
INTRODUCTION
The p53 transcription factor is involved in the regulation of cell proliferation and apoptosis, DNA repair, angiogenesis, and innate immunity [1]. Wild-type p53 (wt p53) functions as a tumor suppressor gene and promotes cell cycle arrest or apoptosis in cancer cells. However, inactivation of p53 by mutation or other mechanisms is a frequent event in tumorigenesis [2].
One of the major endogenous negative regulators of p53 in humans is the human double minute 2 protein (hDM2), which is a ubiquitin E3 ligase that binds to p53 [3]. p53 and hDM2 interact with each other to create an autoregulatory feedback loop that regulates both the activity of p53 and the gene expression of hDM2 [4]. In this feedback loop, p53 activation increases the gene expression level of hDM2. In turn, hDM2 binds to and conceals the N-terminal transactivation domain of p53, and facilitates the nuclear export of p53, leading to p53 degradation by the ubiquitin proteasome pathway. Due to the role of hDM2 in negatively regulating the function of the p53 tumor suppressor protein, the overexpression of hDM2 has been detected in many types of cancer [5]. Many peptide-based or small-molecule disrupters of the p53/hDM2 interaction have been developed through inhibiting a well-defined hydrophobic surface pocket in hDM2 and three key hydrophobic residues of p53 projected from the same face of an α-helix [6]. To date, at least three small-molecule p53/hDM2 disrupters (RO5045337, RO5503781 and MI-888) have been advanced into clinical trials [7–9], however, none has yet been approved for clinical use.
The exploration of organometallic compounds containing transition metals as probes for chemical biology or as lead scaffolds for the design of potent inhibitors of pharmacologically-important molecular targets has received increasing attention in recent years [10–23]. Of particular interest are organometallic iridium compounds due to their tunable chemical and biological reactivities, as well as the distinctive kinetic inertness of the iridium(III) metal center. Another feature of organometallic iridium(III) compounds is that they are always six-coordinate with octahedral geometry [24–28]. Meggers and co-workers have pioneered the development of kinetically-inert organometallic iridium(III) compounds as potent and specific inhibitors of enzyme activity [29]. Moreover, Sadler and co-workers have conducted extensive investigations into organometallic iridium “half-sandwich” compounds as potent anticancer agents [30]. Sheldrick and co-workers have also studied a wide range of iridium(III) polypyridyl compounds that show promising selectivity for cancer cells over normal cells [31]. Meanwhile, our group has previously reported a cyclometallated iridium(III) compound that inhibited the trimerization of tumor necrosis factor alpha (TNF-α) [32], which represented the first example of an organometallic octahedral compound that inhibits PPIs. These studies highlight the increasing interest and importance given to the development of organometallic iridium(III) compounds as potential medicines. However, no metal-based inhibitor of the p53/hDM2 interaction has yet been reported in the literature. In this work, we set out to identify novel p53/hDM2 PPI disrupters via the screening of an in-house collection of kinetically-inert iridium(III) compounds. These efforts culminated in the identification of a novel cyclometallated iridium(III) compound 1 as the first metal-based disrupter of the p53/hDM2 PPI.
RESULTS
Synthesis and characterization of organometallic iridium(III) compounds
A library of structurally diverse, kinetically-inert organometallic iridium(III) compounds 1–6 (Figure 1) with a general structure [Ir(C^N)2(N^N)]+ were designed and synthesized. Among the structural motifs present in compounds 1–6 are aromatic and alkyl motifs, which could potentially form hydrophobic interactions with specific residues of the protein-protein interface, and halogen atoms (chlorine and fluorine), which are key groups in many approved drugs. Compounds 1, 2 and 6 bear 2-phenylpyridine (ppy) C^N ligands substituted with ethyl or fluoro groups, while compound 3 bears 1-phenyl-1H-pyrazole C^N ligands. Compound 5 possesses 2-phenylquinoline C^N ligands, while compound 4 bears 2-phenylbenzothiazole ligands. With regards to the N^N ligand, compound 3 carries the 2, 2′-bipyridine (bpy) moiety, while compound 2 possesses a bpy ligand substituted with tert-butyl groups at the 4 and 4′ positions. Compounds 1 and 4 bear 1, 10-phenanthroline (phen) N^N ligands substituted with one or two chlorine atoms at different positions. Finally, compounds 5 and 6 carry larger N^N ligands with additional fused aromatic rings.
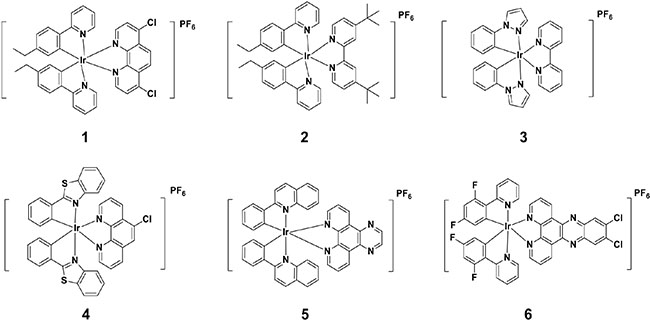
Figure 1: Chemical structures of kinetically-inert organometallic iridium(III) compounds 1–6.
The cyclometallated iridium(III) compound 1 [Ir(eppy)2(dcphen)](PF6) (where eppy = 2-(4-ethylphenyl)pyridine and dcphen = 4, 7-dichloro-1, 10-phenanthroline) was prepared as shown in Scheme 1. 1-Bromo-4-ethylbenzene 7 was converted to aryl boronate 8, which was subsequently coupled to 2-bromopyridine via the Suzuki-Miyaura reaction to give C^N ligand eppy 9. The reaction of 9 with IrCl3·3H2O gave the diiridium compound [(C^N)2Ir(μ-Cl)]2 10. Finally, dimer 10 was reacted with dcphen to generate 1, which was isolated as the hexafluorophosphate salt by the addition of NH4PF6. The crude product was purified by column chromatography on silica to give 1 as a stable orange solid. The stability of 1 was investigated by UV-Vis and 1H NMR experiments (Supplementary Figures S1 and S2), which showed that 1 was stable in 90% [d6] DMSO/10% D2O or 80% acetonitrile/20% Tris-HCl buffer for at least 7 days. The synthesis of the other iridium(III) compounds and ligands are described in the Electronic Supplementary Information. All of the compounds in this work were characterized by 1H-NMR, 13C-NMR, high resolution mass spectrometry (HRMS) and elemental analysis.
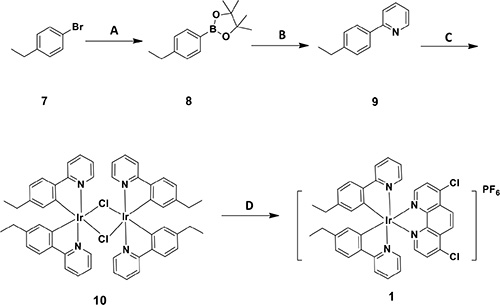
Scheme 1: Synthetic pathway of cyclometallated iridium(III) compound 1. Reagents and conditions: (A) Bis(pinacolato)diboron, Pd(dppf)Cl2, AcONa, toluene, 100°C, N2; (B) 2-bromopyridine, Pd(PPh3)4, K2CO3, EtOH, reflux, N2; (C) IrCl3·3H2O, methoxyethanol/H2O = 3:1, 150°C; (D) 4, 7-dichloro-1, 10-phenanthroline, DCM/MeOH = 1:1, reflux, NH4PF6, H2O, Et2O.
Compound 1 disrupts the p53/hDM2 interaction
To experimentally test the library of the organo-metallic iridium(III) compounds as p53/hDM2 PPI inhibitors, a fluorescence anisotropy (FA) assay was performed as previously described [33]. hDM217–126 (recombinant fragment) was incubated with fluorescein-labelled p53 peptide (p5315–31Flu) and serial dilutions of the organometallic iridium(III) compounds. The results revealed that the fluorescence anisotropy of the system was decreased significantly upon the addition of 1, 5 and 6 (Figure 2 and Supplementary Figure S3, note: higher errors towards the lower asymptote which we ascribe to the absorbance of the iridium organometallic core). In contrast, the p53/hDM2 interaction was not significantly perturbed by the addition of 2–4. The IC50 values of the active cyclometallated compounds against the p53/hDM2 interaction are presented in Table 1. Compound 1 was subsequently selected for further study as a potential disrupter of the p53/hDM2 interaction.
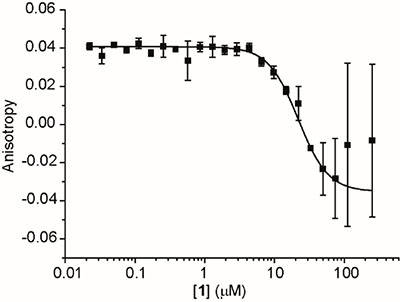
Figure 2: FA titration data of 1. 1 was incubated with 150 nM hDM217–126 recombinant fragment and 50 nM fluorescein-labelled p53 peptide (p5315–31Flu), and fluorescence anisotropy was measured at 480ex/535em.
Table 1: IC50 values of the cyclometallated iridium(III) compounds 1–6 against the p53/hDM2 interaction as determined by a FA assay
Cyclometallated iridium(III) compounds |
IC50/μM |
|---|---|
1 |
16 (± 5) |
2 |
No noticeable binding |
3 |
No noticeable binding |
4 |
No noticeable binding |
5 |
21 (± 5) |
6 |
33 (± 3) |
We next employed the NanoBRET platform [34] to elucidate the effect of 1 on the p53/hDM2 PPI in live cells. Human amelanotic melanoma A375 cells were co-transfected with plasmids expressing p53-HaloTag and NanoLuc-hDM2. Transfected cells were then treated with vehicle, positive control compound NVP-CGM097 and 1 at various concentrations. The results revealed that 1 induced a dose-response decrease of bioluminescence resonance energy transfer (BRET) in treated cells, with an IC50 value of ca. 1 μM (Figure 3A). This result indicates that 1 was able to disrupt the p53/hDM2 PPI in A375 cells.
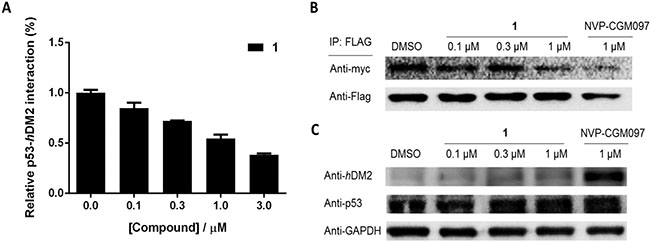
Figure 3: 1 inhibits the interaction of p53/hDM2 in A375 cells without affecting protein expression levels. (A) 1 inhibits the p53/hDM2 NanoBRET protein-protein interaction. (B) 1 inhibits the interaction of p53/hDM2 in A375 cells. The complex of p53/hDM2 in A375 cells was pulled down using anti-Flag magnetic beads and determined by probing with anti-Flag and anti-myc antibodies. (C) 1 has no significant effect on the protein expression levels of p53 and hDM2 in A375 cells. The protein expression levels of p53 and hDM2 determined by probing with anti-p53 and anti-hDM2 antibodies.
A pull-down assay was conducted to further examine the effect of 1 on the p53/hDM2 interaction. A375 cells were co-transfected with cDNAs encoding p53-Flag and hDM2-myc fusion proteins. After 6 h of treatment with NVP-CGM097 or 1, p53/hDM2 complexes were immunoprecipitated using anti-Flag magnetic beads and analysed by immunoblotting. In the absence of 1, hDM2 was precipitated together with p53. However, the amount of precipitated hDM2 was decreased with increasing concentrations of 1 or with NVP-CGM097 (Figure 3B). This result further confirms that 1 antagonized the interaction of p53/hDM2 in cells.
To assess whether 1 suppressed p53 or hDM2 protein expression levels, resulting in a decrease in the levels of precipitated protein detected in the pull-down assay, an immunoblotting experiment was performed. Immunoblotting analysis showed that 1 had no effect on the hDM2 and p53 protein expression levels, even at the highest concentration of 1 μM (Figure 3C). On the other hand, NVP-CGM097 significantly increased the expression of hDM2 protein at 1 μM, consistent with previous reports [35]. We also investigated the possible impact of the isolated ligands of 1 on the p53/hDM2 interaction and protein expression levels. Neither eppy nor dcphen ligands were found to have a significant effect on the p53/hDM2 interaction or protein expression levels (Supplementary Figure S4), suggesting that the inhibition of the p53/hDM2 interaction requires the assembly of the ligands into an intact complex. Taken together, these findings indicate that 1 significantly inhibited the p53/hDM2 interaction in A375 cells without affecting their protein expression.
Compound 1 reactivates p53 transcriptional transactivation and induced apoptosis in cells
We next studied whether 1 could reactivate the transcriptional transactivation function of p53 via blocking the p53/hDM2 interaction. Treatment of A375 cells carrying endogenous wt p53 and a p53-dependent luciferase reporter gene by 1 resulted in an increase in luciferase activity, indicating the activation of p53-direted transcription by 1 (Figure 4A). Furthermore, the expression of p53 target gene products GADD45α and PUMA were significantly increased by 1 in A375 cells (Figure 4B). These results indicate that the activation of GADD45α and PUMA gene expression in A375 cells by 1 is likely driven by its effects on wt p53.
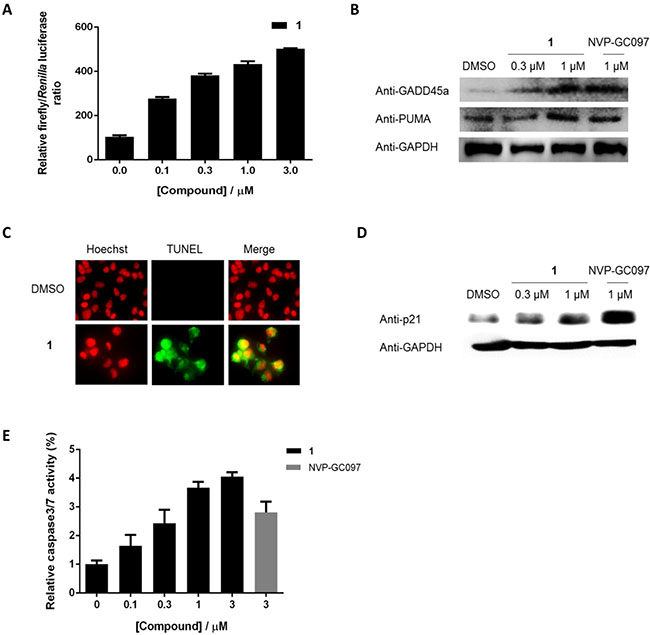
Figure 4: 1 reactivates p53 transcriptional transactivation and induced apoptosis in cells. 1 induces (A) p53-driven luciferase activity, (B) the expression of endogenous p53 targets GADD45α and PUMA, (C) DNA fragmentation, (D) p21 expression, and (E) caspase-3/7 activities in A375 cells.
Transactivation of p53 restricts cellular proliferation by inducing apoptosis and cycle arrest [36]. Therefore, a terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick and labelling (TUNEL) and Hoechst staining assay were then performed to investigate DNA fragmentation, a hallmark of apoptosis, in apoptotic cells. Notably, 1 was found to induce DNA fragmentation at a concentration of 0.3 μM (Figure 4C). Furthermore, 1 induced the expression of p21 (Figure 4D), a cyclin-dependent kinase (CDK) that is transcriptionally activated by p53 in response to DNA damage and acts to induce cell cycle arrest [37]. Finally, the effect of 1 on caspase-3/7, an apoptotic executioner under the control of p53, was investigated in A375 cells using a homogeneous luminescence assay. As shown in Figure 4E, 1 was able to significantly activate caspase-3/7 activities in a dose-dependent manner. Therefore, we reason that the induction of apoptosis by 1 in A375 cells is driven, at least in part, by the activation of p53 resulting from the disruption of the p53/hDM2 interaction.
Compound 1 was toxic to cancer cells
Clinical observations have linked the overexpression of hDM2 with human cancers possessing wild-type p53 status [38, 39]. The cytotoxicity of 1 towards ten cancers cell lines with different p53 statuses (wt, mut or null) was subsequently examined assessed using the MTT assay (Supplementary Table S2). The results showed that 1 inhibited the growth of cancer cells at low micromolar concentration. Notably, 1 exhibited a greater toxicity towards cancer cell lines carrying wt p53 than those carrying mut p53, indicating that toxicity of 1 was wt p53-dependent. Therefore, we hypothesize that the ability of 1 to inhibit cancer cell proliferation may be due, at least in part, to its inhibition of p53/hDM2 interaction, leading to the activation of p53 transcriptional transactivation resulting in cell apoptosis.
DISCUSSION
In this work, we have identified a novel kinetically-inert organometallic iridium(III) compound 1 as the first metal-based p53/hDM2 PPI disrupter. Compound 1 bearing 2-(4-ethylphenyl)pyridine (eppy) and 4, 7-dichloro-1, 10-phenanthroline (dcphen) ligands exhibited the most potent ability to disrupt the interaction between p53 and hDM2 among the six tested iridium(III) compounds, as revealed by a fluorescence anisotropy assay. Moreover, 1 was able to inhibit the interaction of p53/hDM2 cells without affecting their protein expression levels in A375 cells as determined by NanoBRET and pull-down experiments. 1 reactivated p53 transcriptional transactivation in cellulo, and also induced apoptosis and suppressed the growth of cancer cells, which we attribute, at least in part, to the disruption of the p53/hDM2 interaction by 1.
Based on the FA results, a brief structure-activity relationship analysis of the six tested organometallic iridium(III) compounds can be performed. Comparison of compounds 1 and 2 indicates that the dcphen N^N ligand of 1 is superior for p53/hDM2 inhibitory activity compared to the bulky tert-butyl-substituted bpy ligand of 2. Moreover, 3 and 4 had no noticeable effect on the p53/hDM2 interaction, indicating that their specific combinations of N^N and C^N ligands were undesirable for biological activity. Finally, 5 and 6 carrying larger N^N ligands with additional fused aromatic rings also showed moderate p53/hDM2 inhibitory activity, but were less potent than 1. This suggests that the binding site of the target protein may be tolerant to presence of the additional planar aromatic rings of 5 and 6, potentially via stacking interactions with aromatic residues, but not to the tert-butyl substituents of 2, which are more sterically demanding in three dimensions. Taken together, these results suggest that size, electronic properties and steric properties of the organometallic compounds are important in determining their activity against the p53/hDM2 interaction.
The activation of p53 protein is a potential strategy in anti-cancer therapy. High p53 protein levels lead to apoptosis, while moderate p53 protein levels result in cell cycle arrest [40]. In humans, a major negative regulator of p53 is the E3 ubiquitin protein ligase hDM2/mDM2 pathway, the activation of which triggers the degradation of p53 [41]. To date, numerous small-molecule inhibitors of p53/mDM2 interaction have been discovered [40]. The first small-molecule inhibitor of p53/mDM2 interaction, 4, 5-dihydroimidazoline (nutlin, Roche), was demonstrated to bind to mDM2 and induce p53-dependent cell cycle arrest at low micromolar levels in vitro [42]. Several nutlin analogues (nutlin-2 and nutlin-3) and other structural classes of inhibitors, such as spiro-oxindoles and benzodiazepinediones, have demonstrated promising tumor growth inhibition and tumor shrinkage in animal models [40, 43]. mDM2 inhibitors derived from natural products, such as the prenylated xanthones α-mangostin (from the fruit of Garcinia mangostana L.) and gambogic acid (from the resin of Garcinia hanburyi), have also been identified, and were shown to bind in a similar manner to the nutlins [44]. In addition, peptide and stapled peptide inhibitors of the p53/mDM2 interaction have been shown to penetrate cells, bind tightly to mDM2 and induce p53 pathway activation with in vitro IC50 values ranging from 0.005 to 700 μM [45, 46]. However, the most potent peptide inhibitors exhibited only low cellular activities due to their poor cell permeability.
In comparison to known p53/mDM2 protein-protein interaction inhibitors, the iridium(III) compounds developed in this work possess similar abilities to activate p53 signaling and inhibit cancer cell growth in vitro, albeit with reduced potency. Nevertheless, given the status of these compounds as the first metal-based inhibitors of the p53/hDM2 interaction, we consider that compound 1 could be utilized as a structural starting point for the development of more potent anti-cancer compounds. Notably, the simple structure of compound 1 coupled with the modular nature of inorganic synthesis could feasibly allow many iridium(III) compounds to be synthesized and tested, in contrast to the relatively complex small molecule or peptide-based inhibitors of the p53/mDM2 interaction existing in the literature.
MATERIALS AND METHODS
General experimental
Chemicals were purchased from Sigma-Aldrich, J & K Scientific, Armar and TCI, and used as received; all solvents used were reagent grade. TLC was performed on Sorbent Technologies aluminum-backed Silica G TLC plates, and column chromatography was performed on silica gel 60 (Merck, 230–400 mesh). High resolution of mass spectrometry was carried out in the Department of Chemistry, Hong Kong Baptist University. 1H and 13C NMR were recorded on a 400 MHz (1H) and 100 MHz (13C) Bruker instrument using acetone-d6 or DMSO-d6 as the solvent. 1H and 13C chemical shifts are expressed in ppm relative to solvent peak (acetone-d6: 1H δ 2.05,13C δ 206.68, 29.92; DMSO-d6: 1H, 2.50, 13C, 39.5). The following abbreviations are used for signal patterns: s, singlet; d, doublet; dd, doublet of doublets; t, triplet; q, quartet; m, multiplet. All NMR data was acquired and processed using standard Bruker software (Topspin). Elemental analysis of the organometallic compounds was performed in Atlantic Microlab, Inc., USA. The synthetic procedures and photophysical properties of the iridium(III) compounds 1–6 used in this study are provided in the Supporting Information.
Characterization of organometallic iridium(III) compounds
1: [Ir(eppy)2(dcphen)](PF6) (where eppy = 2-(4-ethylphenyl)pyridine and dcphen = 4, 7-dichloro-1, 10-phenanthroline), 4, 7-dichloro-1, 10-phenanthrolinebis(2-(4-ethylphenyl)pyridine)iridium(III) hexafluorophosphate Yield: 68.7%. 1H NMR (400 MHz, Acetone-d6) δ 8.71 (d, J = 6.4 Hz, 2H), 8.43–8.41 (m, 2H), 8.25 (d, J = 5.6 Hz, 2H), 8.17 (d, J = 8.0 Hz, 2H), 7.90–7.83 (m, 4H), 7.76 (d, J = 5.2 Hz, 2H), 6.95–6.91 (m, 4H), 6.28 (d, J = 1.2 Hz, 2H), 2.42 (d, J = 7.8 Hz, 4H), 1.03 (t, J = 7.8 Hz, 6H); 13C NMR (100 MHz, Acetone-d6) 168.7, 152.7, 150.5, 148.9, 147.4, 145.7, 142.8, 139.4, 131.9, 131.0, 128.7, 126.3, 125.8, 123.8, 123.4, 120.4, 29.8, 15.5; MALDI-TOF-HRMS: Calcd. for C38H30Cl2IrN4[M–PF6]+ : 805.1477, found: 805.5739; Anal.: (C38H30Cl2IrN4PF6+2H2O) C, H, N: calcd. 46.25, 3.47, 5.68; found. 46.04, 3.11, 5.67.
2: [Ir(eppy)2(dtbpy)](PF6) (where eppy = 2-(4-ethylphenyl)pyridine and dtbpy = 4, 4′-di-tert-butyl-2, 2′-bipyridine), 4, 4′-di-tert-butyl-2, 2′-bipyridinebis(2-(4-ethylphenyl)pyridine)iridium(III) hexafluorophosphate Yield: 70.3%. 1H NMR (400 MHz, Acetone-d6) δ 8.86 (d, J = 0.8 Hz, 2H), 8.17 (d, J = 8.0 Hz, 2H), 7.97 (d, J = 6.0 Hz, 2H), 7.96–7.90 (m, 2H), 7.79 (d, J = 8.0 Hz, 2H), 7.78–7.75 (m, 2H), 7.70 (d, J = 2.0 Hz, 2H), 7.10–7.07 (m, 2H), 6.90 (d, J = 2.0 Hz, 2H), 6.21 (d, J = 8.0 Hz, 2H), 2.40 (d, J = 7.6 Hz, 4H), 1.41 (s, 18H), 1.01 (t, J = 7.6 Hz, 6H); 13C NMR (100 MHz, Acetone-d6) 169.0, 164.8, 156.8, 152.3, 151.0, 149.8, 147.4, 142.6, 139.3, 131.7, 126.4, 125.8, 123.8, 123.0, 122.8, 120.4, 36.4, 30.2, 29.8, 15.5; MALDI-TOF-HRMS: Calcd. for C44H48IrN4[M–PF6]+: 825.3508, found: 825.1686; Anal.: (C44H48IrN4PF6+H2O) C, H, N: calcd. 53.48, 5.10, 5.67; found. 53.46, 4.86, 5.78.
3: [Ir(ppz)2(bpy)](PF6) (where ppz = 1-phenyl-1H-pyrazole and bpy = 2, 2′-bipyridine), 2, 2′-bipyridinebis(1-phenyl-1H-pyrazole)iridium(III) hexafluorophosphate Reported [47]
4: [Ir(pbt)2(cphen)](PF6) (where pbt = 2-phenylbenzo[d]thiazole and cphen = 5-chloro-1, 10-phenanthroline), 5-chloro-1, 10-phenanthrolinebis(2-phenylbenzo[d]thiazole)iridium(III) hexafluorophosphate Yield: 81.0%. 1H NMR (400 MHz, DMSO-d6) δ 9.07 (d, J = 8.4 Hz, 1H), 8.91 (d, J = 8.0 Hz, 1H), 8.72 (s, 1H), 8.52 (d, J = 1.2 Hz, 1H), 8.43 (d, J = 1.2 Hz, 1H), 8.27 (q, J = 5.2 Hz, 1H), 8.20–8.15 (m, 3H), 8.07 (d, J = 7.6 Hz, 2H), 7.33 (t, J = 8.8 Hz, 2H), 7.21–7.19 (m, 2H), 7.02–6.95 (m, 4H), 6.39 (t, J = 6.4 Hz, 2H), 5.76 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) 181.3, 181.2, 152.1, 151.6, 149.5, 149.2, 148.5, 147.5, 146.0, 140.1, 140.0, 138.8, 136.3, 132.8, 132.7, 132.2, 132.1, 130.9, 130.0, 128.7, 128.4, 128.1, 128.0, 127.9, 127.6, 127.1, 125.9, 124.6, 123.3; MALDI-TOF-HRMS: Calcd. for C38H23ClIrN4S2[M–PF6]+: 827.0681, found: 827.2160; Anal.: (C38H23ClIrN4S2PF6) C, H, N: calcd. 46.94, 2.38, 5.76; found. 46.76, 2.26, 5.76.
5: [Ir(pq)2(pzphen)](PF6) (where pq = 2-phenylquinoline and pzphen = pyrazino[2, 3-f][1, 10]phenanthroline), pyrazino[2, 3-f][1, 10]phenanthrolinebis(2-phenylquinoline)iridium(III) hexafluorophosphate Reported [32]
6: [Ir(dfppy)2(dcdppz)](PF6) (where dfppy = 2-(2, 4-difluorophenyl)pyridine and dcdppz = 11, 12-dichlorodipyrido[3, 2-a:2’,3′-c]phenazine), 11, 12-dichlorodipyrido[3, 2-a:2’,3′-c]phenazinebis(2-(2, 4-difluorophenyl)pyridine)iridium(III) hexafluorophosphate Yield: 78.5%. 1H NMR (400 MHz, Acetone-d6) δ 9.85 (d, J = 7.2 Hz, 2H), 8.72–8.68 (m, 4H), 8.41 (d, J = 8.4 Hz, 2H), 8.30–8.27 (m, 2H), 8.03 (t, J = 8.0 Hz, 2H), 7.95 (t, J = 6.0 Hz, 2H), 7.12–7.09 (m, 2H), 6.85–6.80 (m, 2H), 5.91 (d J = 8.4 Hz, 2H); 13C NMR (100 MHz, Acetone-d6) 165.8, 165.7, 164.8, 164.7, 163.6, 163.3, 163.2, 161.1, 154.5, 154.4, 154.3, 151.2, 151.0, 142.3, 142.0 140.8, 137.3, 137.0, 132.0, 131.2, 129.8, 125.0, 124.7, 124.5, 115.0, 114.8, 100.2, 99.9, 99.6; MALDI-TOF-HRMS: Calcd. for C40H20Cl2F4IrN6[M–PF6]+: 923.0692, found: 923.0699; Anal.: (C40H20Cl2F4IrN6PF6) C, H, N: calcd. 44.95, 1.89, 7.86; found. 44.94, 1.88, 7.68.
Protein expression and fluorescence anisotropy assay
Expression of hDM217–126 recombinant fragment was performed as described previously [33]. Fluorescence anisotropy assays were used to study the displacement of p53 from hDM2 by organometallic compounds. The experiments were performed in triplicate in a volume of 60 μL of the assay buffer (40 mM Na3PO4, pH 7.50, 200 mM NaCl, 0.02 mg/mL BSA) in black flat-bottom 384-well plates. Different concentrations of organometallic compounds were diluted in the assay buffer containing 150 nM hDM2 and 50 nM p5315–31Flu (Ac-SQETFSDLWKLLPENNVC(Flu)-NH2, Peptide Protein Research Ltd.) and incubated at room temperature for 30 min. The intensity and anisotropy was monitored at 480ex/535em (5 nM bandwidth) using a Perkin Elmer EnvisionTM 2103 MultiLabel plate reader. The data was analyzed according to published methods [48, 49].
NanoBRET protein:protein interaction assay
A NanoBRET protein:protein interaction system (Promega) was used to detect p53/hDM2 protein-protein interactions by measuring energy transfer from a bioluminescent protein donor to a fluorescent protein acceptor. A375 cells were incubated with a transfection mixture of 2 μg of p53-HaloTag fusion gene and 0.2 μg of NanoLuc-hDM2 fusion gene in Opti-MEM® I reduced serum medium with TurboFect transfection reagent (Thermo Scientific) in 6-well plates for 20 h at 37°C, 5% CO2. The transfected cells were replated in a white 96-well plate with HaloTag® NanoBRET 618 ligand (100 nM) in Opti-MEM® I reduced serum medium with 4% fetal bovine serum (FBS) and incubated overnight. 1 was the added directly to the culture medium at different concentrations and the plate was incubated for further 6 h. 25 μL of 1X Nano-Glo® substrate in Opti-MEM® I reduced serum medium was added to the wells. The plate was then measured using 460 nm filter and 610 nm filters in SpectraMax M5 microplate reader (Molecular Devices).
Pull-down assay
A375 cells seeded in 6-well plates were co-transfected with pcDNA-Flag-p53 (Addgene) and pCMV-myc3-hDM2 (Addgene) for 6 h using TurboFect transfection reagent following the instruction from manufacturer. Cells were then cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) complete medium overnight. Serial dilutions of organometallic compounds in low FBS buffer were placed into the wells, and incubated with cells for additional 6 h. Protein samples were collected, and the concentration in the supernatant was determined with the bicinchoninic acid (BCA) method. The organometallic compounds of p53/hDM2 in the protein samples were pulled down using anti-Flag magnetic beads to capture the FLAG fusion proteins (Sigma) as previously described [50, 51]. The protein-binding beads were washed three time with TBS buffer (50 mM Tris HCl, 150 mM NaCl, pH 7.4) to remove non-specifically bound proteins, and subsequently subjected to the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) by detection with anti-Flag (1:1000, Sigma-Aldrich) and anti-myc antibodies (1:1000, Beyotime).
Immunoblotting
A375 cells seeded in 6-well plates were treated with vehicle, NVP-CGM097 or different concentrations of 1 for 6 h. Cells were then harvested and the quantity of each protein samples was measured by the BCA method. Same aliquot (30 μg) of each protein sample was separated by SDS-PAGE according to the reported procedure of immunoblotting [50]. The signals were visualized using Enhanced chemiluminescent Plus reagents (GE Healthcare) and analyzed by Image Lab. The primary antibodies used were: anti-PUMA (1:500, Santa Cruz), anti-GADD45α (1:500, Santa Cruz), anti-p21 (1:500, Santa Cruz), anti-GAPDH (1:500, Santa Cruz), anti-p53 (1:500, Santa Cruz) and anti-hDM2 antibodies (1:500, Santa Cruz).
Dual luciferase reporter assay
A375 cells were co-transfected with p53 luciferase reporter gene (Addgene) and Renilla luciferase (Rluc) control reporter vectors at the ratio of 10:1 for 6 h in a 6-well plate. After transfection, cells were cultivated for another 18 h in the complete DMEM medium. Cells were then plated in a 96-well plate overnight. Serial dilutions of 1 were then added to the wells and the plate was incubated for 6 h. Luciferase reporter activity was then measured according to instruction of a dual reporter assay system (Progema) as previous reported [50].
TUNEL assay
The TUNEL assay was performed to measure apoptosis in cells induced by the treatment of 1. Briefly, A375 cells were incubated with 1 for 6 h. Apoptosis in cells was measured using the APO-BrdU™ TUNEL assay kit (Thermo Fisher) according to the manufacturer’s instruction. After fixing cells by the addition of 100 μL of 1% (w/v) paraformaldehyde in phosphate-buffered saline (PBS) and permeabilization with 70% (v/v) ice-cold ethanol, the cells were plated on ice for 30 min. 50 μL of DNA-labeling solution (10 μL of reaction buffer, 0.75 μL of TdT enzyme, 8.0 μL of BrdUTP and 31.25 μL of dH2O) was added to wells. The plate was incubated with the DNA-labeling solution for 1 h at 37°C with shaking every 15 min. Then, the cells were washed with 10 μL of rinse buffer three times before staining with diluted Fluor® 488 dye-labeled anti-BrdU antibody solution (1:20) for 30 min at room temperature. 100 μL of propidium iodide/RNase A staining buffer was then added to wells and the cells were incubated for an additional 30 min. The samples were analyzed using a GE Healthcare Life Sciences IN Cell Analyzer 2000.
Caspase-Glo® 3/7 assay
The activities of caspase 3/7 were detected using Caspase-Glo® 3/7 assay kit (Promega). Following 6 h treatment of different concentrations of 1, a 96-well luminometer plate containing A357 cells was removed from the incubator and equilibrated to room temperature for 30 min. 100 μL of Caspase-Glo® 3/7 reagent was added to each well and the plate was shaking for 30 sec at room temperature. The luminescence signaling of each sample was collected in SpectraMax M5 microplate reader.
Cell proliferation assay
Inhibition of cell growth was measured using the MTT reagent (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide). Different cell lines with variable expression levels of p53 protein (A375, A549, HeLa, A2780, A431, MCF7, MD-MBA-231, MD-MBA-468, T47D and H1299 cell lines) were plated onto 96-well plates at a density of 4, 000 cells/well overnight prior to the addition of 1 at different concentrations ranging from 0.01–100 μM for a further 72 h. 10 μL MTT reagent (5 mg/mL) was added to the wells and the cells were incubated for an additional 4 h before 100 μL of DMSO was added to the wells. The absorbance of each well was read on a SpectraMax M5 microplate reader (Molecular Devices) at the wavelength of 570 nm.
ACKNOWLEDGMENTS AND FUNDING
This work is supported by Hong Kong Baptist University (FRG2/14–15/004), the Health and Medical Research Fund (HMRF/13121482 and HMRF/14130522), the Research Grants Council (HKBU/201913), National Natural Science Foundation of China (21575121), Guangdong Province Natural Science Foundation (2015A030313816), Hong Kong Baptist University Century Club Sponsorship Scheme 2015, the European Research Council [ERC-StG-240324] and [ERC-PoC 632207], the French National Research Agency/Research Grants Council Joint Research Scheme (A-HKBU201/12; Oligoswitch ANR-12-IS07-0001), Interdisciplinary Research Matching Scheme (RC-IRMS/14–15/06), the Science and Technology Development Fund, Macao SAR (103/2012/A3), the University of Macau (MYRG2015-00137-ICMS-QRCM, MRG023/LCH/2013/ICMS and MRG044/LCH/2015/ICMS).
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000; 408:307–310.
2. Carry JC, Garcia-Echeverria C. Inhibitors of the p53/hdm2 protein-protein interaction-path to the clinic. Bioorg Med Chem Lett. 2013; 23:2480–2485.
3. Deb SP. Cell cycle regulatory functions of the human oncoprotein MDM2. Mol Cancer Res. 2003; 1:1009–1016.
4. Chene P. Inhibiting the p53-MDM2 interaction: An important target for cancer therapy. Nat Rev Cancer. 2003; 3:102–109.
5. Chene P. Inhibition of the p53-MDM2 interaction: Targeting a protein-protein interface. Mol Cancer Res. 2004; 2:20–28.
6. Fischer PM, Lane DP. Small-molecule inhibitors of the p53 suppressor HDM2: have protein-protein interactions come of age as drug targets? Trends Pharmacol Sci. 2004; 25:343–346.
7. Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding QJ, Jiang N, Liu JJ, Zhao CL, Glenn K, Wen Y, Tovar C, Packman K, Vassilev L, et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. Acs Med Chem Lett. 2013; 4:466–469.
8. Y Z, D B, S W. Small Molecule inhibitors of MDM2-p53 and MDMX-p53 interaction as new cancer therapeutics. BioDiscovery. 2013; 4–18.
9. Shangary S, Wang SM. Small-Molecule Inhibitors of the MDM2-p53 Protein-Protein Interaction to Reactivate p53 Function: A Novel Approach for Cancer Therapy. Annu Rev Pharmacol Toxicol. 2009; 49:223–241.
10. Leung CH, He HZ, Liu LJ, Wang MD, Chan DSH, Ma DL. Metal complexes as inhibitors of transcription factor activity. Coord Chem Rev. 2013; 257:3139–3151.
11. Leung CH, Lin S, Zhong HJ, Ma DL. Metal complexes as potential modulators of inflammatory and autoimmune responses. Chem Sci. 2015; 6:871–884.
12. Meier SM, Novak MS, Kandioller W, Jakupec MA, Roller A, Keppler BK, Hartinger CG. Aqueous chemistry and antiproliferative activity of a pyrone-based phosphoramidate Ru (arene) anticancer agent. Dalton Trans. 2014; 43:9851–9855.
13. Serebryanskaya TV, Lyakhov AS, Ivashkevich LS, Schur J, Frias C, Prokop A, Ott I. Gold(I) thiotetrazolates as thioredoxin reductase inhibitors and antiproliferative agents. Dalton Trans. 2015; 44:1161–1169.
14. Leonidova A, Pierroz V, Adams LA, Barlow N, Ferrari S, Graham B, Gasser G. Enhanced Cytotoxicity through Conjugation of a “Clickable” Luminescent Re(I) Complex to a Cell-Penetrating Lipopeptide. ACS Med Chem Lett. 2014; 5:809–814.
15. Rilak A, Bratsos I, Zangrando E, Kljun J, Turel I, Bugarcic ZD, Alessio E. New Water-Soluble Ruthenium(II) Terpyridine Complexes for Anticancer Activity: Synthesis, Characterization, Activation Kinetics, and Interaction with Guanine Derivatives. Inorg Chem. 2014; 53:6113–6126.
16. McConnell AJ, Lim MH, Olmon ED, Song H, Dervan EE, Barton JK. Luminescent Properties of Ruthenium(II) Complexes with Sterically Expansive Ligands Bound to DNA Defects. Inorg Chem. 2012; 51:12511–12520.
17. Ma DL, He HZ, Leung KH, Chan DSH, Leung CH. Bioactive Luminescent Transition-Metal Complexes for Biomedical Applications. Angew Chem Int Ed. 2013; 52:7666–7682.
18. Barragan F, Lopez-Senin P, Salassa L, Betanzos-Lara S, Habtemariam A, Moreno V, Sadler PJ, Marchan V. Photocontrolled DNA Binding of a Receptor-Targeted Organometallic Ruthenium(II) Complex. J Am Chem Soc. 2011; 133:14098–14108.
19. Hindo SS, Mancino AM, Braymer JJ, Liu YH, Vivekanandan S, Ramamoorthy A, Lim MH. Small Molecule Modulators of Copper-Induced A beta Aggregation. J Am Chem Soc. 2009; 131:16663–16665.
20. Muldoon J, Ashcroft AE, Wilson AJ. Selective Protein-Surface Sensing Using Ruthenium(II) Tris(bipyridine) Complexes. Chem-Eur J. 2010; 16:100–103.
21. Leung KH, He HZ, He BY, Zhong HJ, Lin S, Wang YT, Ma DL, Leung CH. Label-free luminescence switch-on detection of hepatitis C virus NS3 helicase activity using a G-quadruplex-selective probe. Chem Sci. 2015; 6:2166–2171.
22. Lu LH, Chan DSH, Kwong DWJ, He HZ, Leung CH, Ma DL. Detection of nicking endonuclease activity using a G-quadruplex-selective luminescent switch-on probe. Chem Sci. 2014; 5:4561–4568.
23. Leung CH, Chan DSH, He HZ, Cheng Z, Yang H, Ma DL. Luminescent detection of DNA-binding proteins. Nucleic Acids Res. 2012; 40:941–955.
24. Ma DL, Chan DSH, Leung CH. Group 9 Organometallic Compounds for Therapeutic and Bioanalytical Applications. Acc Chem Res. 2014; 47:3614–3631.
25. Lo KKW, Zhang KY. Iridium(III) complexes as therapeutic and bioimaging reagents for cellular applications. Rsc Adv. 2012; 2:12069–12083.
26. Leung CH, Liu LJ, Lu LH, He BY, Kwong DWJ, Wong CY, Ma DL. A metal-based tumour necrosis factor-alpha converting enzyme inhibitor. Chem Commun. 2015; 51:3973–3976.
27. Zhong HJ, L. L, Leung KH, Wong CL, Peng C, Yan SC, Ma DL, Cai Z, Wang HM, Leung CH. An iridium(III)-based irreversible protein-protein interaction inhibitor of BRD4 as a potent anticancer agent. Chem Sci. 2015; 6:5400–5408.
28. Liu LJ, Lu L, Zhong HJ, He B, Kwong D, Ma DL, Leung CH. An Iridium(III) Complex Inhibits JMJD2 Activities and Acts as A Potential Epigenetic Modulator. J Med Chem. 2015; 58:6697–6673.
29. Kastl A, Wilbuer A, Merkel AL, Feng L, Di Fazio P, Ocker M, Meggers E. Dual anticancer activity in a single compound: visible-light-induced apoptosis by an antiangiogenic iridium complex. Chem Commun. 2012; 48:1863–1865.
30. Hearn JM, Romero-Canelon I, Qamar B, Liu Z, Hands-Portman I, Sadler PJ. Organometallic Iridium(III) Anticancer Complexes with New Mechanisms of Action: NCI-60 Screening, Mitochondrial Targeting, and Apoptosis. ACS Chem Biol. 2013; 8:1335–1343.
31. Geldmacher Y, Oleszak M, Sheldrick WS. Rhodium(III) and iridium(III) complexes as anticancer agents. Inorg Chim Acta. 2012; 393:84–102.
32. Leung CH, Zhong HJ, Yang H, Cheng Z, Chan DSH, Ma VPY, Abagyan R, Wong CY, Ma DL. A Metal-Based Inhibitor of Tumor Necrosis Factor-alpha. Angew Chem Int Ed. 2012; 51:9010–9014.
33. Barnard A, Long K, Martin HL, Miles JA, Edwards TA, Tomlinson DC, Macdonald A, Wilson AJ. Selective and Potent Proteomimetic Inhibitors of Intracellular Protein-Protein Interactions. Angew Chem Int Ed. 2015; 54:2960–2965.
34. Machleidt T, Woodroofe CC, Schwinn MK, Mendez J, Robers MB, Zirnmerman K, Otto P, Daniels DL, Kirkland TA, Wood KV. NanoBRET-A Novel BRET Platform for the Analysis of Protein-Protein Interactions. ACS Chem Biol. 2015; 10:1797–1804.
35. Holzer P, Masuya K, Furet P, Kallen J, Valat-Stachyra T, Ferretti S, Berghausen J, Bouisset-Leonard M, Buschmann N, Pissot-Soldermann C, Rynn C, Ruetz S, Stutz S, et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J Med Chem. 2015; 58:6348–6358.
36. Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003; 22:9030–9040.
37. Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005; 65:3980–3985.
38. Kitagaki J, Agama KK, Pommier Y, Yang YL, Weissman AM. Targeting tumor cells expressing p53 with a water-soluble inhibitor of Hdm2. Mol Cancer Ther. 2008; 7:2445–2454.
39. Koblish HK, Zhao SY, Franks CF, Donatelli RR, Tominovich RM, LaFrance LV, Leonard KA, Gushue JM, Parks DJ, Calvo RR, Milkiewicz KL, Marugan JJ, Raboisson P, et al. Benzodiazepinedione inhibitors of the Hdm2 : p53 complex suppress human tumor cell proliferation in vitro and sensitize tumors to doxorubicin in vivo. Mol Cancer Ther. 2006; 5:160–169.
40. Hoe KK, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014; 13:217–236.
41. Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res. 2003; 1:1001–1008.
42. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004; 303:844–848.
43. Patel S, Player MR. Small-molecule inhibitors of the p53-HDM2 interaction for the treatment of cancer. Expert Opin Inv Drug. 2008; 17:1865–1882.
44. Leao M, Pereira C, Bisio A, Ciribilli Y, Paiva AM, Machado N, Palmeira A, Fernandes MX, Sousa E, Pinto M, Inga A, Saraiva L. Discovery of a new small-molecule inhibitor of p53-MDM2 interaction using a yeast-based approach. Biochem Pharmacol. 2013; 85:1234–1245.
45. Brown CJ, Quah ST, Jong J, Goh AM, Chiam PC, Khoo KH, Choong ML, Lee MA, Yurlova L, Zolghadr K, Joseph TL, Verma CS, Lane DP. Stapled Peptides with Improved Potency and Specificity That Activate p53. ACS Chem Biol. 2013; 8:506–512.
46. Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, et al. Stapled alpha-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A. 2013; 110:E3445–E3454.
47. Tamayo AB, Garon S, Sajoto T, Djurovich PI, Tsyba IM, Bau R, Thompson ME. Cationic bis-cyclometalated iridium(III) diimine complexes and their use in efficient blue, green, and red electroluminescent devices. Inorg Chem. 2005; 44:8723–8732.
48. Plante JP, Burnley T, Malkova B, Webb ME, Warriner SL, Edwards TA, Wilson AJ. Oligobenzamide proteomimetic inhibitors of the p53-hDM2 protein-protein interaction. Chem Commun. 2009; 5091–5093.
49. Barnard A, Long K, Yeo DJ, Miles JA, Azzarito V, Burslem GM, Prabhakaran P, Edwards TA, Wilson AJ. Orthogonal functionalisation of alpha-helix mimetics. Org Biomol Chem. 2014; 12:6794–6799.
50. Ma DL, Liu LJ, Leung KH, Chen YT, Zhong HJ, Chan DSH, Wang HMD, Leung CH. Antagonizing STAT3 Dimerization with a Rhodium(III) Complex. Angew Chem Int Ed. 2014; 53:9178–9182.
51. Liu LJ, Leung KH, Chan DSH, Wang YT, Ma DL, Leung CH. Identification of a natural product-like STAT3 dimerization inhibitor by structure-based virtual screening. Cell Death Dis. 2014; 5:e1293.