment of single and combination pulse-treated AML blasts at an optimum ratio of 1:10 (Tefinostat: AraC) revealed a significant reduction in colonies in combination assays compared to with each agent used alone (p < 0.04).
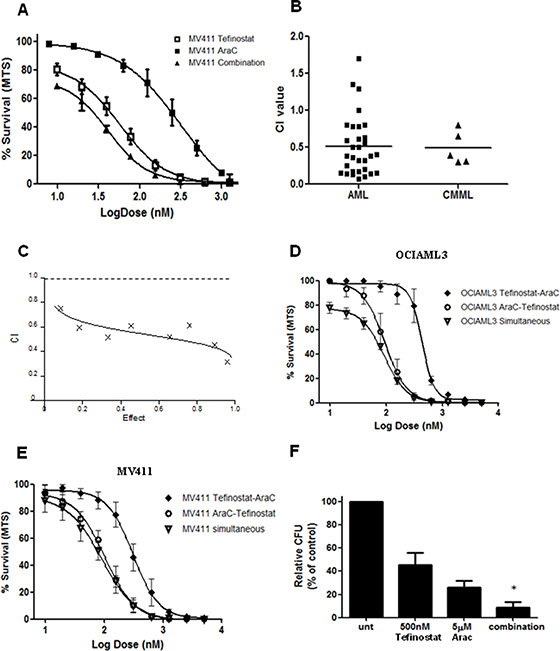
Figure 5: Tefinostat is synergistic with Cytarabine. (A) Dose response curves for MV411 cells treated both singly and in combination for Tefinostat and AraC (dose ratio 1:10, Tefinostat:AraC). (B) Synergy analysis of tefinostat and AraC displayed as combination index (CI) values at EC50 dose affected at an optimal ratio of 1:10 (Tefinostat:AraC) in primary patient samples set up in triplicate (AML Mean combination index (CI) = 0.51, n = 31, CMML mean CI = 0.49, n = 5). (C) Representative CI response plot from primary AML showing CI values across a range of dose effects (fraction affected). Dose response curves for simultaneous and sequential dosing of drugs in (D) OCIAML3 and (E) MV411 cell lines. (F) Clonogenic assessment of synergy interaction in primary AML blasts (n = 4) following single agent and 1:10 ratio combination pulse treatment (*p < 0.04 MWU, between both single agents and combination colony growth).
DISCUSSION
Although overall survival rates in AML have improved gradually over the last four decades this has been achieved largely through incremental improvements in supportive care and stem cell transplantation strategies with very little change in standard chemotherapy strategies over this time; minimal impact has been seen in clinical outcomes for older patients who represent the majority of newly-diagnosed cases. Similarly in CMML, a disease with an even higher median age of presentation, there is a considerable unmet need for non-toxic therapeutic strategies that are capable of altering the natural history of the disease. There is a strong pre-clinical rationale for HDAC inhibition in haematological malignancies, but systemic toxicities including gastrointestinal disturbances, myelosuppression, fatigue and insomnia have so far hindered the clinical update of HDAC inhibitors.
Here we report a series of in vitro experiments in which we confirm the monocytoid-lineage specificity of tefinostat in leukaemic cells obtained from patients with AML and CMML. We observed a dose-dependent induction of apoptotic response in myelo-monocytic cell types and a significantly greater drug sensitivity at sub-micromolar concentrations in this group compared to that seen in undifferentiated or non-monocytic patient samples. Esterase Sensitive Motif-mediated targeting of tefinostat and the resulting CHR-2847 accumulation in these cell types resulted in a 3.5–6.5 fold increase in anti-proliferative potency compared to the that seen using the untargeted t-butyl tefinostat analogue CHR-8185, in line with the previously-reported improved efficacy of tefinostat in comparison with other ‘non-targeted’ HDAC inhibitors such as vorinostat [23]. Lesser responses to tefinostat were seen in undifferentiated AML patient subgroups M0/M1; although sensitivity was observed in occasional undifferentiated samples with inherent HDAC inhibitor sensitivity this manifested as equal sensitivity to tefinostat and CHR-8185. Similarly, responses in M2 subtypes were variable and this correlated with the diversity of cellular sub-populations and CD14+ expression seen within this group.
Although mechanisms of action of HDAC inhibitors remain to be fully characterised, sensitivity to these agents has been linked to the apoptotic capability of each cell type. Differing combinations of apoptotic mediators, cell cycle abnormalities and differentiation blocks between patient samples make it difficult to predict cellular outcome to HDAC inhibitor treatment, although previous studies have shown that monocytic AML cell types such as the M5 cell line THP1 have greater apoptotic sensitivity to the HDAC inhibitor vorinostat than other non-monocytic AML cell lines [24]; this has been potentially linked to activity of Bcl-2 family members [25]. Previous studies of the HDAC inhibitor panobinostat have shown induction of growth arrest and increase in myeloid transcription factor expression associated with differentiation in AML cells [26]. We found, however, that tefinostat induces apoptosis at sub-EC50 level doses, indicating a different mechanism of action to that of panobinostat. Other potential mechanisms of action include acetylation and degradation of key chaperone molecules such as HSP90 [27] which supports a multitude of leukaemic oncoproteins. We find total HSP90 protein levels to be unaffected by tefinostat exposure in primary AML cells (data not shown), although this does not exclude the possibility of a functional change in HSP90 activity.
High levels of hCE-1 expression were found to drive a significant increase in tefinostat efficacy as measured by growth inhibition assays (p = 0.001), and also strongly correlated with expression of the mature monocytoid marker CD14+. CD14+ is widely used to define a mature monocytic cell type; we and others find, however, that CD14 may also be low or absent from leukaemic monocytes as part of the heterogeneity associated with this disease [28]. We observed that whilst, in the majority of patients, CD14 levels correlated with increased hCE-1 expression, this was not exclusive, particularly in M2 AMLs where, as previously reported, high hCE-1 levels were occasionally observed in the absence of significant CD14 expression [29].
Induction of intracellular H3/H4 histone acetylation was observed in all tefinostat-responsive AML and CMML patient samples in contrast to previous studies where other agents such as entinostat, vorinostat and panabinostat induced only modest levels of H3/H4 acetylation and monocytic populations were found to be resistant to HDAC inhibitory treatment [23], [30], [31]. hCE-linked targeting of tefinostat appears to circumvent this issue, with CD14+ monocytic AML sub-populations and CMML patient samples, as expected, showing the highest levels of drug response. Additionally, we observed preservation of normal bone marrow CD34+ cells and clonogenic function at AML-toxic doses of tefinostat in contrast to other HDAC inhibitors used in similar studies where significant increases in acetylation were observed in normal CD34+ cells [32, 33]. This finding emphasises the potential for limiting off-target systemic toxicities by harnessing hCE-1-dependent HDAC inhibitory activity in malignancies involving cells of monocytoid lineage (AML-M4, AML-M5 and CMML).
As yet a strong correlation between hyperacetylation and clinical response to HDAC inhibitors remains to be fully established. HDAC inhibitory efficacy is also reported to be dependent on accumulation of DNA damage [15], replication stalling [17], and apoptotic induction [34]. AML cases with leukaemia-associated fusion proteins are DNA damage repair deficient and therefore may be more resistant to HDAC inhibition [35]. We found no association, however, between tefinostat sensitivity and any patient characteristics such as cytogenetic group. We demonstrated that tefinostat induces the DNA damage marker γ-H2A.X in the majority of responsive patient samples similarly to that reported for other HDAC inhibitors such as vorinostat [36], suggesting that this may be a potential biomarker of patient response. Our data suggest that tefinostat induces both apoptosis and DNA damage accumulation within 24 hours of treatment in a majority of samples which may predict greater clinical response to this HDAC inhibitor. HDAC inhibition has also previously been demonstrated to signal pro-survival pathway feedback in the form of hyperacetylation of p65 NFκB signalling [37]; dual inhibition of HDAC and NFκB may potentiate efficacy in AML [38]. We observed a transient increase in NFκB p65 induction following tefinostat treatment, although this did not appear to impair γ-H2A.X or apoptotic induction in our sample cohort.
Another putative mechanism of action of HDAC inhibitors in single and combination studies is that of oxidative injury [16, 37, 39], This process is driven through NOX-mediated ROS induction which has been shown by our group to be constitutively active in myelomonocytic AML subtypes [40], and which may, in part, contribute to the greater efficacy of tefinostat in these patients. Although HDACis have been previously reported to selectively target FLT3 ITD-mutant proteins for degradation in AML [41] and to enhance FLT3 inhibition in cell lines [42, 43], we found no association between in vitro sensitivity to tefinostat and FLT3-ITD mutation status in our primary sample cohort (Supplementary Table S1).
As HDAC inhibition may result in a reduction of cells in S-phase, concerns have arisen that combination with traditional chemotherapeutic agents such as AraC may be antagonistic and that sequential administration may be required to optimise response [30, 37]. Our combination studies suggested that simultaneous dosing with tefinostat and AraC may be the most effective therapeutic regime and clonogenic synergy assays suggest this combination is also capable of targeting proliferative subsets within monocytoid AMLs. Pre-dosing with AraC did not significantly improve efficacy of the combination studies and pre-treatment with tefinostat produced an antagonistic response. Primary AML blasts demonstrated consistently high degrees of synergy in all primary samples tested (CI = 0.5, n = 36). This synergistic interaction with AraC (a principal component of AML cytotoxic regimens for many years) warrants further larger scale evaluation as a prelude to formal clinical evaluation.
The effective in vitro targeting of HDAC inhibition in monocytoid AML and CMML described above, combined with encouraging early signs of clinical activity (coupled to selective HDAC inhibition) in the absence of tefinostat-related toxicity in the Ossenkoppele study, provides a compelling case for further clinical evaluation of tefinostat in larger studies in monocytoid-lineage leukaemias. Rapid identification of suitable ‘monocytoid’ patients at diagnosis will present some challenges for trial design and will place an emphasis on close interactions between laboratory and clinic. Of the two CMML patients treated in the phase I study, one achieved a bone marrow complete response (CR) at relatively small doses and moving forward MONOCLE, a single-arm phase 2 study of tefinostat monotherapy in CMML (EudraCT 2015-002281-23) is shortly to commence recruitment at sites within the UK.
METHODS
Cell culture reagents and patient samples
AML samples were collected with informed consent from newly-diagnosed patients entering UK NCRI AML15, 16 and 17 studies (Ethics Reference Numbers 2005-001149/40, 2005-002847/14, 2007-003798/16) (Supplementary Tables S1 and S2) in accordance with the Declaration of Helsinki. CMML samples were obtained with informed consent under the MDSBio Sample Collection Study (Ethics Reference Number 06/Q1606/110). Mononuclear cells were purified by Ficoll density gradient followed by CD45 staining (AML samples with < 70% blast content following purification were excluded from further analysis). AML/CMML cells were cultured in IMDM (Sigma) with 10% FCS (Biosera). Normal bone marrow MNCs were purchased from Lonza and CD34+ cells were isolated using MiniMACS CD34 columns (Miltenyi Biotec) according to the manufacturer’s instructions. HL60, MV411, OCIAML3 and THP-1 cell lines were cultured as recommended by ATCCLGC (http://www.lgcstandardsatcc.org). All cultures were incubated at 37°C, 5% CO2. Tefinostat (CHR-2845) and its N-butyl analogue control CHR8185 were supplied by Chroma Therapeutics (Abingdon, UK). Cytarabine was obtained from Sigma.
Western blot analysis
Sample preparation of lysates and western blotting was performed as previously described [47]. Blots were probed with: γ-H2A.X Ser139 (Cell Signalling), hCE-1 (Lifespan biosciences). Equal loading was confirmed by Actin (Abcam). Quantification of proteins was carried out using AIDA image analyser v4.22 (Raytest, Straubenhardt, Germany).
Drug response and cell viability assays
In vitro cell survival/drug response assays were set up with serial dilution of drugs in 96 well plates with 8 × 104 cells per well. Cell viability was assessed by Cell Titre 96 Aqueous One MTS reagent (Promega) at 48 Hrs according to the manufacturer’s instructions. Samples with less than 70% viability at 48 hours in untreated samples were excluded from analysis. For cytosine arabinoside (AraC) synergy experiments, dose response assays were set up in triplicate for single and fixed ratio combination treated AML cell lines and primary samples using clinically-relevant AraC concentrations. For sequential drug dosing cells were pre-treated for 24 Hrs with either Tefinostat or AraC prior to addition of the second drug. For sub-population analysis of drug response assays, cells were harvested after 48 h, surface stained with anti-CD45, CD14, CD64 and resuspended in 1 μg/ml 7aminoactinomycin D (7AAD) to determine viable cells remaining by flow cytometry using a FACSCalibur® cytometer (BD Biosciences, Oxford, UK). Triplicates were averaged and dose response curves inputted to Calcusyn to generate EC50 and combination index values (Calcusyn v2.0 software,Biosoft, Cambridge, UK). Annexin V positivity was measured using the Annexin V Apoptosis Detection Kit (eBioscience, Hatford, UK) according to the manufacturer’s instructions.
Intracellular flow cytometry
Cells were prestained for 20 min with surface markers CD14, CD64, CD45 (BD Biosciences), then fixed in phosflow lyse buffer (BD biosciences) for 10 mins at 37°C before washing (PBS/0.5%BSA/0.1 NaAzide) and permeabilising in phosflow permeabilisation buffer II (BD biosciences) for 30 mins on ice. Primary antibodies were added for 45 mins at room temperature (RT) anti-hCE-1 (Lifespan biosciences LS-105283) or IgG control (Novous biological NB810-56910). For assessment of modulation of intracellular acetylation, cells were pre-incubated for 6 hours with Tefinostat prior to surface staining and fixation. Primary antibody Ac-k-103 (Cell signaling #9681) and IgG control (Mouse IgG2a ×0943 DAKO) were used at 200 ng/ml. Finally, cells were washed twice and incubated for 45 mins at RT with PE conjugated secondary antibodies goat-anti-rabbit or goat-anti-mouse (hCE-1 and Ak-k-103 respectively) prior to FACS analysis.
Colony assays
CD34+ NBM cells and AML samples with > 70% CD14+ were plated in triplicate at 1 × 103 (NBM) and 5 × 104 (AML) cells per dish in semi-solid medium (Methocult Optimum H4034, Stem Cell Technologies, Location) in the presence of increasing doses of Tefinostat or vehicle control and cultured for 14 days at 37°C, 5% CO2 prior to colony counting. For synergistic assays, AML blasts were pulse treated with single agent (1 μM tefinostat, 10 μM AraC) or a 1:10 ratio (Tefinostat:AraC) prior to plating out in methocult.
Statistical analysis
Multivariable analyses were performed using either logistic or Cox’s proportional hazard regression methods adjusted for age, white blood cell count, cytogenetic group, performance status, de novo/secondary disease and sex. For all other results, differences between mean values were compared by Minitab v13 (Minitab Inc. PA, USA) using Mann WhitneyU or paired t-test.
ACKNOWLEDGMENTS
This study was carried out with funding from Chroma Therapeutics and Cancer Research UK.
Authors’ contributions
SK was the principal investigator; JZ, MG designed and performed the laboratory work for this study; RKH participated in the statistical analysis; JZ, SK co-ordinated the research; JZ and SK wrote the paper.
CONFLICTS OF INTEREST
The authors declare that there are no competing financial interests in relation to the work described.
REFERENCES
1. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976; 33:451–8.
2. Swerdlow SH, Campo E, Lee Harris N. WHO classification of tumours of hematopoietic and lymphoid tissues. 2015.
3. DiNardo CD, Cortes JE. New treatment for acute myelogenous leukemia. Expert Opin Pharmacother. 2015; 16:95–106.
4. Bacher U, Haferlach T, Schnittger S, Kreipe H, Kroger N. Recent advances in diagnosis, molecular pathology and therapy of chronic myelomonocytic leukaemia. Br J Haematol. 2011; 153:149–67.
5. Parikh SA, Tefferi A. Chronic myelomonocytic leukemia: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013; 88:967–74.
6. Shi B, Xu W. The development and potential clinical utility of biomarkers for HDAC inhibitors. Drug Discov Ther. 2013; 7:129–36.
7. Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009; 280:168–76.
8. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006; 5:769–84.
9. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014; 13:673–91.
10. Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, Craddock C, Turner BM. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005; 19:1751–9.
11. Bruserud O, Stapnes C, Tronstad KJ, Ryningen A, Anensen N, Gjertsen BT. Protein lysine acetylation in normal and leukaemic haematopoiesis: HDACs as possible therapeutic targets in adult AML. Expert Opin Ther Targets. 2006; 10:51–68.
12. Hackanson B, Rimmele L, Benkisser M, Abdelkarim M, Fliegauf M, Jung M, Lubbert M. HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leuk Res. 2012; 36:1055–62.
13. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011; 11:726–34.
14. Bacher U, Kern W, Schnittger S, Hiddemann W, Schoch C, Haferlach T. Further correlations of morphology according to FAB and WHO classification to cytogenetics in de novo acute myeloid leukemia: a study on 2,235 patients. Ann Hematol. 2005; 84:785–91.
15. Shivarov V, Gueorguieva R, Stoimenov A, Tiu R. DNMT3A mutation is a poor prognosis biomarker in AML: results of a meta-analysis of 4500 AML patients. Leuk Res. 2013; 37:1445–50.
16. Yang L, Liu Y, Zhu L, Xiao M. DNMT3A R882 mutation is associated with elevated expression of MAFB and M4/M5 immunophenotype of acute myeloid leukemia blasts. Leuk Lymphoma. 2015; 1–9.
17. Bojang P, Jr., Ramos KS. The promise and failures of epigenetic therapies for cancer treatment. Cancer Treat Rev. 2014; 40:153–69.
18. Shubassi G, Robert T, Vanoli F, Minucci S, Foiani M. Acetylation: a novel link between double-strand break repair and autophagy. Cancer Res. 2012; 72:1332–5.
19. Petruccelli LA, Dupere-Richer D, Pettersson F, Retrouvey H, Skoulikas S, Miller WH, Jr. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One. 2011; 6:e20987.
20. Conti C, Leo E, Eichler GS, Sordet O, Martin MM, Fan A, Aladjem MI, Pommier Y. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010; 70:4470–80.
21. Cashen A, Juckett M, Jumonville A, Litzow M, Flynn PJ, Eckardt J, LaPlant B, Laumann K, Erlichman C, DiPersio J. Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS). Ann Hematol. 2012; 91:33–8.
22. Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, Faderl S, Koller C, Morris G, Rosner G, Loboda A, Fantin VR, Randolph SS, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008; 111:1060–6.
23. Klimek VM, Fircanis S, Maslak P, Guernah I, Baum M, Wu N, Panageas K, Wright JJ, Pandolfi PP, Nimer SD. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008; 14:826–32.
24. Kuendgen A, Knipp S, Fox F, Strupp C, Hildebrandt B, Steidl C, Germing U, Haas R, Gattermann N. Results of a phase 2 study of valproic acid alone or in combination with all-trans retinoic acid in 75 patients with myelodysplastic syndrome and relapsed or refractory acute myeloid leukemia. Ann Hematol. 2005; 84:61–6.
25. Platzbecker U, Al-Ali HK, Gattermann N, Haase D, Janzen V, Krauter J, Gotze K, Schlenk R, Nolte F, Letsch A, Ottmann OG, Kundgen A, Lubbert M, et al. Phase 2 study of oral panobinostat (LBH589) with or without erythropoietin in heavily transfusion-dependent IPSS low or int-1 MDS patients. Leukemia. 2014; 28:696–8.
26. Ossenkoppele GJ, Lowenberg B, Zachee P, Vey N, Breems D, Van de Loosdrecht AA, Davidson AH, Wells G, Needham L, Bawden L, Toal M, Hooftman L, Debnam PM. A phase I first-in-human study with tefinostat - a monocyte/macrophage targeted histone deacetylase inhibitor - in patients with advanced haematological malignancies. Br J Haematol. 2013; 162:191–201.
27. Silva G, Cardoso BA, Belo H, Almeida AM. Vorinostat induces apoptosis and differentiation in myeloid malignancies: genetic and molecular mechanisms. PLoS One. 2013; 8:e53766.
28. Pierceall WE, Lena RJ, Medeiros BC, Blake N, Doykan C, Elashoff M, Cardone MH, Walter RB. Mcl-1 dependence predicts response to vorinostat and gemtuzumab ozogamicin in acute myeloid leukemia. Leuk Res. 2014; 38:564–8.
29. Bots M, Verbrugge I, Martin BP, Salmon JM, Ghisi M, Baker A, Stanley K, Shortt J, Ossenkoppele GJ, Zuber J, Rappaport AR, Atadja P, Lowe SW, et al. Differentiation therapy for the treatment of t(8; 21) acute myeloid leukemia using histone deacetylase inhibitors. Blood. 2014; 123:1341–52.
30. Bansal H, Bansal S, Rao M, Foley KP, Sang J, Proia DA, Blackman RK, Ying W, Barsoum J, Baer MR, Kelly K, Swords R, Tomlinson GE, et al. Heat shock protein 90 regulates the expression of Wilms tumor 1 protein in myeloid leukemias. Blood. 2010; 116:4591–9.
31. Krasinskas AM, Wasik MA, Kamoun M, Schretzenmair R, Moore J, Salhany KE. The usefulness of CD64, other monocyte-associated antigens, and CD45 gating in the subclassification of acute myeloid leukemias with monocytic differentiation. Am J Clin Pathol. 1998; 110:797–805.
32. Klobusicka M, Kusenda J, Babusikova O. Myeloid enzymes profile related to the immunophenotypic characteristics of blast cells from patients with acute myeloid leukemia (AML) at diagnosis. Neoplasma. 2005; 52:211–8.
33. Gojo I, Tan M, Fang HB, Sadowska M, Lapidus R, Baer MR, Carrier F, Beumer JH, Anyang BN, Srivastava RK, Espinoza-Delgado I, Ross DD. Translational phase I trial of vorinostat (suberoylanilide hydroxamic acid) combined with cytarabine and etoposide in patients with relapsed, refractory, or high-risk acute myeloid leukemia. Clin Cancer Res. 2013; 19:1838–51.
34. Kummar S, Gutierrez M, Gardner ER, Donovan E, Hwang K, Chung EJ, Lee MJ, Maynard K, Kalnitskiy M, Chen A, Melillo G, Ryan QC, Conley B, et al. Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clin Cancer Res. 2007; 13:5411–7.
35. Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, Tidwell ML, Greer J, Chung EJ, Lee MJ, Gore SD, Sausville EA, Zwiebel J, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007; 109:2781–90.
36. Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G, Sharma S, Kantarjian H, Dugan M, Albitar MA, et al. phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res. 2006; 12:4628–35.
37. Lee CK, Wang S, Huang X, Ryder J, Liu B. HDAC inhibition synergistically enhances alkylator-induced DNA damage responses and apoptosis in multiple myeloma cells. Cancer Lett. 2010; 296:233–40.
38. Petruccelli LA, Pettersson F, Del Rincon SV, Guilbert C, Licht JD, Miller WH, Jr. Expression of leukemia-associated fusion proteins increases sensitivity to histone deacetylase inhibitor-induced DNA damage and apoptosis. Mol Cancer Ther. 2013; 12:1591–604.
39. Dai Y, Chen S, Kmieciak M, Zhou L, Lin H, Pei XY, Grant S. The novel Chk1 inhibitor MK-8776 sensitizes human leukemia cells to HDAC inhibitors by targeting the intra-S checkpoint and DNA replication and repair. Mol Cancer Ther. 2013; 12:878–89.
40. Yaseen A, Chen S, Hock S, Rosato R, Dent P, Dai Y, Grant S. Resveratrol sensitizes acute myelogenous leukemia cells to histone deacetylase inhibitors through reactive oxygen species-mediated activation of the extrinsic apoptotic pathway. Mol Pharmacol. 2012; 82:1030–41.
41. Dai YS, Xu J, Molkentin JD. The DnaJ-related factor Mrj interacts with nuclear factor of activated T cells c3 and mediates transcriptional repression through class II histone deacetylase recruitment. Mol Cell Biol. 2005; 25:9936–48.
42. Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R, Smyth MJ, Johnstone RW. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A. 2001; 98:10833–8.
43. Hole PS, Zabkiewicz J, Munje C, Newton Z, Pearn L, White P, Marquez N, Hills RK, Burnett AK, Tonks A, Darley RL. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood. 2013; 122:3322–30.
44. Buchwald M, Pietschmann K, Muller JP, Bohmer FD, Heinzel T, Kramer OH. Ubiquitin conjugase UBCH8 targets active FMS-like tyrosine kinase 3 for proteasomal degradation. Leukemia. 2010; 24:1412–21.
45. Pietschmann K, Bolck HA, Buchwald M, Spielberg S, Polzer H, Spiekermann K, Bug G, Heinzel T, Bohmer FD, Kramer OH. Breakdown of the FLT3-ITD/STAT5 axis and synergistic apoptosis induction by the histone deacetylase inhibitor panobinostat and FLT3-specific inhibitors. Mol Cancer Ther. 2012; 11:2373–83.
46. Lin WH, Yeh TK, Jiaang WT, et al. Evaluation of the antitumor effects of BPR1J-340, a potent and selective FLT3 inhibitor, alone or in combination with an HDAC inhibitor, vorinostat, in AML cancer. PLoS One. 2014; 9:e83160.
47. Zabkiewicz J, Pearn L, Hills RK, Morgan RG, Tonks A, Burnett AK, Darley RL. The PDK1 master kinase is over-expressed in acute myeloid leukemia and promotes PKC-mediated survival of leukemic blasts. Haematologica. 2014; 99:858–64.