
INTRODUCTION
TP53 is a tumor suppressor gene that encodes the p53 protein, which acts as a transcription factor and induces cell cycle arrest, apoptosis, and cellular senescence. [1] Mutations in the coding sequence of TP53 gene are one of the most common mechanisms of p53 deregulation, and they can be detected in more than 50% of all cancers. [2]
In myelodysplastic syndromes (MDS), TP53 mutations are detected in approximately 5-20% of cases when modern deep sequencing methods are used. [3-8] Previous studies have consistently shown that TP53 mutations are associated with higher-risk MDS, therapy-related disease, complex cytogenetics (including chromosome 5, 7 and 17 abnormalities), and poor overall survival. [4, 7-10] With regards to treatment response, TP53 mutations are associated with resistance to cytarabine-based chemotherapy in MDS and acute myeloid leukemia (AML). [11, 12] In recent years, hypomethylating agents (HMA) such as 5-azacitidine or decitabine, has become the standard of care in higher risk MDS patients. [13, 14] However, it is not well understood whether TP53 mutation status also predicts resistance to HMA therapy in patients with MDS. Further, while prognostic impact of TP53 mutations has been well described, because of the strong correlation with other poor prognostic factors, such as complex karyotype, the independent prognostic value of TP53 mutations is not clearly understood.
To better understand independent prognostic and predictive value of TP53 mutations in the context of HMA therapy, we sequenced TP53 in a large cohort of MDS patients who were treated with HMA therapy.
RESULTS
Patient characteristics
Clinical characteristics of the 168 patients at the time of sequencing are listed in Table 1. More than half of the patients were classified as having RAEB and 11% of them were RAEB-T. Compared to other published MDS genomics studies, our cohort included fewer patients with refractory anemia with ringed sideroblasts (RARS). [7, 10, 22] Majority of the patients had higher risk MDS and 56% of the patients were classified as high or very high risk by IPSS-R.
Table 1: Baseline clinical characteristics of the 168 patients with MDS who were screened for TP53 mutation and were treated with HMA therapy
Characteristics |
N or median (% or range) |
|---|---|
Median age, range, y |
67 (17-89) |
Female |
64 (38) |
Pathological classification |
|
5q- syndrome |
2 (1) |
RA |
16 (9) |
RCMD |
27 (16) |
RCMD-RS |
1 (<1) |
RAEB-1 |
37 (22) |
RAEB-2 |
32 (19) |
RAEB-T |
19 (11) |
RARS |
3 (2) |
MDS-U |
1 (<1) |
MDS/MPD |
1 (<1) |
CMML-1 |
21 (13) |
CMML-2 |
8 (5) |
Therapy-related MN |
40 (24) |
Median WBC count, range, x 109/L |
3.2 (0.6-162.0) |
Median ANC count, range, x 109/L |
1.3 (0.0-103.7) |
Median HGB count, range, g/dL |
9.3 (6.0-15.8) |
Median PLT count, range, x 109/L |
62 (2-655) |
Median BM blast count, range, % |
7 (0-29) |
Normal karyotype |
54 (32) |
Complex karyotype |
48 (29) |
Monosomal karyotype |
43 (26) |
Deletion 17p/monosomy 17 |
15 (9) |
IPSS-R cytogenetic risk category |
|
Very good |
12 (7) |
Good |
53 (32) |
Intermediate |
51 (30) |
Poor |
3 (2) |
Very Poor |
47 (28) |
Unknown |
2 (1) |
Overall IPSS-R risk |
|
Very good |
9 (5) |
Good |
18 (11) |
Intermediate |
44 (26) |
High |
41 (24) |
Very high |
53 (32) |
Unknown |
3 (2) |
Abbreviations. RA: refractory anemia, RCMD: refractory cytopenia with multilineage dysplasia, RCMD-RS: refractory cytopenia with multilineage dysplasia with ringed sideroblast, RAEB: refractory anemia with excess blast, RARS: refractory anemia with ringed sideroblast, MDS-U: myelodysplastic syndromes unclassified, MDS/MPD: myelodysplastic syndromes/myeloproliferative disease, MN: myeloid neoplasia, WBC: white blood cell, ANC: absolute neutrophil cells, HGB: hemoglobin, PLT: platelet, BM bone marrow, IPSS-R: Revised International Prognostic Scoring System.
*complex karyotype is defined as having > 3 chromosomal abnormalities.
† monosomal karyotype is defined as having at least 2 autosomal monosomies or a single autosomal monosomy associated with at least one structural chromosomal abnormality.
†† IPSS-R cytogenetic risk stratification is described as previously. [17]
Landscape of TP53 mutations
In total, 45 TP53 mutations were detected in 38 patient samples (23%). Thirty-one patients had single TP53 mutations, and 7 had double TP53 mutations identified. Eighty-seven percent of the detected mutations were missense, 9% were nonsense, and 4% were frameshift indels. Among the missense and nonsense mutations, 69% were transition and 31% were transversion, and C/G>T/A alteration was most common. Mutations in TP53 were predominantly detected in the core DNA-binding domain (93%), and only 2 mutations were detected in the tetramerization domain (Figure 1). The most frequently mutated codon was codon 272 (9%), followed by codons 273 (7%) and 248 (7%). Ninety-four percent of the detected missense mutations were predicted to be non-functional for transcriptional activity. [23] Ninety-five percent of them were predicted to be deleterious according to the SIFT algorithm. [24] The median variant allele frequency (VAF) of the TP53 mutations was 35.4% (range: 8.9-93.3), and 95% of the detected TP53 mutations had VAF ≥ 10% (Supplemental Figure 1).
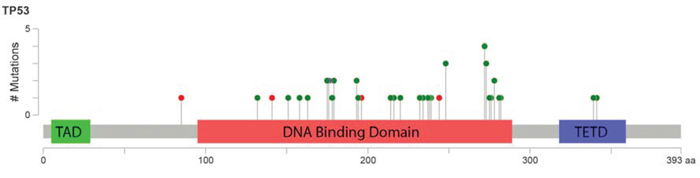
Figure 1: Lollipop figure of TP53 mutations detected in 168 patients with MDS and CMML. Green dots indicate missense mutations and red dots indicate nonsense mutations. The figure for was created using cBioPortal website (http://www.cbioportal.org/).
Correlation with other mutations
For the patients whose WES data were available (N = 53), we investigated degree of co-occurrence and mutual exclusivity with other previously well-characterized myeloid driver mutations (Figure 2). Seven of 10 TP53 mutated cases did not have any other co-occurring driver mutations, and this is consistent with other genomic studies that TP53 mutated cases carry few co-occurring mutations. [3, 6, 10] Although statistically not significant, TP53 mutation tended to be mutually exclusive to mutations in one of the RNA splicing pathway genes (U2AF1, SRSF2, SF3B1, and ZRSR2, P = 0.07).
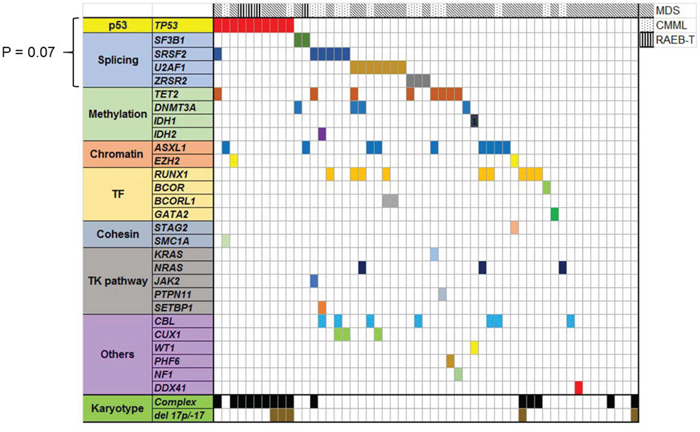
Figure 2: Landscape of well characterized myeloid driver mutations in 53 MDS/CMML patients whose bone marrow samples were sequenced by WES. TP53 mutated cases had less co-occuring mutations. TP53 mutation and splicing gene mutations had trend to be mutually exclusive (P = 0.07).
TP53 mutations and clinical characteristics
Table 2 compares clinical characteristics of MDS patients based on TP53 mutational status. By pathological classification, patients with RAEB-T had the highest frequency of TP53 mutations (47%). TP53 mutations were more frequently detected in therapy-related disease than in de novo disease. Patients with TP53 mutations were also significantly more neutropenic, thrombocytopenic, and had higher bone marrow blasts at presentation. Furthermore, TP53 mutation status was significantly associated with complex and monosomal karyotypes and 17p deletion / monosomy 17. Thirteen patients had both TP53 mutations and 17p deletion / monosomy 17. The majority of the TP53 mutations were detected in patients with IPSS-R high or very high-risk disease.
Table 2: Comparison of clinical characteristics between TP53 mutated patients and wild type (WT) patients
|
TP53 mutated |
TP53 WT |
|
|---|---|---|---|
N = 38 (%) |
N = 130 (%) |
P value |
|
WHO classification |
|
|
|
5q- syndrome |
1 (3) |
1 (<1) |
NA |
RA |
3 (8) |
13 (1) |
NA |
RCMD |
2 (5) |
25 (19) |
NA |
RCMD-RS |
0 (0) |
1 (<1) |
NA |
RAEB-1 |
10 (26) |
27 (21) |
NA |
RAEB-2 |
10 (26) |
22 (17) |
NA |
RAEB-T |
9 (24) |
10 (8) |
NA |
RARS |
1 (3) |
2 (2) |
NA |
MDS-U |
0 (0) |
1 (<1) |
NA |
MDS/MPD |
0 (0) |
1 (<1) |
NA |
CMML-1 |
1 (3) |
20 (15) |
NA |
CMML-2 |
1 (3) |
7 (5) |
NA |
Therapy-related MN, (%) |
16 (40)* |
24 (60)* |
0.003 |
De novo disease, (%) |
22 (17)† |
106 (83)† |
|
Median WBC count, range, x 109/L |
2.9 (1.0-29.5) |
3.7 (0.6-162.0) |
0.02 |
Median ANC count, range, x 109/L |
0.9 (0.05-21.5) |
1.4 (0.0-103.7) |
0.02 |
Median HGB count, range, g/dL |
9.3 (6.8-12.8) |
9.4 (6.0-15.8) |
0.76 |
Median PLT count, range, x 109/L |
47 (9-290) |
73 (2-655) |
0.008 |
Median BM blast count, range, % |
10 (0-29) |
6 (0-30) |
0.006 |
Cytogenetics |
|
|
|
Complex karyotype |
32 (84) |
16 (12) |
<0.001 |
Deletion 17p/-17 |
13 (34) |
2 (2) |
<0.001 |
Monosomal karyotype |
33 (87) |
10 (8) |
<0.001 |
IPSS-R |
|
|
|
Very Low |
1 (3) |
8 (6) |
<0.001 |
Low |
0 (0) |
18 (14) |
|
Intermediate |
1 (3) |
43 (33) |
|
High |
5 (13) |
36 (28) |
|
Very high |
30 (79) |
23 (18) |
|
Unknown |
1 (3) |
2 (2) |
|
Abbreviations. RA: refractory anemia, RCMD: refractory cytopenia with multilineage dysplasia, RCMD-RS: refractory cytopenia with multilineage dysplasia with ringed sideroblast, RAEB: refractory anemia with excess blast, RARS: refractory anemia with ringed sideroblast, MDS-U: myelodysplastic syndromes unclassified, MDS/MPD: myelodysplastic syndromes/myeloproliferative disease, MN: myeloid neoplasia, WBC: white blood cell, ANC: absolute neutrophil cells, HGB: hemoglobin, PLT: platelet, BM bone marrow, IPSS-R: Revised International Prognostic Scoring System.
*Denominator is total number of therapy-related MN.
†Denominator is total number of de novo disease.
Treatment response to HMA therapy
Complete response (CR) and overall response (OR) were observed in 49 patients (29%) and 57 patients (34%), respectively. Table 3 summarizes the association between various clinical characteristics and response to HMA therapy. Patients with thrombocytopenia at baseline had a significantly worse OR rate than patients with higher platelet count (23% versus 41%, P = 0.01). There was a non-significant trend toward better OR rate in patients with neutropenia than patients with higher neutrophil count (43% versus 28%, P = 0.06). No significant difference was observed in response rate by ages, type of therapy, extent of anemia, IPSS-R risk groups, and cytogenetic abnormalities. The HMA response rates were similar between patients with TP53 mutations and WT TP53 (TP53 mutated vs. WT, CR: 34% vs. 27%, P = 0.38, OR: 45% vs. 32%, P = 0.13). Response rate was not different by TP53 mutation status in patients sub-grouped by treatment types (SOC HMA versus HMA combination with investigational agents) (Supplemental Table 3). Correlations between treatment response and other myeloid driver mutations are described in Supplemental Table 4. In patients who were tested for TET2 mutation (N = 79), there was no significant difference observed in response to HMA therapy by TET2 mutation status (TET2 mutated vs. WT, CR: 17% vs. 36%, P = 0.10, OR: 22% vs. 38%, P = 0.18). We also tested response rate based on both TET2 and ASXL1 mutation status because previous publication suggested high response rate in TET2 mutated but ASXL1 WT patients. [3] However, we did not see significant difference in response rate based on TET2 and ASXL1 mutation status (TET2 mutated/ASXL1 WT vs. other, CR: 20% vs. 34%, P = 0.23, OR: 27% vs. 36%, P = 0.36). In the current cohort, patients with mutation in RAS (KRAS and/or NRAS) had significantly worse response to HMA therapy (RAS mutated vs. WT, CR: 8% vs. 31%, P = 0.06, OR: 8% vs. 36%, P = 0.03). Although statistically not significant, DNMT3A mutation and mutation in one of the splicing pathway gene were associated with trend toward worse OR to HMA (Supplemental Table 4).
Table 3: Various clinical factors including TP53 mutation status and response to HMA therapy
Variables |
Number |
CR rate (%) |
P value |
OR rate (%) |
P value |
|---|---|---|---|---|---|
All patients |
168 |
49 (29) |
NA |
57 (34) |
NA |
Age < 70 |
102 |
34 (33) |
0.14 |
39 (38) |
0.14 |
Age ≥ 70 |
66 |
15 (23) |
|
18 (27) |
|
MDS (including RAEB-T) |
139 |
40 (29) |
0.81 |
47 (34) |
0.95 |
CMML |
29 |
9 (31) |
|
10 (35) |
|
WHO classification |
|
|
0.14 |
|
0.06 |
5q- syndrome |
2 |
1 (50) |
|
1 (50) |
|
RA |
16 |
3 (19) |
|
3 (19) |
|
RCMD |
27 |
5 (19) |
|
5 (19) |
|
RCMD-RS |
1 |
0 (0) |
|
0 (0) |
|
RAEB-1 |
37 |
11 (30) |
|
14 (38) |
|
RAEB-2 |
32 |
7 (22) |
|
9 (28) |
|
RAEB-T |
19 |
10 (53) |
|
11 (58) |
|
RARS |
3 |
2 (67) |
|
2 (67) |
|
MDS-U |
1 |
0 (0) |
|
1 (100) |
|
MDS/MPD |
1 |
1 (100) |
|
1 (100) |
|
CMML-1 |
21 |
5 (24) |
|
6 (29) |
|
CMML-2 |
8 |
4(50) |
|
4 (50) |
|
Therapy-related MN |
128 |
38 (30) |
0.79 |
44 (34) |
0.83 |
De novo disease |
40 |
11 (28) |
|
13 (33) |
|
ANC ≥ 0.8 x 109/L |
109 |
26 (24) |
0.06 |
31 (28) |
0.06 |
ANC < 0.8 x 109/L |
58 |
22 (38) |
|
25 (43) |
|
HGB ≥ 8 g/dL |
148 |
44 (30) |
0.66 |
52 (35) |
0.37 |
HGB < 8 g/dL |
20 |
5 (25) |
|
5 (25) |
|
PLT ≥ 50 x 109/L |
102 |
35 (34) |
0.07 |
42 (41) |
0.01 |
PLT < 50 x 109/L |
66 |
14 (21) |
|
15 (23) |
|
BM blast ≤ 10% |
111 |
29 (26) |
0.17 |
33 (30) |
0.12 |
BM blast > 10% |
55 |
20 (36) |
|
23 (42) |
|
Cytogenetics |
|
|
|
|
|
Non complex |
114 |
34 (30) |
0.93 |
39 (34) |
0.88 |
Complex |
48 |
14 (30) |
|
17 (35) |
|
Non-monosmal |
119 |
34 (29) |
0.62 |
39 (33) |
0.42 |
Monosomal |
43 |
14 (33) |
|
17 (40) |
|
IPSS-R |
|
|
|
|
|
Very Low/Low/Int |
72 |
19 (26) |
0.60 |
23 (32) |
0.74 |
High/Very high |
93 |
28 (30) |
|
32 (34) |
|
Therapy types |
|
|
|
|
|
SOC Aza/DAC |
78 |
22 (28) |
0.88 |
25 (32) |
0.71 |
HMA+investigational |
90 |
27 (30) |
|
32 (35) |
|
TP53-mutated |
38 |
13 (34) |
0.38 |
15 (45) |
0.13 |
TP53 WT |
130 |
35 (27) |
|
41 (32) |
|
Abbreviations. CR: complete response, OR: overall response, RA: refractory anemia, RCMD: refractory cytopenia with multilineage dysplasia, RCMD-RS: refractory cytopenia with multilineage dysplasia with ringed sideroblast, RAEB: refractory anemia with excess blast, RARS: refractory
Time to response and response duration in TP53 mutated patients
For responders, there was no difference in time to response between TP53-mutated patients and WT patients (1.9 month vs. 2.3 month in TP53-mutated and WT patients, respectively, P = 0.08; Figure 3A). However, TP53-mutated patients had significantly shorter response duration compared to WT patients (5.7 months vs. 28.5 months in TP53-mutated and WT patients, respectively, P = 0.003, Figure 3B).
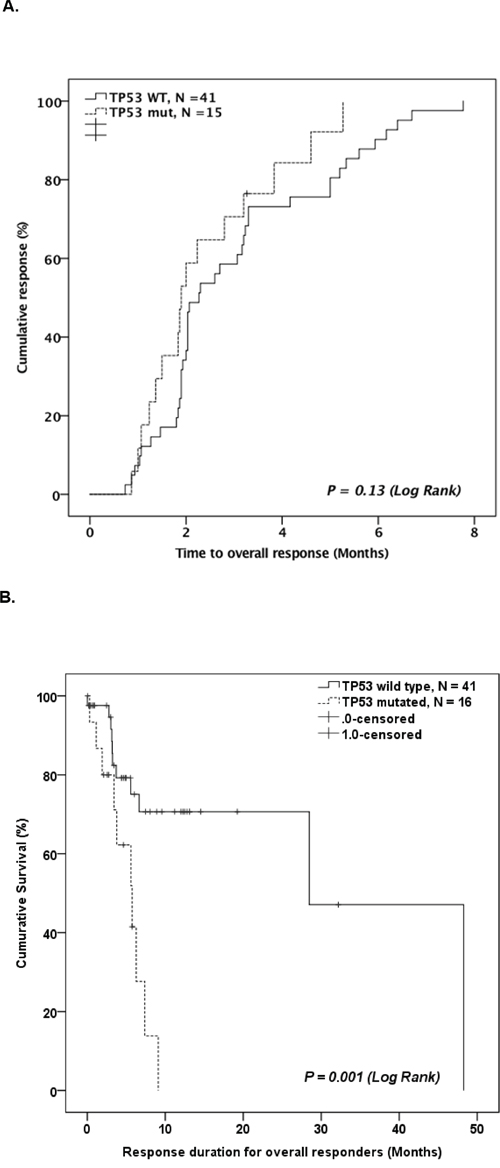
Figure 3: A. Kaplan-Meier curve comparing time to best response for TP53 mutated patients and TP53 WT patients who responded to HMA therapy. B. Kaplan-Meier curve comparing duration of response for TP53 mutated patients and TP53 WT patients who responded to HMA therapy.
Survival outcome
The median OS of the studied patients was 14.8 months (95% CI: 11.8-17.7 months). TP53 mutated patients had significantly worse OS compared to WT patients in both entire cohort (median 9.4 months [95% CI: 6.9-11.9] vs. 20.7 months [95% CI: 16.4-25.0], P <0.001; Figure 4A) and in patients subgrouped by treatment type (Supplemental Figure 2). TP53 mutation status identified a distinct prognostic group of patients among the higher-risk patient groups. In the IPSS-R very high-risk group, TP53-mutated patients had significantly worse OS compared to WT patients (Figure 4B). Further, in a group of patients who had complex karyotypes, survival of TP53 mutated patients was significantly worse than that of WT patients (Figure 4C). On the other hand, TP53 mutation status did not differentiate survival outcome among monosomal karyotype patients (Figure 4D). Other clinical and mutational factors that were significantly prognostic to OS in univariate analysis are therapy-related disease, complex karyotype, monosomal karyotype, and IPSS-R high or very high risk group (Supplemental Table 5).
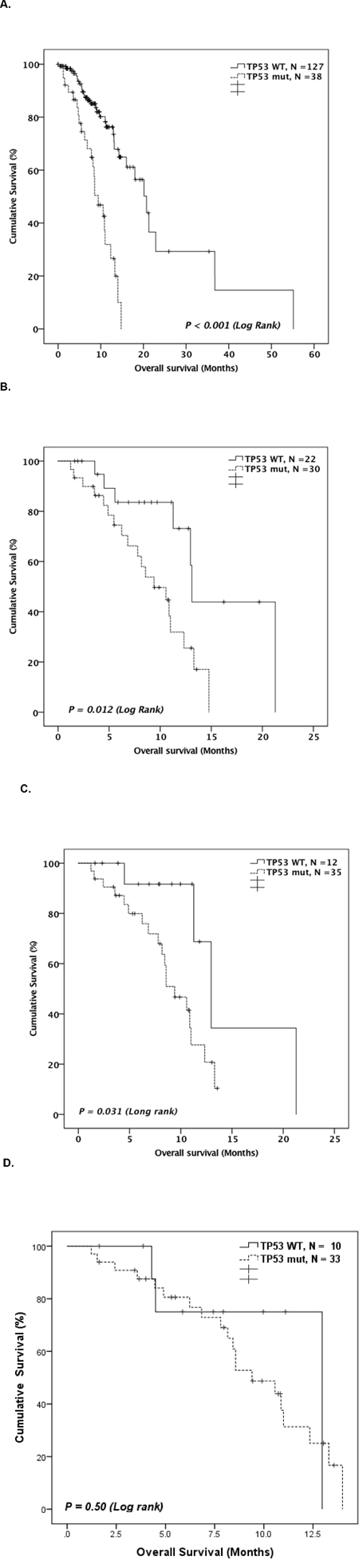
Figure 4: A. Kaplan-Meier curve comparing OS of TP53 mutated patients and TP53 WT patients. Kaplan-Meier curve comparing overall OS of TP53 mutated patients and TP53 WT patients among. B. patients with IPSS-R high or very high risk (N = 52). C. patients with complex karyotypes (N = 47), D. patients with monosomal karyotypes (N = 43).
Multivariate analysis considering variables IPSS-R high and very high-risk (vs. others), TP53 mutation (vs. WT), complex karyotype (vs. others), monosomal karyotype (vs. others), and therapy-related disease (vs. de novo) were conducted. Because complex karyotype and monosomal karyotype are strongly correlated, they were tested in separate models. In both models, TP53 mutation status showed statistically significant negative impact on OS (Table 4). The number of patients who underwent HSCT was not statistically different between TP53-mutated and WT patients (9 patients for TP53-mutated [24%] versus 26 patients for TP53 WT [20%], P = 0.62). For patients who underwent HSCT, survival outcome after HSCT was not statistically different between TP53-mutated and WT patients (median OS after HSCT, TP53-mutated vs. WT, 6.3 months vs. 8.8 months, P = 0.95).
Table 4: Multivariate analysis for overall survival in MDS patients treated with HMA therapy
Model 1 |
|
|
|
Reduced Model |
|
|
|---|---|---|---|---|---|---|
|
HR |
95% CI |
P –value |
HR |
95% CI |
P –value |
TP53 mutation (mutated vs. WT) |
3.31 |
1.20-9.08 |
0.02 |
3.01 |
1.58-5.69 |
0.0007 |
IPSS-R risk (high/very high vs. very low/low/intermediate) |
2.24 |
1.04-4.83 |
0.04 |
2.31 |
1.08-4.94 |
0.03 |
Therapy-related (yes vs. de novo) |
1.79 |
0.98-3.28 |
0.06 |
|
|
|
Monosomal karyotype (monosomal vs. non-monosomal) |
0.86 |
0.31-2.40 |
0.77 |
|
|
|
Model 2 |
||||||
|
HR |
95% CI |
P –value |
|
|
|
TP53 mutation (mutated vs. WT) |
3.66 |
1.53-8.75 |
0.004 |
|
|
|
IPSS-R risk (high/very high vs. very low/low/intermediate) |
2.35 |
1.08-5.12 |
0.03 |
|
|
|
Therapy-related (yes vs. de novo) |
1.77 |
0.97-3.23 |
0.06 |
|
|
|
Complex karyotype (complex vs. non-complex) |
0.73 |
0.3-1.75 |
0.47 |
|
|
|
Longitudinal follow up of TP53 mutation
Of the 38 patients with TP53 mutations, 13 had at least one longitudinal sample sequenced for TP53 mutations after HMA therapy. Clinical course and TP53 mutation follow-up of these patients is summarized in Figure 5 and Supplemental Figure 3. Seven of 13 patients achieved initial CR with HMA therapy. All 7 eventually lost response and progressed and at progression all 7 of these patients were identified to have the same TP53 mutations. Two patients who underwent HSCT later relapsed. At the time of relapse, the same TP53 mutations were also detected in those patients’ bone marrow. Overall, except for two patients, the same TP53 mutations were persistently detected at the time of disease progression or relapse.
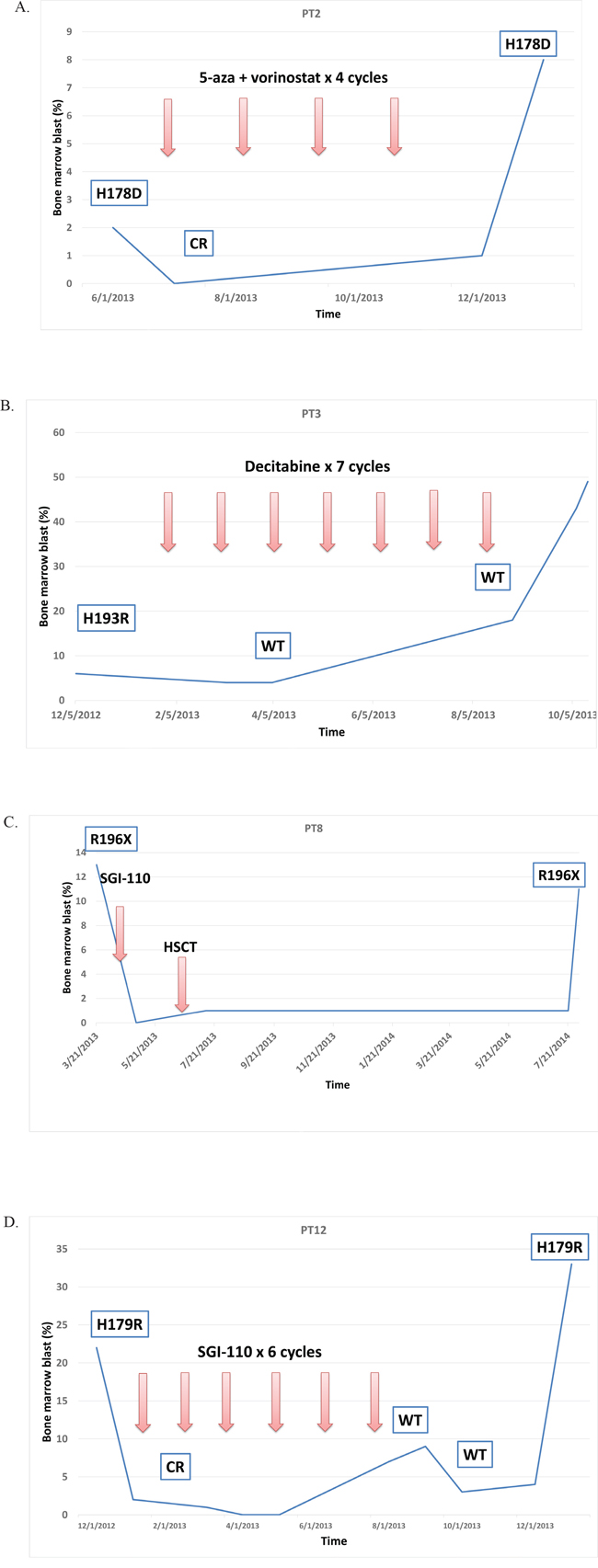
Figure 5: Representative cases of longitudinal TP53 follow up. Except for PT3 case, the same TP53 mutations persisted at times of disease progression or relapse. A. The patient PT2 had TP53 p.H178D mutation. Received 4 cycles of 5-azacitidine and vorinostat achieving complete response (CR). When disease progressed, the same TP53 p.H178D mutation was detected. B. The patient PT3 had TP53 p.H193R mutation Received 7 cycles of standard of care decitabine and after 3 cycles, TP53 mutation became negative on bone marrow. When disease transformed to AML, TP53 was still wild type (WT). C. The patient PT8 had TP53 p.R196* nonsense mutation and received 1 cycle of guadecitabine (SGI-110) followed by allogeneic hematopoietic stem cell transplant (HSCT). One year later, when disease relapsed, the same TP53 p.R196* mutation was detected in bone marrow. D. The patient PT12 had TP53 p. H179R mutation. Received 6 cycles of guadecitabine (SGI-110) and achieved CR. TP53 sequencing showed WT. However, when disease relapsed, the same TP53 p. H179R was detected in bone marrow.
DISCUSSION
In this study, we investigated mutational landscape, clinical correlation, prognostic and predictive value of TP53 mutations in MDS patients who were treated with HMA therapy. Several important findings emerged from this study.
First, we confirmed the strong independent prognostic significance of TP53 mutations in MDS. Because TP53 mutations are strongly associated with other poor prognostic factors, such as complex karyotypes, monosomal karyotype, and therapy-related disease, their independent prognostic value has been debated. In this study, TP53 mutation status identified a distinct prognostic group among patients with complex karyotypes or IPSS-R-defined very high-risk patients. In multivariate analysis for OS, the hazard ratio of TP53 mutation was the strongest among other predictors of poor prognosis, such as complex karyotype and IPSS-R high/very high risk. Similar findings were confirmed in AML patients with complex karyotype. [12] Taken together, these findings suggest that screening for TP53 mutations in addition to conventional karyotyping helps identifying the highest risk groups of MDS patients. Although, majority of TP53 mutated cases already have other poor prognostic markers and additional impact on prognosis is somewhat limited.
Second, despite their poor prognosis, TP53-mutated MDS patients responded to HMA therapy as well as WT patients. This is in contrast to the resistance observed in patients with TP53-mutated AML and MDS when they are treated with cytarabine-based cytotoxic chemotherapy. [11, 12] Recently, Bejar et al. analyzed the association between various somatic mutations and response to HMA therapy in MDS. They found that TET2 mutations were associated with favorable response to HMA therapy but TP53 mutations did not predict response to HMA therapy. [3] Our data confirms that TP53 mutations do not predict for HMA response.
Despite equivalent response to HMA therapy, duration of response was significantly shorter in TP53-mutated patients compared to WT patients. The median response duration was approximately 6 months in TP53-mutated patients, and virtually all patients who responded to HMA therapy lost response within 10 months. In a subset of patients who had longitudinal follow-up for TP53 mutations, the same TP53 mutations almost always persisted at the time of disease progression. These findings suggest that despite its efficacy, HMA therapy is not capable of eliminating abnormal hematopoietic clones with TP53 mutations. Similar findings of persistent TP53 mutations after therapy have also been reported in cases with 5q- syndrome. [5]
From a clinical perspective, our findings do not discourage the use of HMA therapy in TP53-mutated cases but rather support its use. Considering that TP53 mutations have been associated with resistance to cytarabine based chemotherapy [11, 12], better response is expected with HMA therapy. However, based on our findings, most responders will lose their responses within the first year of therapy. A novel strategy to extend response is clearly needed for TP53-mutated MDS patients. Unfortunately, current HSCT strategies have not proven to be the answer as recent studies have indicated that TP53-mutated MDS patients have dismal outcomes after HSCT [22], although in our limited number of patients who underwent HSCT, survival after HSCT was not different between TP53-mutated and WT patients. Patients did not receive pre-HSCT HMA therapy in the previous study and all of our studied patients received HMA therapy prior to HSCT, so more analysis may be needed to fully understand the outcomes of TP53-mutated MDS patients who receive HSCT. Nevertheless, understanding the molecular mechanism of acquired resistance to HMA therapy and developing novel therapeutic strategies are urgently needed to improve outcomes for TP53-mutated MDS patients.
We note that some of the findings of our study are in opposition to previous studies. Notably, we did not see favorable response rate to HMA therapy in TET2 mutated patients. Only a part of our patients were sequenced for TET2, which may have decreased statistical power. In addition, although we strictly followed the response criteria defined by IWG, treatment response evaluation in MDS sustains some subjectivity. Further, there is a significant heterogeneity in the treatment regimen in our study cohort, both of which may have biased the result. Of note, even in the prior study, association between TET2 mutations and HMA response was not robust and statistical significance became apparent only when TET2 mutations were restricted to VAF > 10%. [3] These results suggest that application of molecular data to treatment decision-making still requires caution in MDS patients.
In summary, our study suggests that TP53 mutation status is the strongest predictor of prognosis in MDS patients treated with HMA therapy. Its prognostic value is significant after adjusting prognostic effect of other factors such as complex karyotypes and IPSS-R risk. Despite the poor prognosis, TP53-mutated MDS patients respond equally as well to HMA therapy as WT TP53 patients. However, their duration of response is significantly shorter, and TP53 mutations almost always persist at the time of disease progression. Novel therapeutic strategies to improve duration of response in TP53-mutated MDS are urgently needed.
MATERIALS AND METHODS
Studied patients and treatment
We identified 321 patients with previously untreated MDS who were referred to The University of Texas MD Anderson Cancer Center (MDACC) between 2012 and 2014. Of the 321 patients, 168 patients (52%) were treated with HMA therapy and were therefore eligible for further analysis. Diagnosis of MDS was classified using the WHO classification system. [15] We also included patients in this study that were historically classified as refractory anemia with excess blasts in transformation (RAEB-T). [16] This is because HMA therapy has been recognized as one of the standard care for this subgroup of patients. [13] Cytogenetic and overall prognostic risks were calculated by Revised International Scoring System (IPSS-R). [17] Although IPSS-R was generated based on the data from de novo MDS patients, we also applied IPSS-R to therapy-related MDS patients in the current study because previous studies have shown that IPSS-R retains power in this group of patients. [18]
Seventy-eight patients (46%) received standard of care (SOC) 5-azacitidine or decitabine (38 patients received 5-azacitidine alone, and 40 patients received decitabine alone), 79 patients (47%) received either 5-azacitidine or decitabine in combination with other investigational agents under various clinical trials (68 patients received 5-azacitidine combinations, and 11 patients received decitabine combinations), and 11 patients (7%) received guadecitabine (SGI-110) [19] as a single agent under a clinical trial. Details of therapy regimens are described in Supplemental Table 1. The median interval from original diagnosis at outside institutions to presentation to our institution was 0.6 months (range: 0-62 months). All bone marrow samples analyzed in this study were obtained at the time of presentation to MDACC. Written informed consent was provided by all studied patients, and the study protocol was approved by the institutional review board (IRB) at MD Anderson Cancer Center. The study was conducted in accordance with the Declaration of Helsinki.
Sample processing, DNA sequencing, and variant calling
TP53 analysis in all 168 patients was performed on the initial bone marrow sample by one of the following methods: whole-exome sequencing (WES, N = 53), targeted gene capture deep sequencing using a next-generation sequencing (NGS) platform (28-gene panel, N = 26 or 53-gene panel, N = 89). For WES, Agilent SureSelect All Exon V4 was used for exome capture hybridization, and an Illumina HiSeq 2000 sequencer was used for sequencing with 75 base pair paired-end reads. Both the 28-gene and 53-gene panel sequencing was performed on Illumina MiSeq platform as previously described. [20] The panels of genes sequenced by the 28- and 53-gene panels are listed in Supplemental Table 2. WES and the 28-gene panel covered the entire coding sequence of TP53, and the 53-gene panel covered the entire coding sequence of exons 4-8 and part of exons 2 and 10. The median coverage within the targeted region was 124x and 4,000x with WES and targeted gene capture deep sequencing, respectively. Some of the TP53-mutated patients had longitudinal assessment of TP53 mutations by polymerase chain reaction (PCR)-based Sanger sequencing. Methods for variant calling and filtering process are described in Supplemental Method.
Definition of response and survival outcome
Definition of response to HMA therapy followed the 2006 International Working Group (IWG) criteria. [21] Overall response (OR) was defined as having either complete response (CR), partial response (PR), or hematological improvement (HI). Duration of response was calculated from the time of response to the time of loss of response or last follow-up, whichever occurred first. Patients who underwent hematopoietic stem cell transplant (HSCT) were censored at the time of transplant when calculating response duration. Time to achieve best response (TTR) was calculated from the date of HMA therapy initiation to the date of best response. Overall survival (OS) was calculated from the date of HMA therapy initiation to death or the last follow-up date.
Statistical methods
The chi-square or Fisher exact test was used to assess differences in categorical variables, and the Mann-Whitney U test was used to analyze continuous variables difference. The log-rank test was used to examine between-group differences in survival outcome. Multivariate analysis was conducted using Cox proportional hazards regression. Statistical analyses were performed using SPSS (version 22; IBM Corporation, Armonk NY).
ACKNOWLEDGMENTS
This work was supported in part by MD Anderson Cancer Center Support Grant P30 CA016672, Red and Charline McCombs Institute Center for Global Cancer Early Detection at The University of Texas MD Anderson Cancer Center (K.T.), Dr. Kenneth B. McCredie Chair in Clinical Leukemia Research endowment (G.G.M), Edward P. Evans Foundation (G.G.M), Fundacion Ramon Areces (G.G.M), Cancer Prevention & Research Institute of Texas (CPRIT, RP100202, G.G.M), and by generous philanthropic contributions to MD Anderson’s MDS/AML Moon Shot Program (G.G.M. and H.K.).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Authors’ contributions
K.T. designed the study, collected and analyzed data, and wrote the manuscript. K.P. performed the targeted gene sequencing. C.B.R. performed the pathological diagnosis. K.T., and J.Z. performed the bioinformatics analysis. C.G., and A.F. performed the WES. K.T., E.J., T.K., M.A., M.K., C.D., N.D., J.C., Z.E., H.K., and G.G.M. treated patients, and provided clinical samples. A.F., H.K. and G.G.M. provided leadership. All authors reviewed and approved the manuscript.
REFERENCES
1. Soussi T. p53 alterations in human cancer: more questions than answers. Oncogene. 2007; 26:2145-2156.
2. Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer cell. 2007; 12:303-312.
3. Bejar R, Lord A, Stevenson K, Bar-Natan M, Perez-Ladaga A, Zaneveld J, Wang H, Caughey B, Stojanov P, Getz G, Garcia-Manero G, Kantarjian H, Chen R, Stone RM, Neuberg D, Steensma DP, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014; 124:2705-2712.
4. Kulasekararaj AG, Smith AE, Mian SA, Mohamedali AM, Krishnamurthy P, Lea NC, Gaken J, Pennaneach C, Ireland R, Czepulkowski B, Pomplun S, Marsh JC, Mufti GJ. TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. British journal of haematology. 2013; 160:660-672.
5. Jadersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Gohring G, Hedlund A, Hast R, Schlegelberger B, Porwit A, Hellstrom-Lindberg E, Mufti GJ. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. Journal of clinical oncology. 2011; 29:1971-1979.
6. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, Shlien A, Groves MJ, Forbes SA, Raine K, Hinton J, Mudie LJ, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013; 122:3616-3627; quiz 3699.
7. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N, Shiraishi Y, Shiozawa Y, Chiba K, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014; 28:241-247.
8. Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL. Clinical effect of point mutations in myelodysplastic syndromes. The New England journal of medicine. 2011; 364:2496-2506.
9. Bally C, Ades L, Renneville A, Sebert M, Eclache V, Preudhomme C, Mozziconacci MJ, de The H, Lehmann-Che J, Fenaux P. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leukemia research. 2014; 38:751-755.
10. Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, Miller CA, Niu B, McLellan MD, Dees ND, Fulton R, Elliot K, Heath S, Grillot M, Westervelt P, Link DC, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013; 27:1275-1282.
11. Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, Morel P, Fenaux P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994; 84:3148-3157.
12. Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M, Kugler CM, Holzmann K, Gaidzik VI, Paschka P, Held G, von Lilienfeld-Toal M, Lubbert M, Frohling S, Zenz T, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012; 119:2114-2121.
13. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Gattermann N, Germing U, Sanz G, List AF, Gore S, Seymour JF, Dombret H, Backstrom J, Zimmerman L, McKenzie D, Beach CL, Silverman LR. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. Journal of clinical oncology. 2010; 28:562-569.
14. Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, Helmer R, 3rd, Shen L, Nimer SD, Leavitt R, Raza A, Saba H. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006; 106:1794-1803.
15. Swerdlow SH, International Agency for Research on Cancer. and World Health Organization. (2008). WHO classification of tumours of haematopoietic and lymphoid tissues. (Lyon, France: International Agency for Research on Cancer).
16. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposals for the classification of the myelodysplastic syndromes. British journal of haematology. 1982; 51:189-199.
17. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, Bennett JM, Bowen D, Fenaux P, Dreyfus F, Kantarjian H, Kuendgen A, Levis A, Malcovati L, Cazzola M, Cermak J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012; 120:2454-2465.
18. Ok CY, Hasserjian RP, Fox PS, Stingo F, Zuo Z, Young KH, Patel K, Medeiros LJ, Garcia-Manero G, Wang SA. Application of the international prognostic scoring system-revised in therapy-related myelodysplastic syndromes and oligoblastic acute myeloid leukemia. Leukemia. 2014; 28:185-189.
19. Issa JP, Roboz G, Rizzieri D, Jabbour E, Stock W, O’Connell C, Yee K, Tibes R, Griffiths EA, Walsh K, Daver N, Chung W, Naim S, Taverna P, Oganesian A, Hao Y, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015; 16:1099-1110.
20. Patel KP, Newberry KJ, Luthra R, Jabbour E, Pierce S, Cortes J, Singh R, Mehrotra M, Routbort MJ, Luthra M, Manshouri T, Santos FP, Kantarjian H, Verstovsek S. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood. 2015; 126:790-797.
21. Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD, Pinto A, Beran M, de Witte TM, Stone RM, Mittelman M, Sanz GF, Gore SD, Schiffer CA, Kantarjian H. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006; 108:419-425.
22. Bejar R, Stevenson KE, Caughey B, Lindsley RC, Mar BG, Stojanov P, Getz G, Steensma DP, Ritz J, Soiffer R, Antin JH, Alyea E, Armand P, Ho V, Koreth J, Neuberg D, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. Journal of clinical oncology. 2014; 32:2691-2698.
23. Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100:8424-8429.
24. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009; 4:1073-1081.