
INTRODUCTION
TEF3-1 (transcriptional enhancer factor 3 isoform 1), also known as TEAD4 (TEA domain family member 4), belongs to a class of transcription factors, which are all widely expressed in eukaryotic cells. This transcription factor was found to bind to the MCAT motif of gene promoters [1, 2]. This gene product is a member of the transcriptional enhancer factor (TEF) family of transcription factors, which contain the TEA/ATTS DNA-binding domain. It is also called related transcriptional enhancer factor 1 (RTEF-1), transcription factor 13-like 1 (TCF13 L1), and related transcription enhancer factor 1B (hRTEF-1B). This gene is also critical for the establishment of the trophectoderm (TE)-specific transcriptional program in the preimplantation mouse embryo [3].
However, integrative genomics analyses have revealed the involvement of TEF3-1 in the multi-level dysregulation and oncogenic characteristics that are observed in gastric cancer (GC) and in the development of pancreatic cancer [4, 5]. In addition, the transcriptional coactivator Yes-associated protein (YAP) is a major regulator of organ size and proliferation, and increased YAP/TEF3-1 activity plays a causal role in cancer progression and metastasis [6–8]. As coactivators, YAP1 or TAZ interacts with the TEAD/TEF family of transcription factors (TEAD1-4) to transcriptionally regulate the target genes in tumor tissue [9–11]. Moreover, as a negative regulator of the YAP-TEAD transcriptional complex, VGL might be a new tumor suppressor target in the treatment of lung cancer [12, 13].
Tumor angiogenesis is critical for the formation of vascular structures and tumor metastasis. One of the hallmarks of tumor angiogenesis is the proliferation of endothelial cells, which is also regulated by cell cycle arrest. As a transcriptional stimulator of vascular endothelial growth factor (VEGF) in hypoxic endothelial cells, TEF3-1 induces an increase in angiogenic processes including the proliferation of endothelial cells and the formation of the vasculature [14–16]. This brought to our attention several recent clinical studies that have reported on the YAP/TEAD complex, which is activated in solid tumors such as liver, breast, and ovarian cancers [11, 17, 18]. Particularly, TEF3-1 is also found to be preferentially expressed in malignant mesothelioma (MM) and has been shown to promote the expression of specific genes that regulate cell proliferation and the cell cycle [19]. We presume that an understanding of the molecular mechanisms by which tumor angiogenesis is induced by TEF3-1 in endothelial cells.
Here, we show that nuclear TEF3-1 promoted G1/S cell cycle transition, but that a deficiency of TEF3-1 induced G1/S arrest and a decrease in the proliferation of HUVECs. A microarray analysis also indicated that TEF3-1 overexpression upregulated the expression of genes related to cancer, as most of the upregulated genes are involved in tumor angiogenesis. In addition, our data showed the presence of nuclear TEF3-1 protein in the tumor vasculature of gastric cancer, which suggests that the nuclear location of TEF3-1 in GC may be a biomarker for tumor progression and diagnosis. Finally, we observed that the knock down of TEF3-1 inhibited tumor angiogenesis in an animal model.
RESULTS
Overexpression of TEF3-1 and TEF3-1ΔNLS with a lentiviral vector in HUVECs
In previous experiments, the nuclear localization of TEF3-1 was first established by the induction of vascular endothelial growth factor (VEGF) and was found to contribute to proliferation, migration, tube formation, and angiogenesis [20]. However, the way in which TEF3-1 is regulated in tumor angiogenesis remains poorly understood. To explore the mechanism of TEF3-1 in HUVEC cells, this gene was overexpressed using a lentiviral system. After the HUVECs were infected with the Flag-tagged lentivirus, the protein levels of TEF3-1 and TEF3-1ΔNLS were examined by western blot analysis with antibodies against TEF3-1 or Flag; these antibodies recognize the tags that were fused with full-length TEF3-1 and TEF3-1ΔNLS, in which the nuclear location sequence (NLS) was deleted, respectively. This demonstrates that both TEF3-1 and TEF3-1ΔNLS were overexpressed in HUVECs, as expected (Figure 1A). Interestingly, TEF3-1 changed the morphology of HUVECs, which became more elongated than normal cells. Notably, when TEF3-1ΔNLS deleted the nuclear localization sequence, the mutant gene did not significantly change the morphology of the cells (Figure 1B). As shown in Figure 1C, the Flag-fused TEF3-1 was located in the nuclei, while the Flag-fused TEF3-1ΔNLS remained in the cytoplasm. This finding suggests that the function of this gene is associated with its cellular localization. To be consistent with these results, we also wanted to demonstrate the mechanism of the nuclear localization of TEF3-1 in HUVECs.
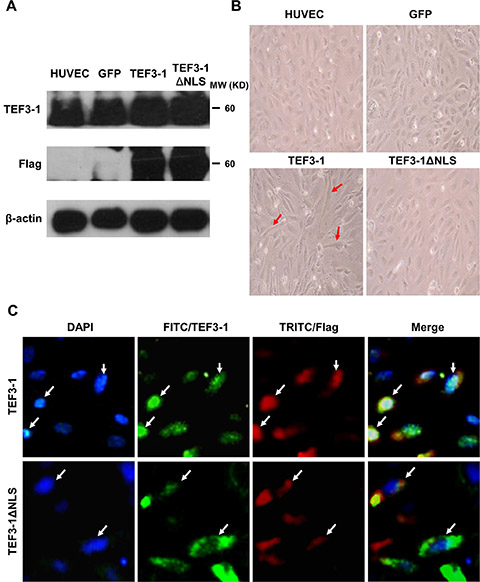
Figure 1: Expression of TEF3-1 or TEF3-1ΔNLS in HUVECs. The mocked HUVECs were as the negative control. GFP was also expressed using lentiviral system to infect HUVECs as the other negative control. (A) Immunoblot analysis of the stable expression of the indicated proteins with Flag-tagged lentiviral vector in HUVECs. Full-length TEF3-1 or TEF3-1ΔNLS was detected using anti-TEF3-1 antibodies and anti-Flag antibodies. (B) Phase-contrast images of HUVECs with TEF3-1 or TEF3-1ΔNLS stable expression. Magnification × 200. (C) Immunofluorescence of HUVECs stained with Hoechst or labeled with anti-TEF3-1 antibodies or anti-Flag antibodies. Magnification × 200. The results shown are the sum of two independent experiments.
TEF3-1 and its localization to the nucleus are required to promote G1/S transition
First, to uncover how TEF3-1 expression would impact the cell cycle regulation of HUVECs, cell proliferation was observed after TEF3-1 overexpression. The stable expression of TEF3-1 was significantly increased in comparison with mock cells, while the expression of TEF3-1ΔNLS was not increased (Figure 2A). Furthermore, according to a flow cytometric analysis, we found that TEF3-1 expression promoted the transition of cell cycle from G1 to S phase and that the TEF3-1 nuclear localization was important for the promotion of cell cycle progress (Figure 2B and 2C). Additionally, a western blot analysis again demonstrated that TEF3-1 could increase the expression levels of cyclin E and CDKs, which are related to G1/S cell cycle transition. At the same time, TEF3-1ΔNLS expression did not clearly increase the expression levels of cyclin D and CDKs (Figure 2D). Together, these results indicate that TEF3-1 expression promotes G1/S transition in HUVECs and that the nuclear localization signal of TEF3-1 is also necessary for gene function. These results suggest a significant proliferative effect of the overexpression of TEF3-1 on cell cycle progress in vitro.
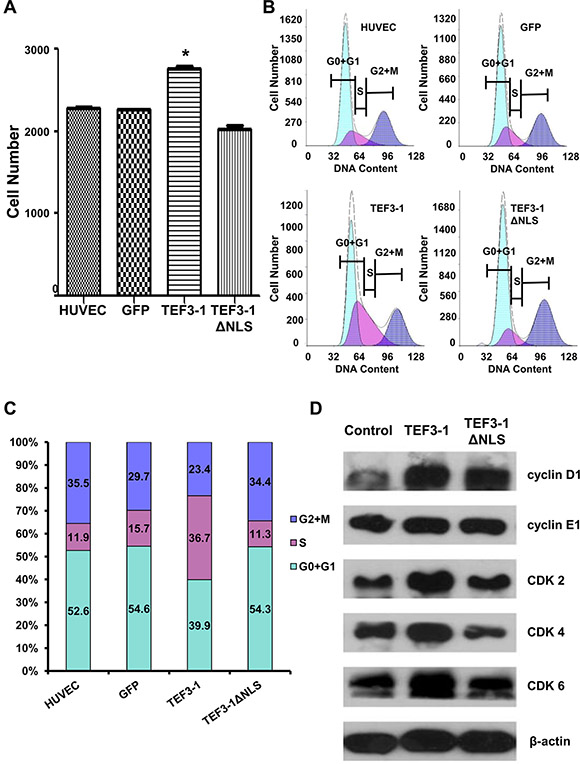
Figure 2: TEF3-1 and its localization to the nucleus affect the expression of G1/S cell cycle regulatory proteins in HUVECs. The data represent an average of at least three independent experiments. The mocked HUVECs were as the negative control. GFP was also expressed using lentiviral system to infect HUVECs as the other negative control. (A) TEF3-1 increases the proliferation of HUVECs. HUVECs were treated with lentiviruses that express control vector, TEF3-1 and TEF3-1ΔNLS, respectively. The cell numbers were counted in a cell proliferation assay. *indicates P < 0.05 compared with the control. (B and C) The percentage of cells in each phase was determined by flow cytometric analysis. RNase and PI were added to the cell suspension after HUVECs were treated with lentiviruses that express GFP, TEF3-1 and TEF3-1ΔNLS, respectively. (D) Western blot assays were performed to detect the levels of cell cycle-related proteins. β-actin was used as the loading control.
Stably downregulated TEF3-1 expression suppresses cell proliferation and reduces the G1/S cell cycle signal in vitro in HUVECs
To demonstrate that the suppression of TEF3-1 expression could inhibit cell proliferation and G1 to S cell cycle transition, we used lentiviral-mediated SiRNA that specifically targets TEF3-1 to stably inhibit the expression of TEF3-1 in HUVECs. In this study, three types of lentiviral-mediated SiRNAs (LV-SiTEF3-1#1, LV-SiTEF3-1#2 and LV-SiTEF3-1#3) were used to suppress TEF3-1 protein in HUVECs and were compared with a mock vector. As shown in Figure 3A, LV-SiTEF3-1#2 and LV-SiTEF3-1#3 clearly downregulated TEF3-1 expression, especially LV-SiTEF3-1#3. Subsequently, we observed that cell proliferation ability was significantly reduced in comparison with the mock-treated cells (Figure 3B). Furthermore, we found that the knock down of TEF3-1 delayed G1 to S phase transition (Figure 3C and 3D). A western blot analysis demonstrated that the downregulation of TEF3-1 decreased the expression levels of cyclin E, CDKs and P21 (Figure 3E). These results also suggest an obvious effect of the downregulation of TEF3-1 on cell cycle progress in HUVECs.
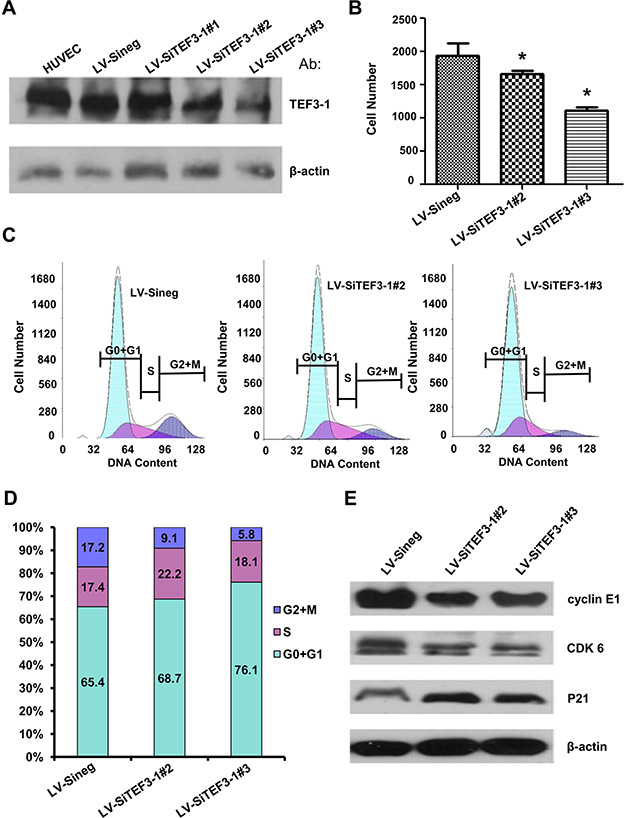
Figure 3: Depletion of TEF3-1 induces G1/S cell cycle arrest in vitro. (A) Immunoblot analysis of TEF3-1 and actin expression in HUVECs infected with control lentivirus, LV-SiTEF3-1#2 and LV-SiTEF3-1#3, respectively.(B) HUVECs were treated with lentiviruses that express control vector and TEF3-1 SiRNAs, respectively. The cell numbers were counted in a cell proliferation assay. *indicates P < 0.05 compared with the control. (C and D) The percentage of cells in each phase was determined by flow cytometric analysis. RNase and PI were added to the cell suspension after HUVECs were treated with lentiviruses that express control, LV-SiTEF3-1#2 and LV-SiTEF3-1#3, respectively. (E) Immunoblot analysis of cell cycle-related proteins in HUVECs infected with control lentivirus, LV-SiTEF3-1#2 and LV-SiTEF3-1#3, respectively.
Microarray data indicates that TEF3-1 affects cell cycle- and angiogenesis-related gene expression in endothelial cells
TEF3-1 was recently reported to possess functions in endothelial cells [15, 21], but additional mechanisms still need to be defined. In order to understand the mechanism by which TEF3-1 regulates cell cycle progression and angiogenesis in endothelial cells, we performed a microarray analysis. Then a 2-fold cut-off threshold for our microarray data were chosen to screen the different expressed genes. With the analysis of Cluster 3.0, Hierarchical clustering confirmed the different genes and their expression levels in the TEF3-1-infected HUVEC cells compared with the control-treated cells (Figure 4A). When we further analyzed the microarray results, we found that 108 genes were downregulated and 248 genes were upregulated (Figure 4B and Supplementary Table S1). Using these results and a KEGG pathway analysis [22], we explored signal pathways that may be regulated by TEF3-1 in HUVEC cells. As expected, one of the major pathways that was identified as the cell cycle-associated pathway (Figure 4C). To analyze signal pathways by GO analysis, the rank of cell cycle-associated pathways was considerably higher (Figure 4D and Supplementary Table S2). This suggests that one of the major functions of TEF3-1 in HUVECs is regulation of the cell cycle. In all, the genes including cyclins, cyclin-dependent kinases (CDKs), and cell division control (CDC) proteins, were directly associated with the cell cycle and were identified in the microarray.
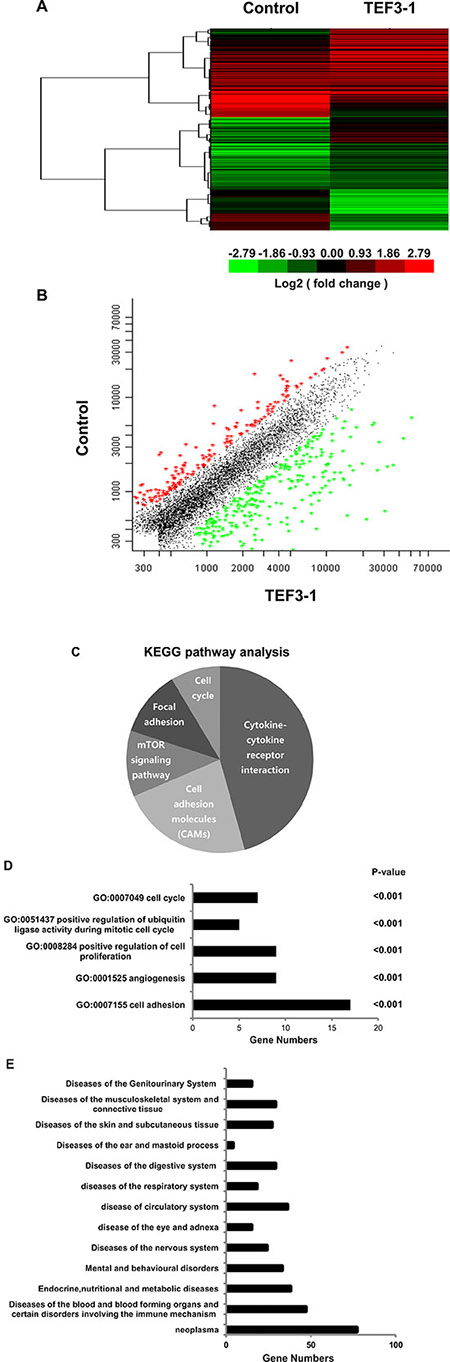
Figure 4: Microarray analysis reveals the functional importance of TEF3-1 in the cell cycle and in angiogenesis. (A) Hierarchical clustering analysis of differentially expressed genes between LV-TEF3-1- and control lentivirus-infected HUVECs. Control lentivirus were packaged with empty plasmids. (Red) higher level of gene expression; (green) lower level of gene expression; (black) no change from the control. A color scale is shown below. (B) A scatter plot shows genes that are upregulated and downregulated compared with gene regulation observed with the control lentivirus. The different genes were selected based on a 2-fold (upregulated) or a 0.5-fold (downregulated) change threshold. (C) The KEGG pathway analysis shows that cell cycle regulation was one of the most frequently identified pathways. (D) Gene ontology of biological processes for differentially expressed genes using a molecular annotation system (MAS). Notably, these GOs are related to the cell cycle and angiogenesis with a P-value < 0.001 compared with control cells. (E) The different genes are involved in different diseases according to the ICD (International Classification of Disease) and 77 genes are involved in neoplastic diseases.
Importantly, when TEF3-1 was overexpressed in HUVECs, angiogenesis was one of the important pathways in these cells. Moreover, compared with the other pathways, the rank of the adhesion pathway was the highest because it is one of the vital steps of tumor angiogenesis (Figure 4E). Importantly, when we sorted the different genes that are involved in different diseases according to the ICD (International Classification of Disease) and the disease database (http://www.malacards.org), we identified approximately 77 genes that are involved in neoplastic diseases (Figure 4E and Supplementary Table S3). These results strongly imply that TEF3-1 regulates the cell cycle in HUVECs and that this protein might play a role in tumor angiogenesis.
Enhanced nuclear localization of TEF3-1 in endothelial cells in GC
Because nuclear TEF3-1 promotes the proliferation of endothelial cells, we wanted to confirm the specific localization of TEF3-1 in GC, thus, 46 samples were examined for TEF3-1 expression. Because CD31 is a marker of endothelial cells in tissue samples, an immunohistochemical analysis was performed with antibodies to CD31 and TEF3-1 in paired GC/normal tissues. In these tissues, we examined the protein expression levels and patterns of TEF3-1 expression in endothelial cells. We had 23 GC tumor samples available, and 9 out of 23 featured high expression of TEF3-1. According to IHC analysis, in 55% of the tumor tissues, TEF3-1 was located in the nuclei of both endothelial cells and tumor cells (Figure 5A, panels a and b). However, in the 23 matched normal tissues, only 3 out of 23 expressed TEF3-1, and in most cases, the expression was not in the nucleus (Figure 5A, panels c and d; P < 0.05).
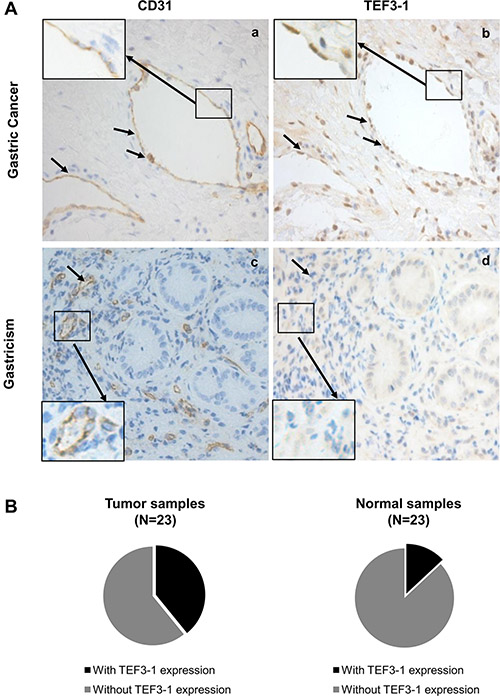
Figure 5: Frequent nuclear localization of TEF3-1 accompanied by CD31 expression in gastric cancer. (A) Representative images from immunohistochemical staining for TEF3-1 and CD31 are presented for the GC and the matched normal tissues. 400x magnified figures are shown. The arrows indicate notable correlations between the expression of TEF3-1 and CD31. (B) Expression of TEF3-1 in 46 samples of GC and normal tissues.
In addition, the cancer tissues with a higher expression level of TEF3-1 also exhibited higher amounts of TEF3-1 in endothelial cells compared with normal tissues (Figure 5B). TEF3-1 was also found in most of the nuclei of GC tissues with TEF3-1 expression, which was consistent with a previous report that showed that TEF3-1 accumulates in the nuclei of HUVECs [20] (Supplementary Table S4). Therefore, TEF3-1 is indicated that was more frequently localized to the nucleus and accompanies angiogenesis in GC. Given that angiogenesis may be considered a required step for late-stage GC, TEF3-1 might be involved in the progression of GC.
Knock down of TEF3-1 inhibits angiogenesis in Tumor Xenografts in vivo
As TEF3-1 can promotes endothelial cell proliferation, and the knock down of TEF3-1 inhibits G1/S cell cycle transition, we generated a colon cancer model to test whether LV-SiTEF3-1 could inhibit tumor vessel growth in nude mice. Nude mice bearing HCT116 tumors (approximate volume: 50 mm3) were randomly divided into two groups (5 mice/group) and were treated with LV-SiTEF3-1 or LV-Sineg control each day. The body weight of the mice and the tumor size were measured daily. As shown as Figure 6A, LV-SiTEF3-1 inhibited tumor growth. Treatment with LV-SiTEF3-1 also led to a slower increase in tumor weight, which is consistent with the increase in tumor volume (Figure 6B). Notably, we found no additional weight loss or other signs of toxicity, even in mice that were treated with LV-SiTEF3-1 for 31 days (Figure 6C).
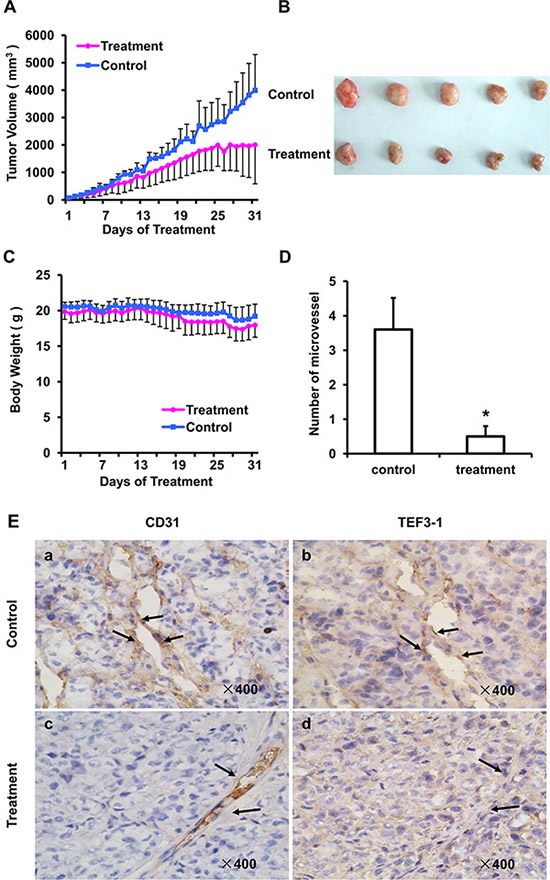
Figure 6: SiRNA of TEF3-1 inhibits angiogenesis in colon cancer in vivo. HCT116 cells were inoculated into mice to establish a tumor model, as indicated in the materials and methods. Mice bearing tumors were randomly placed into two groups (5 mice/group) and were treated daily with LV-Sineg (control) or LV-SiTEF3-1#3 (treatment) for 31 days. The dose of injection was 1 × 106 TU/ml, and animal weight and tumor volume were measured daily. (A) Mean tumor volumes after treatment. Values represent the means ± SD. *P < 0.05. (B) After 31 days of treatment, the in vivo tumors are shown in the images. (C) The mean values of the body weights of the mice. (D) Mean microvessel density of five fields that were randomly selected in each section after treatment; n = 5 for each group. Representative images of normal mouse tissues; values represent the means ± SD. (E) CD31 and TEF3-1 were evaluated by immunohistochemistry in tumor tissues derived from the SiRNA lentivirus-treated and the control-treated mice. Magnification: × 400.
Moreover, as vascular density could be measured in anti-CD31-stained vasculatures, the density of the vessels inside the tumors was observed in tumor sections after 31 days of treatment. The results of the immunohistochemical analysis showed a significant decrease in microvessel density in the LV-SiTEF3-1-treated group (Figure 6D), which further demonstrates the inhibition of vascular growth in colon cancer in nude mice. In order to evaluate the relationship between TEF3-1 expression and microvessel growth in the LV-SiTEF3-1-treated group and in the control group, an immunohistochemical analysis of mouse tumor tissues was performed using a specific anti-TEF3-1 antibody in the same field. As shown in Figure 6E, normal mouse tumors exhibited high levels of positive staining for both CD31 and TEF3-1 (Figure 6E, panels a and b). Notably, the majority of mouse tumors in the LV-SiTEF3-1-treated group expressed low levels of TEF3-1, even when they expressed CD31 (Figure 6E, panels c and d). Therefore, LV-SiTEF3-1 also exerts antitumor effects, and one of the important consequences is the inhibition of endothelial cell angiogenesis.
DISCUSSION
Remarkably, we found that TEF3-1 plays roles in cell cycle regulation in endothelial cells, and a microarray analysis demonstrated that TEF3-1 overexpression promoted cell adhesion, cell cycle progression and angiogenesis. The inhibition of TEF3-1 was also confirmed in an animal model of colon cancer, where mice that were treated with the corresponding lentiviral vector demonstrated a significant reduction in tumor growth and vascular formation.
Previous studies have suggested that TEF3-1 is required for proliferation, migration, tube formation and angiogenesis of endothelial cells [14]. This may be because TEF3-1 is the transcription factor for VEGF, in addition to TEF3-1 can upregulate downstream genes such as DSCR1-1 L, EDG1, and FGF to induce angiogenesis in endothelial cells [15, 23–25]. Moreover, our data also indicate that VEGFC, VCAM and FOXC were upregulated with nuclear TEF3-1 overexpression in HUVECs, these genes are all involved in angiogenesis, promoting vascular and tumor growth (Supplementary Table S1) [26–28].
As an active transcription factor, TEF3-1 and its family members are also considered as part of the Hippo signaling pathway, which recently has been reported to be a pervasive oncogenic pathway [8, 29]. In this pathway, YAP (Yes-associated protein) promotes cell proliferation and angiogenesis and especially promotes tube formation of human microvascular endothelial cells in human cholangiocarcinoma via TEF3-1 transcription factors [30]. In pancreatic cancer, YAP1 also portends a novel mechanism for an oncogenic gene [5]. For the same reason that the YAP/TEAD complex is an important pathway in gastric cancer, VGLL4 and other peptides that specifically inhibit the YAP/TEAD complex are used to treat GC [4, 13, 31, 32]. Moreover, miRNAs that target the complex may also be developed as a promising therapeutic strategy [33]. For example, in one study, it was found that TEF3-1 interacted with KLF5 and suppressed p27 gene expression in triple negative breast cancers (TNBC) cell lines [34]. Therefore, TEF3-1 might also be a potential target and biomarker for the treatment of breast cancer.
Notably, although YAP was also recently reported to be an efficient anticancer target [5, 35, 36], it was noted that YAP could inhibit the growth of human malignant cells via apoptosis [37]. This indicated that YAP might have a dual function in human malignancies [38]. As a binding partner of YAP, TEF3-1 also exhibited functions both in the regulation of preimplantation mouse embryos [39] and in the regulation of malignant tumor cells [30, 40]. Usually, TEF3-1 and YAP family members are oncogenes [41], then the effect of nuclear location for TEF3-1 also can be cogitated in further study.
Our results suggest that TEF3-1 overexpression is important for its accumulation in the nucleus. IHC experiments support the notion that TEF3-1 remains in the nucleus in GC tumor tissues, while normal tissues did not show the same location and expression patterns of TEF3-1. These results implied that the cellular distribution of TEF3-1 during tumor angiogenesis is regulated by nuclear retention. If TEF3-1 is expressed in the cytoplasm and not in the nucleus, then TEF3-1 cannot regulate tumor angiogenesis. If TEF3-1 was overexpressed and activated, nuclear TEF3-1 would then activate most of the downstream genes that induce cell cycle promotion and angiogenesis (Figure 7).
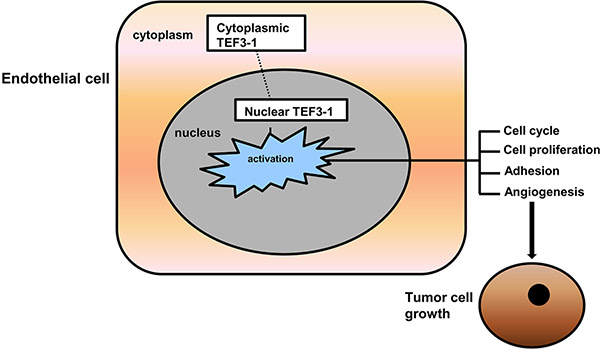
Figure 7: Putative model for the signaling pathway that mediates TEF3-1 induced angiogenesis. It indicates nuclear TEF3-1 regulates HUVECs cell cycle, proliferation, adhesion and angiogenesis, thus promotes tumor growth.
In summary, we have now unveiled a novel role for nuclear TEF3-1 in tumor angiogenesis and in the control of the cell cycle in endothelial cells. Thus, it would be very interesting to further explore the potential of using TEF3-1 nuclear expression as a mechanism-based prognostic biomarker or in combination with other potential markers in prognostic studies. Those data also suggest that the knock down of TEF3-1 may be an attractive antiangiogenic therapeutic target for the treatment of tumors.
MATERIALS AND METHODS
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the authors’ institutional review board in College of Pharmacy, Wuhan University.
Materials
EBM medium for HUVECs was purchased from Lonza (Walkersville, MD, USA). Antibodies against β-tubulin, cyclin D, cyclin E1, CDK2, CDK4, and CDK6 were obtained from Cell Signaling Technology (Boston, MA, USA). Experiments were repeated at least three times.
Cell culture and lentiviral expression
HUVECs were cultured and transduced with lentiviruses that carry various genes, as previously described [16]. Briefly, the lentiviral vector was generated in HEK 293T cells by transfection with three plasmids. HEK 293T cells (5 × 106) were transfected with 5 μg of the transfer plasmid, which encodes the expressed gene, 1.25 μg of the pMD2. G plasmid and 3.75 μg of the psPAX2 plasmid.
Cloning and expression of TEF3-1 and SiRNAs
The TEF3-1 isoform was subjected to DNA sequence analysis. TEF3-1 and its mutations were subcloned into a pHAGE-Flag vector with primers BamHI-TEF3-1F (5′ CGCGGATCCTTGGAGGGCACGGCCGG 3′) and XholI-TEF3-1R (5′ CCGCTCGAGTCATTCTTTCA CCAGCCTGTAGA 3′). The TEF3-1ΔNLS contains the whole TEF3-1 cDNA without the CTGGCTCGTCGCAAA sequence, as described previously [20].
SiRNAs were designed using the website http://www.sirnawizard.com/design.php and were cloned into a pLKO.1 vector. The TEF3-1 SiRNA sequences were as follows: SiTEF3-1#1, GAGAATGGACACTACTCTTAC; SiTEF3-1#2, GACAGAGTATGCTCGCTATGA; SiTEF3-1#3, GCTGTGCATTGCCTATGTCTT.
Cell proliferation assay
The effects of TEF3-1 overexpression on cell proliferation were characterized by cell counts. To assess cellular proliferation, a growth curve was generated after 0.5 × 106 cells/well were seeded into a 6-well tissue culture plate. The cells were counted daily using a hemocytometer.
Cell cycle analysis
After an incubation period of 22 h, various lentiviral vectors were added. HUVEC cells were harvested and treated with 70% ice-cold ethanol overnight. The cells were treated with RNase (50 μg/mL) at 37°C for 1 h and were then treated with PI (20 μg/mL) for 30 min at 4°C in the dark. The DNA content was analyzed by flow cytometry (Beckman Coulter).
Western blot analysis
The cells were harvested and lysed in 1% SDS on ice. The supernatant was collected, and the protein concentration was determined with the Pierce BCA Protein Assay Kit (Thermo Scientific). Equivalent amounts of protein (20 μg) from each sample were transferred and incubated with the specific antibodies; the immunoblots were visualized by chemiluminescence. GAPDH was probed to ensure equal protein loading.
Immunofluorescence
Briefly, cells were fixed in 4% paraformaldehyde, and the slides were blocked with 3% serum in phosphate buffered saline (PBS) with 0.1% Triton-X100. Cells were incubated with the primary antibodies overnight at 4°C and were then washed three times with 0.1% Tween-20 in PBS. Next, the slides were incubated with fluorescent-labeled secondary antibodies, and after several washes, the staining was visualized by fluorescence microscopy.
Immunohistochemistry
Human tissue sections from the tissue microarray were deparaffinized, rehydrated through graded alcohol solutions. Antigen retrieval was performed by heating the sections in a 99°C water bath for 40 minutes. After endogenous peroxidase activity was quenched and nonspecific binding was blocked, the sections were incubated with antibodies to TEF3-1 (Abcam, Cambridge, MA) or CD31 (DAKO, Glostrup, Denmark) at room temperature for 30 minutes. Slides were incubated with the secondary antibody (Flex HRP) for 30 minutes. After the slides were washed, they were incubated with Flex DAB Chromogen and counterstained with Flex Hematoxylin.
Microarray procedures
The cells were harvested and the total RNA was extracted. The commercially available 22 K Human Genome Array was purchased from CapitalBio Corporation (Beijing, China). Labeling, hybridization, washing and scanning were performed according to the standard operating procedure of CapitalBio Corporation. The obtained images were analyzed with LuxScan Version 3.0 (CapitalBio Corp.), which employed the LOWESS normalization method. The different expressed genes were analyzed using Molecular annotation system (MAS) and software Cluster 3.0 [42].
Tumor xenografts
Five-week-old female BALB/c nude mice were obtained from the Animal Model Centre of Hunan Province (Changsha, Hunan, China). The HCT116 cells were implanted into the right flank of each mouse. When the tumor volume reached 50–100 mm3, the mice were randomly distributed into control and treatment groups (n = 5). The control group received the control lentiviral vector (LV-control) in PBS, whereas the treatment group was given LV-SiTEF3-1#3 for 31 days, after which the tumors were dissected, weighed and frozen.
Microvessel staining analysis
As previously described, CD31 was used to stain microvessels in mouse tissue. Frozen sections were stained with rat anti-mCD31 antibody (BD Biosciences, Pharmingen) [43, 44], and the number of vessels was counted [45, 46].
Statistical analysis
Data shown are presented as the means ± S.D. of at least three independent experiments. Statistical significance was considered at P < 0.05 according to Student’s t test.
ACKNOWLEDGMENTS
This study was supported by the National Program on Key Basic Research Project (973 Program, No. 2010CB529804), the National Natural Science Foundation of China (Grant No. 30971456, 81273540, 81472550 and 31400155), and by the Innovation Seed Fund of Wuhan University School of Medicine. We would like to thank Professor Zan Huang, College of Life Science at Wuhan University for his support in the production of recombinant lentivirus. The authors also wish to thank the Center for Medical Research at Wuhan University for help with the flow cytometric analysis.
Abbreviations
TEF3-1, Transcription Enhancer Factor 3 Isoform 1; TEAD4, TEA Domain Family Member 4; YAP1, Yes-associated Protein 1; CDK, Cyclin-dependent Kinase; GC, Gastric Cancer; HUVEC, Human Umbilical Vein Endothelial Cells; HEK 293T cells, Human Embryonic Kidney 293T Cells; HCT116, Human Colon Cancer Cell 116; LV, Lentiviral Vector; NLS, Nuclear Localization Sequence; KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology; DSCR1-1L, Down Syndrome Candidate Region 1 Isoform 1; IHC, Immunohistochemistry.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
1. Yockey CE, Smith G, Izumo S, Shimizu N. cDNA cloning and characterization of murine transcriptional enhancer factor-1-related protein 1, a transcription factor that binds to the M-CAT motif. The Journal of biological chemistry. 1996; 271:3727–3736.
2. Mahoney WM, Jr., Hong JH, Yaffe MB, Farrance IK. The transcriptional co-activator TAZ interacts differentially with transcriptional enhancer factor-1 (TEF-1) family members. The Biochemical journal. 2005; 388:217–225.
3. Home P, Saha B, Ray S, Dutta D, Gunewardena S, Yoo B, Pal A, Vivian JL, Larson M, Petroff M, Gallagher PG, Schulz VP, White KL, et al. Altered subcellular localization of transcription factor TEAD4 regulates first mammalian cell lineage commitment. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:7362–7367.
4. Lim B, Park JL, Kim HJ, Park YK, Kim JH, Sohn HA, Noh SM, Song KS, Kim WH, Kim YS, Kim SY. Integrative genomics analysis reveals the multilevel dysregulation and oncogenic characteristics of TEAD4 in gastric cancer. Carcinogenesis. 2014; 35:1020–1027.
5. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, Chang Q, Chu GC, Al-Khalil R, et al. Yap1 activation enables bypass of oncogenic kras addiction in pancreatic cancer. Cell. 2014; 158:185–197.
6. Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:E2441–2450.
7. Xie Q, Chen J, Feng H, Peng S, Adams U, Bai Y, Huang L, Li J, Huang J, Meng S, Yuan Z. YAP/TEAD-mediated transcription controls cellular senescence. Cancer research. 2013; 73:3615–3624.
8. Stanger BZ. Quit your YAPing: a new target for cancer therapy. Genes & development. 2012; 26:1263–1267.
9. Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nature cell biology. 2011; 13:877–883.
10. Pan D. The hippo signaling pathway in development and cancer. Developmental cell. 2010; 19:491–505.
11. Hiemer SE, Szymaniak AD, Varelas X. The Transcriptional Regulators TAZ and YAP Direct Transforming Growth Factor beta-induced Tumorigenic Phenotypes in Breast Cancer Cells. The Journal of biological chemistry. 2014; 289:13461–13474.
12. Serrano I, McDonald PC, Lock F, Muller WJ, Dedhar S. Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nature communications. 2013; 4:2976.
13. Zhang W, Gao Y, Li P, Shi Z, Guo T, Li F, Han X, Feng Y, Zheng C, Wang Z, Li F, Chen H, Zhou Z, et al. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell research. 2014; 24:331–343.
14. Shie JL, Wu G, Wu J, Liu FF, Laham RJ, Oettgen P, Li J. RTEF-1, a novel transcriptional stimulator of vascular endothelial growth factor in hypoxic endothelial cells. The Journal of biological chemistry. 2004; 279:25010–25016.
15. Messmer-Blust AF, Zhang C, Shie JL, Song Q, He P, Lubenec I, Liu Y, Sellke F, Li J. Related transcriptional enhancer factor 1 increases endothelial-dependent microvascular relaxation and proliferation. Journal of vascular research. 2012; 49:249–259.
16. Qiao C, Jiang Y, Deng C, Huang Z, Teng K, Chen L, Liu X. Characterization of the transcriptional activation domains of human TEF3–1 (transcription enhancer factor 3 isoform 1). Archives of biochemistry and biophysics. 2015; 569:54–61.
17. Xia Y, Chang T, Wang Y, Liu Y, Li W, Li M, Fan HY. YAP promotes ovarian cancer cell tumorigenesis and is indicative of a poor prognosis for ovarian cancer patients. PloS one. 2014; 9:e91770.
18. Tao J, Calvisi DF, Ranganathan S, Cigliano A, Zhou L, Singh S, Jiang L, Fan B, Terracciano L, Armeanu-Ebinger S, Ribback S, Dombrowski F, Evert M, et al. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology. 2014; 147:690–701.
19. Fujii M, Nakanishi H, Toyoda T, Tanaka I, Kondo Y, Osada H, Sekido Y. Convergent signaling in the regulation of connective tissue growth factor in malignant mesothelioma: TGFbeta signaling and defects in the Hippo signaling cascade. Cell Cycle. 2012; 11:3373–3379.
20. Liu X, Zhao D, James L, Li J, Zeng H. Requirement of the nuclear localization of transcription enhancer factor 3 for proliferation, migration, tube formation, and angiogenesis induced by vascular endothelial growth factor. FASEB journal. 2011; 25:1188–1197.
21. Messmer-Blust AF, Philbrick MJ, Guo S, Wu J, He P, Guo S, Li J. RTEF-1 attenuates blood glucose levels by regulating insulin-like growth factor binding protein-1 in the endothelium. Circulation research. 2012; 111:991–1001.
22. Suzuki M, Kobayashi H, Tanaka Y, Hirashima Y, Kanayama N, Takei Y, Saga Y, Suzuki M, Itoh H, Terao T. Bikunin target genes in ovarian cancer cells identified by microarray analysis. The Journal of biological chemistry. 2003; 278:14640–14646.
23. Liu X, Zhao D, Qin L, Li J, Zeng H. Transcription enhancer factor 3 (TEF3) mediates the expression of Down syndrome candidate region 1 isoform 1 (DSCR1–1L) in endothelial cells. The Journal of biological chemistry. 2008; 283:34159–34167.
24. He P, Philbrick MJ, An X, Wu J, Messmer-Blust AF, Li J. Endothelial differentiation gene-1, a new downstream gene is involved in RTEF-1 induced angiogenesis in endothelial cells. PloS one. 2014; 9:e88143.
25. Tabata S, Ikeda R, Yamamoto M, Shimaoka S, Mukaida N, Takeda Y, Yamada K, Soga T, Furukawa T, Akiyama S. Thymidine phosphorylase activates NFkappaB and stimulates the expression of angiogenic and metastatic factors in human cancer cells. Oncotarget. 2014; 5:10473–10485. doi: 10.18632/oncotarget.2242.
26. Schlesinger M, Bendas G. Vascular cell adhesion molecule-1 (VCAM-1)—an increasing insight into its role in tumorigenicity and metastasis. International journal of cancer. 2015; 136:2504–2514.
27. Cai J, Tian AX, Wang QS, Kong PZ, Du X, Li XQ, Feng YM. FOXF2 suppresses the FOXC2-mediated epithelial-mesenchymal transition and multidrug resistance of basal-like breast cancer. Cancer letters. 2015; 367:129–137.
28. Carpenter RL, Paw I, Zhu H, Sirkisoon S, Xing F, Watabe K, Debinski W, Lo HW. The gain-of-function GLI1 transcription factor TGLI1 enhances expression of VEGF-C and TEM7 to promote glioblastoma angiogenesis. Oncotarget. 2015; 6:22653–22665. doi: 10.18632/oncotarget.4248.
29. Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L, Claerhout S, Fiers M, Journe F, et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nature communications. 2015; 6:6683.
30. Marti P, Stein C, Blumer T, Abraham Y, Dill MT, Pikiolek M, Orsini V, Jurisic G, Megel P, Makowska Z, Agarinis C, Tornillo L, Bouwmeester T, et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology. 2015; 62:1497–1510.
31. Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W, Wang X, Guo T, Li P, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer cell. 2014; 25:166–180.
32. Clattenburg L, Wigerius M, Qi J, Rainey JK, Rourke JL, Muruganandan S, Sinal CJ, Fawcett JP. NOS1AP Functionally Associates with YAP To Regulate Hippo Signaling. Molecular and cellular biology. 2015; 35:2265–2277.
33. Li N, Yu N, Wang J, Xi H, Lu W, Xu H, Deng M, Zheng G, Liu H. miR-222/VGLL4/YAP-TEAD1 regulatory loop promotes proliferation and invasion of gastric cancer cells. American journal of cancer research. 2015; 5:1158–1168.
34. Wang C, Nie Z, Zhou Z, Zhang H, Liu R, Wu J, Qin J, Ma Y, Chen L, Li S, Chen W, Li F, Shi P, et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget. 2015; 6:17685–17697. doi: 10.18632/oncotarget.3779.
35. Perra A, Kowalik MA, Ghiso E, Ledda-Columbano GM, Di Tommaso L, Angioni MM, Raschioni C, Testore E, Roncalli M, Giordano S, Columbano A. YAP activation is an early event and a potential therapeutic target in liver cancer development. Journal of hepatology. 2014; 61:1088–1096.
36. Cai J, Maitra A, Anders RA, Taketo MM, Pan D. beta-Catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes & development. 2015; 29:1493–1506.
37. Lapi E, Di Agostino S, Donzelli S, Gal H, Domany E, Rechavi G, Pandolfi PP, Givol D, Strano S, Lu X, Blandino G. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Molecular cell. 2008; 32:803–814.
38. Wang H, Du YC, Zhou XJ, Liu H, Tang SC. The dual functions of YAP-1 to promote and inhibit cell growth in human malignancy. Cancer metastasis reviews. 2014; 33:173–181.
39. Nishioka N, Yamamoto S, Kiyonari H, Sato H, Sawada A, Ota M, Nakao K, Sasaki H. Tead4 is required for specification of trophectoderm in pre-implantation mouse embryos. Mechanisms of development. 2008; 125:270–283.
40. Rodriguez-Segui SA, Bessa J. Gatekeepers of pancreas: TEAD and YAP. Oncotarget. 2015; 6:15736–15737. doi: 10.18632/oncotarget.4607.
41. Perra A, Kowalik MA, Giordano S, Columbano A. Reply to: “YAP in tumorigenesis: Friend or foe?”. Journal of hepatology. 2015; 62:1445.
42. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005; 436:518–524.
43. Koukourakis MI, Giatromanolaki A, Thorpe PE, Brekken RA, Sivridis E, Kakolyris S, Georgoulias V, Gatter KC, Harris AL. Vascular endothelial growth factor/KDR activated microvessel density versus CD31 standard microvessel density in non-small cell lung cancer. Cancer research. 2000; 60:3088–3095.
44. Qin L, Zhao D, Liu X, Nagy JA, Hoang MV, Brown LF, Dvorak HF, Zeng H. Down syndrome candidate region 1 isoform 1 mediates angiogenesis through the calcineurin-NFAT pathway. Mol Cancer Res. 2006; 4:811–820.
45. Davani S, Marandin A, Mersin N, Royer B, Kantelip B, Herve P, Etievent JP, Kantelip JP. Mesenchymal progenitor cells differentiate into an endothelial phenotype, enhance vascular density, and improve heart function in a rat cellular cardiomyoplasty model. Circulation. 2003; 108 Suppl 1:II253–258.
46. Xiao W, Jiang Y, Men Q, Yuan L, Huang Z, Liu T, Li W, Liu X. Tetrandrine induces G1/S cell cycle arrest through the ROS/Akt pathway in EOMA cells and inhibits angiogenesis in vivo. International journal of oncology. 2015; 46:360–368.