
INTRODUCTION
MPNSTs are an aggressive subtype of soft-tissue sarcoma that develops in association with peripheral nerves or nerve roots. Composed of neoplastic Schwann cells, these malignancies are thought to arise from benign nerve sheath tumors, termed plexiform neurofibromas. Plexiform neurofibromas are frequently detected during early childhood, raising the possibility that they are congenital tumors that initiate during fetal or early postnatal life. While transformation of these plexiform neurofibromas in childhood is uncommon, the development of pain or motor weakness associated with a pre-existing plexiform neurofibroma in a teenager or young adult should prompt evaluation for a MPNST. In patients with these malignant tumors, overall survival is poor, and successful treatment options are limited. Even when surgery is employed in combination with radiation or chemotherapy, ~50% of individuals will experience local recurrence as well as distant metastases, and the majority will succumb to their cancer within 5 years [1-3].
MPNSTs occur most frequently in the setting of the Neurofibromatosis 1 (NF1) tumor predisposition syndrome, affecting approximately 8-13% of people with NF1 [4]. Individuals with NF1 are born with a germline mutation in one copy of the NF1 gene, such that all cells in their bodies have one dysfunctional NF1 allele. However, tumorigenesis requires somatic loss of the other NF1 allele in the appropriate cell of origin. Consistent with a key role for the NF1 gene in both NF1-associated and sporadic MNPST pathogenesis, bi-allelic NF1 gene inactivation has been reported in approximately 60-90% of NF1-associated MPNSTs and 40-60% of sporadic cases [5, 6]. While loss of NF1 gene expression is required for MPNST development, it is not sufficient. As such, MPNST formation requires additional cooperating genetic events, the most frequent of which is mutational inactivation of the TP53 tumor suppressor gene, occurring in approximately 75% of cases [7-9].
In this regard, genetically-engineered mouse (GEM) lines with conditional Nf1 gene inactivation in Schwann cell precursors do not develop MPNSTs [10-14] unless coupled with concomitant Trp53 loss [15, 16], epidermal growth factor receptor (Egfr) amplification [7, 17], Pten loss [18], or Ink4a deletion [19, 20]. In each of these GEM strains, the cooperating genetic mutation was introduced simultaneously with Schwann cell precursor Nf1 gene inactivation throughout the body. While each of these models has been informative for understanding MPNST pathogenesis, they do not permit temporal (timing of the cooperating somatic genetic change) or spatial (specific nerve location) control over MPNST development. To generate a model in which temporal control of the transforming genetic alteration can be achieved in a single nerve location, we employed two GEM strains in which Cre-mediated Nf1 inactivation occurs in Schwann cell precursor cells during embryogenesis and p53 knockdown is somatically acquired at 6-8 weeks of age in cells within the right sciatic nerve. Using this approach, 60-70% of mice develop sciatic nerve MPNSTs associated with focal neurological dysfunction. The coupling of somatic retroviral knockdown and embryonic Nf1 gene inactivation establishes an experimentally-manipulable platform to evaluate other cooperating genetic changes in MPNST pathogenesis as well as preclinical mouse strains in which clinical symptomatology can be used to monitor tumor progression.
RESULTS AND DISCUSSION
Leveraging whole exome sequencing methodologies, a recent study from our laboratory revealed a temporal sequence of genetic changes in a single patient with progression of an NF1-associated plexiform neurofibroma to MPNST over a 14-year period. Analysis of these pathological specimens demonstrated an increasing proportion of cells with a somatic NF1 gene mutation (second hit) as the tumor progressed from a benign plexiform neurofibroma to a MPNST. In addition, loss of one copy of the TP53 gene was only detected at the MPNST stage [21]. These observations in a single patient support a model in which NF1 gene inactivation precedes TP53 mutation in the pathogenesis of NF1-associated MPNST.
To model this temporal sequence in mice, we leveraged two different GEM strains in which bi-allelic Nf1 gene inactivation occurred in Schwann cell precursors during embryonic development. For these experiments, Nf1flox/flox mice were intercrossed with either Periostin-Cre [22, 23] or GFAP-Cre [24] mice to eliminate Nf1 protein (neurofibromin) expression in Schwann cell precursors. We first sought to generate mice with a germline null Nf1 gene mutation and a conditional Nf1 allele; however, the majority of Periostin-Cre; Nf1flox/null mice did not survive to weaning age (~3-4 weeks), as previously reported by others [23]. As such, Periostin-Cre; Nf1flox/flox mice and GFAP-Cre; Nf1flox/null mice, were used for these experiments. Importantly, neither strain develops MPNSTs without the introduction of additional genetic alterations (e.g., somatic p53 knockdown).
In the Periostin-Cre model, promoter activity is detected as early as embryonic day 10 (E10) within post-migratory Schwann cell progenitor cells. Based on published reporter activity studies, recombination occurs in the enteric nervous system, peripheral nervous system (including Schwann cells), and within a subpopulation of cardiac outflow tract mesenchymal cells [22]. As expected, intercrossing Periostin-Cre mice with a Rosa-GREEN reporter strain [25] revealed green fluorescent protein (GFP) expression in sciatic nerves (Figure 1a). Similarly, in the GFAP-Cre model, where promoter activity has been reported as early as E13.5 in the brain [24], we observed robust GFP expression in the sciatic nerves of adult GFAP-Cre mice following intercrossing with Rosa-GREEN reporter mice (Figure 1a).
To determine whether Cre-mediated excision might also occur in the hematopoietic cells that populate the tumor microenvironment [12, 26-28] (e.g., mast cells and macrophages), both Cre driver lines were intercrossed with Rosa-GREEN reporter mice [25]. Whereas GFP expression was observed within the adult sciatic nerve, there was no GFP labeling within the bone marrow (Figure 1a). The lack of GFP expression in the bone parenchyma demonstrates that Cre-mediated excision is largely limited to the Schwann cell precursors within the sciatic nerve, rather than infiltrating bone marrow-derived stromal cells in the tumor microenvironment.
Western blot analysis of the sciatic nerves of Periostin-Cre; Nf1flox/flox mice revealed loss of neurofibromin expression, but intact p53 expression (Figure 1b). The absence of neurofibromin expression demonstrates that early Nf1 gene inactivation in Schwann cell precursors occurs prior to the induction of shRNA retrovirus-mediated p53 knockdown. Similar to Periostin-Cre; Nf1flox/flox mice, no neurofibromin expression was detected in the sciatic nerves of GFAP-Cre; Nf1flox/null mice by western blot (Figure 1b).
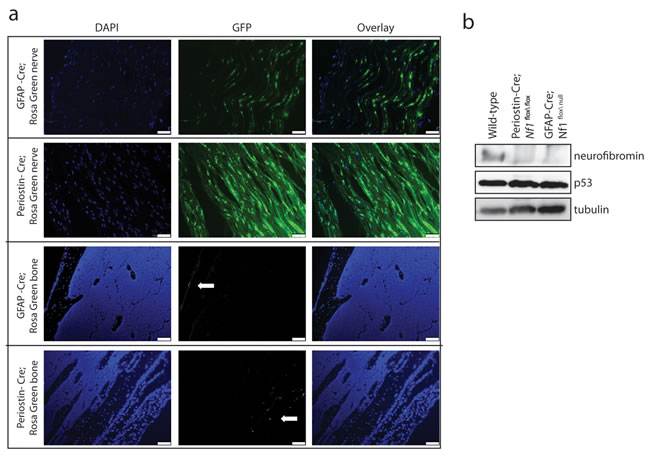
Figure 1: Murine MPNST model system employed. a. Representative images of sciatic nerves and femurs (bone) from GFAP-Cre (FVB-Tg(GFAP-cre)25Mes/J) and Periostin-Cre mice crossed to Rosa-Green reporter mice. Green fluorescent protein (GFP)-positive cells were observed in the sciatic nerve and bone periosteum (white arrows), but not in the hematopoietic cells within the bone parenchyma. Scale bar, 50µm. b. Western blotting reveals complete loss of neurofibromin expression and intact p53 expression in Periostin-Cre; Nf1flox/flox and GFAP-Cre; Nf1flox/null sciatic nerves compared to intact neurofibromin and p53 expression in the wild-type sciatic nerve.
Next, to introduce p53 knockdown specifically in cells within the sciatic nerve, we leveraged a lentiviral approach in which Nf1 and Trp53 knockdown occurs following p53 shRNA virus injection (Figure 2a). This construct has previously been employed to reduce Nf1 and Trp53 gene expression in the brain in vitro and in vivo, resulting in high-grade gliomas [29]. In this regard, we observed 40% and 70% reductions in neurofibromin and p53 expression, respectively, in NIH-3T3 cells 96 hours post-lentiviral infection in vitro (Figure 2b). The choice to employ a lentivirus containing shRNAs targeting both the Nf1 and Trp53 genes reflected a lack of reproducible knockdown (KD) when the identical Trp53 shRNA was employed alone (data not shown). Importantly, since Nf1 expression has already been silenced in GFAP-Cre; Nf1flox/null, GFAP-Cre; Nf1flox/flox, and Periostin-Cre; Nf1flox/flox mice, the effects of shRNA Nf1 KD on the preneoplastic Schwann cell component should be negligible.
For all MPNST induction experiments, adult sciatic nerves were surgically isolated and injected with p53 shRNA lentivirus when the mice reached 6-8 weeks of age. Mice were then monitored by serial examination until they demonstrated neurological abnormalities (e.g., right leg weakness), at which time they were euthanized for gross pathological and histological analyses (Figure 2c).
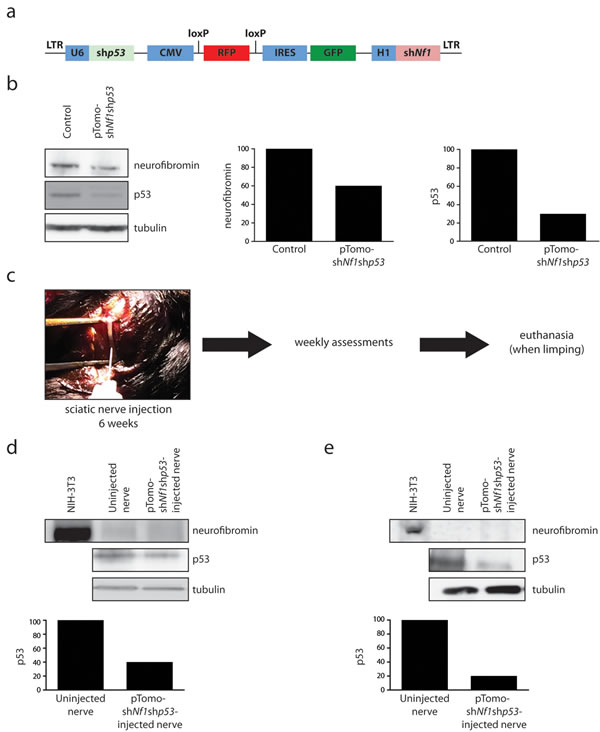
Figure 2: Lentiviral introduction of pTomo-shNf1;shp53 into the sciatic nerve. a. Schematic of the pTomo-shNf1;shp53 lentivirus construct used in these studies [29]. b. Western blotting demonstrates a 40% reduction in neurofibromin expression and a 70% reduction in p53 expression 96 hours after pTomo-shNf1;shp53 lentiviral infection of NIH-3T3 cells. c. Schematic of study design. d. Western blotting reveals complete loss of neurofibromin expression coupled with a 60% reduction in p53 expression within the sciatic nerves of Periostin-Cre; Nf1flox/flox mice injected with pTomo-shNf1;shp53 virus relative to uninjected control nerves. e. Western blotting reveals complete loss of neurofibromin expression coupled with an 80% reduction in p53 expression within the sciatic nerves of GFAP-Cre; Nf1flox/flox mice injected with pTomo-shNf1;shp53 virus relative to uninjected control nerves.
Initial experiments used mice in which neurofibromin expression was ablated in periostin-expressing cells, including Schwann cell precursors within the peripheral nerves (Periostin-Cre; Nf1flox/flox mice), consistent with previous lineage tracing studies [30]. While the precise cell of origin is not known in this model, Cre-driven reporter expression has been observed in brain lipid binding protein (BLBP)-immunoreactive cells, which represent Schwann cell precursors or immature Schwann cells [30]. To induce MPNSTs, pTomo-shNf1;shp53 lentivirus or vehicle was injected into the right sciatic nerves of 6-8 week old Periostin-Cre; Nf1flox/flox mice, resulting in GFP expression following Cre-mediated recombination (Supplemental Figure 1a). Following the injection of this lentivirus into the right sciatic nerves of 6-8 week old mice, the pre-existing complete loss of neurofibromin expression in these nerves was now accompanied by a 60% reduction in p53 protein expression within the tumors (Figure 2d). With an average latency of 183 days post-injection (Table 1), ~60% of the pTomo-shNf1;shp53 lentivirus-injected Periostin-Cre; Nf1flox/flox mice exhibited paresis of the affected leg, as evidenced by pronounced right-sided lower extremity limping. At this time, these mice were euthanized and their sciatic nerves evaluated for the presence of a malignant tumor. In 10/18 of these mice, grossly-appearing mass lesions were appreciated in the nerves of lentiviral-injected animals, but not in the vehicle-injected controls (Figure 3a).
Table 1: MPNST development in Nf1 mutant mice following p53 knockdown
Mouse genotype (injection) |
Hyperplasia |
MPNST |
MPNST latency, mean days (range) |
Periostin-Cre; Nf1flox/flox (vehicle) |
2/11 |
0/11 |
No tumors: 83-304d* |
Periostin-Cre; Nf1flox/flox (pTomo-shNf1;shp53) |
8/18 |
10/18 |
183d (144-278d) |
GFAP-Cre; Nf1flox/null (vehicle) |
2/6 |
0/6 |
No tumors: 49-217d* |
GFAP-Cre; Nf1flox/null (pTomo-shNf1;shp53) |
3/11 |
8/11 |
91d (25-211d) |
GFAP-Cre; Nf1flox/flox (vehicle) |
0/4 |
0/4 |
No tumors:129-153d* |
GFAP-Cre; Nf1flox/flox (pTomo-shNf1;shp53) |
2/5 |
3/5 |
176d (117-298d) |
Wild-type (pTomo-shNf1;shp53) |
0/4 |
0/4 |
No tumors: 182-365d# |
Nf1+/- (pTomo-shNf1;shp53) |
0/4 |
0/4 |
No tumors: 182-365d# |
*Vehicle mice were euthanized and analyzed at time points equivalent to those when p53 shRNA-injected mice were euthanized or at a time when they became ill due to non-tumor-related issues (rectal prolapse or hydrocephalus). The details are provided below:
Vehicle-injected GFAP-Cre; Nf1flox/null mice: 2 mice at 49 days, 2 mice at 154 days and 2 mice at 217 days.
Vehicle-injected Periostin-Cre; Nf1flox/flox mice: 1 mouse at 83 days, 3 mice at 144 days, 2 mice at 154 days, 2 mice at 230 days, 2 mice at 259 days, and 1 mouse at 304 days.
Vehicle-injected GFAP-Cre; Nf1flox/flox mice: 2 mice at 129 days, 1 mouse at 138 days, and 1 mouse at 158 days.
Wild-type and Nf1+/- mice: 2 mice at 183 days and 2 mice at 365 days for each group.
#Wild-type and Nf1+/- mice were euthanized and analyzed at 6 month or 12 month time points, as no mice developed symptoms or non-tumor-related issues.
Neuropathological analyses of the resulting tumors revealed striking hypercellularity, mild to moderate nuclear pleiomorphism, reduced S100β immunopositivity, increased Ki67 labeling (Figure 3b), infiltration of adjacent fibroadipose tissue, and mitotic figures (Supplemental Figure 1b), as seen in their human low-grade MPNST counterparts. Additionally, these murine MPNSTs retained basement membrane staining (Collagen 4A; Col4a immunoreactivity) and demonstrated mast cell infiltration visualized by tryptase staining, as reported in other murine MPNST-like tumors [31] (Figure 3b). Based on the observed increased cellularity, nuclear atypia, and mitotic activity, these lesions are most accurately classified as low-grade MPNST, as suggested by the Comparative Pathology of Nerve Sheath Tumors in Mouse Models and Humans Committee Consensus Report [32]. The remaining 8 mice exhibited hyperplasia only, similar to some of the vehicle-injected mice.
The development of MPNSTs in mice without complete loss of p53 expression (Figure 2d) suggests that bi-allelic (complete) inactivation of the Trp53 gene might not be necessary for malignant transformation. While we cannot exclude the acquisition of additional genetic changes that effectively abrogate p53 signaling, previous reports in both mouse models and human pathological MPNST specimens have likewise support the concept that TP53 haploinsufficiency may be sufficient for MPNST formation [7, 9]. Similarly, our recent whole exome sequencing analysis of a single patient with NF1-related plexiform neurofibroma malignant transformation revealed only heterozygous TP53 loss [21].
The development of MPNSTs in Periostin-Cre; Nf1flox/flox mice raises the intriguing possibility that malignant transformation does not require heterozygous Nf1 loss in cells within the tumor microenvironment. While previous studies from one of our laboratories (D.W.C.) have clearly demonstrated a critical role for infiltrating Nf1+/- stromal cells in benign murine plexiform neurofibroma formation and growth [12, 27, 28], the apparent stromal independence in MPNST pathogenesis could reflect differences between low-grade tumor growth requirements and those operative in malignant cancers. In this respect, similar differences have also been reported for murine Nf1 brain tumors: Nf1+/- stromal cells are required for low-grade murine optic glioma formation and continued growth in vivo [33-35], but high-grade glioblastoma development can occur following Nf1 and p53 inactivation in nestin+ neural stem cells alone in vivo [36].
Since the early lethality (pre-weaning) observed in Nf1+/- mice with periostin-Cre-mediated Nf1 gene inactivation precluded an examination of the contribution of the Nf1+/- tumor microenvironment to MPNST biology, we sought to reduce the latency of MPNST development by coupling somatic retrovirus-mediated p53 knockdown and embryonic Schwann cell precursor Nf1 gene inactivation in the setting of an Nf1+/- microenvironment. For these experiments, we employed GFAP-Cre mice [24], in which Cre-mediated recombination occurs in the sciatic nerve (Figure 1a). In contrast to the Periostin-Cre; Nf1flox/null mice, the majority of which die by 4 weeks of life, Nf1flox/null; GFAP-Cre mice are viable into adulthood. Similar to the experiments performed with the Periostin-Cre; Nf1flox/flox mice, pTomo-shNf1;shp53 lentivirus or vehicle was injected into the sciatic nerves of 6-8 week old GFAP-Cre; Nf1flox/null mice. Following the injection of this lentivirus into the right sciatic nerves of 6-8 week old mice, the pre-existing complete loss of neurofibromin expression in these nerves was now accompanied by an 80% reduction in p53 protein expression within the tumors (Figure 2e). While none of the vehicle-injected mice exhibited any neurological deficits, eight of eleven GFAP-Cre; Nf1flox/null mice injected with pTomo-shNf1;shp53 lentivirus exhibited right leg weakness. Following euthanasia, gross morphological features of MPNST were observed (Figure 4a). As observed with the pTomo-shNf1;shp53 lentivirus-injected Periostin-Cre; Nf1flox/flox mice, MPNSTs in the GFAP-Cre; Nf1flox/null mice with somatic p53 knockdown exhibited striking hypercellularity, mild to moderate nuclear pleiomorphism, reduced S100β immunoreactivity, increased Ki67 labeling, mast cell infiltrates (Tryptase staining), infiltration of adjacent fibroadipose tissue, and Col4A immunoreactivity (Figure 3b). However, in contrast to pTomo-shNf1;shp53 lentivirus-injected Periostin-Cre; Nf1flox/flox mice, the average time to MPNST formation was only ~90 days as opposed to ~183 days.
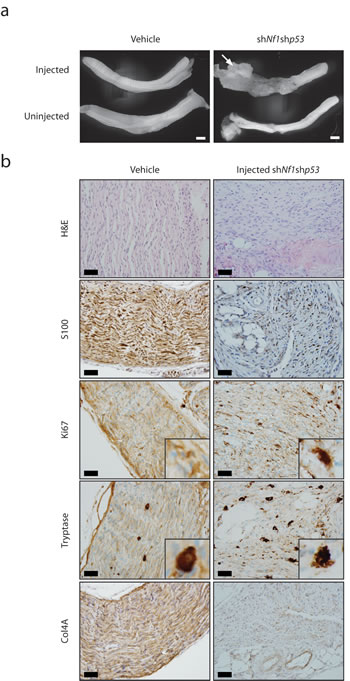
Figure 3: MPNST development in mice with embryonic Schwann cell precursor Nf1 loss and postnatal somatic Trp53 reduction. a. Gross images of the sciatic nerves from Periostin-Cre; Nf1flox/flox mice injected with vehicle or pTomo-shNf1;shp53 virus. Low-grade MPNSTs were only observed in the sciatic nerves of Periostin-Cre; Nf1flox/flox mice injected with pTomo-shNf1;shp53 virus. Scale bar, 1000µm. b. Sciatic nerve sections from Periostin-Cre; Nf1flox/flox mice injected with pTomo-shNf1;shp53 virus demonstrate increased cellularity, nuclear pleiomorphism, and mitotic figures, consistent with low-grade MPNST (H&E). Induced MPNSTs exhibit reduced S100 β-staining, increased Ki67 labeling, increased mast cell infiltration (tryptase staining), and collagen-4A (Col4A) basement membrane immunoreactivity. Scale bar, 40µm.
It is possible that the shortened MPNST latency observed in injected GFAP-Cre; Nf1flox/null mice reflected growth-promoting contributions from the heterozygous tumor microenvironment. Support for this idea derives from early findings in GFAP-Cre; Nf1flox/flox mice (harboring wild-type stromal cells) injected with pTomo-shNf1;shp53 lentivirus. In this respect, the 3 mice that developed MPNSTs exhibited longer latencies than observed in GFAP-Cre; Nf1flox/null mice (average of 176 days, Table 1). However, in striking contrast, 50% of the GFAP-Cre; Nf1flox/null mice injected with pTomo-shNf1;shp53 lentivirus had developed a limp and exhibited low-grade MPNST on pathological examination by 50 days post-injection. Coupled with the results obtained using Periostin-Cre; Nf1flox/flox mice, it is most likely that cells in the Nf1+/- tumor microenvironment provide additional growth factors or chemokines (e.g., CXCL12 [37]) that accelerate the growth of the newly-formed MPNST, leading to a reduced time to clinical symptomatology.
Since the lentivirus employed contained shRNAs that targeted both the Trp53 and Nf1 genes, we next sought to determine whether Nf1 shRNA-mediated KD was sufficient to generate MPNSTs in wild-type or Nf1+/- mice. Following injection of pTomo-shNf1;shp53 lentivirus at 6 weeks of age, mice were examined 6 months or 12 months later. No mice developed a limp and no tumors were appreciated on gross or histologic examination (Table 1). These findings lend further support to the hypothesis that loss of Nf1 gene expression in embryonic Schwann cell precursors is required for plexiform neurofibroma development and subsequent MPNST formation, as previously demonstrated by others [38-40]
The high frequency of MPNSTs in individuals with NF1 coupled with genetic analyses of human MPNSTs argues that NF1 loss is an obligate genetic event in MPNST development. Moreover, the fact that bi-allelic inactivation of the NF1 gene is observed in most NF1-associated [5, 6, 41] and sporadic [42] MNPSTs supports a critical role for this tumor suppressor gene in MPNST pathogenesis. While neurofibromin loss in cells of the Schwann cell lineage is required for MPNST formation, it is clearly not sufficient. This lack of sufficiency is nicely illustrated in Nf1 GEM strains, where mice with conditional Nf1 gene inactivation in Schwann cell precursors alone also do not develop MPNSTs [10-14], including the GFAP-Cre; Nf1flox/null and Periostin-Cre; Nf1flox/flox mouse strains used in the current study. However, the co-existence of other cooperating genetic events in combination with Nf1 loss does result in MPNST formation (summarized in Supplemental Table 1). In most of these mouse models, complex intercrosses of transgenic and conditional knockout strains are required, and there is no control over the timing of the acquired somatic genetic event or the location in which the malignancy will arise [7, 15-20, 43-45]. Using a combination of conditional knockout mice and lentivirus-mediated somatic p53 inactivation, we describe an efficient platform for the generation of MPNSTs. As such, this model is ideally suited to define the biological importance of somatically-acquired cooperating genetic events to MPNST pathogenesis, thus allowing investigators to compare the effects of known somatic genetic alterations (e.g., p53 versus p16 inactivation) or those identified in large-scale genomic discovery efforts [46]. In addition, this experimental system provides both spatial and temporal control. Future uses of this platform provide opportunities to determine the impact of introducing cooperating genetic mutations as a function of mouse age or in different peripheral nerves. Moreover, since somatic p53 knockdown is directed to the sciatic nerve, these mice develop a neurologic deficit (limp), similar to some patients with MPNSTs. The ability to monitor clinical signs will be important if these models are employed for preclinical studies.
In summary, the implementation of this experimental MPNST model system has allowed us to make two important observations. First, we demonstrate that somatic reduction, but not absence, of Trp53 expression following Nf1 gene inactivation is sufficient for MPNST formation. Second, we demonstrate that MPNST formation can occur in the absence of a microenvironment composed of cells heterozygous for a germline Nf1 gene mutation. Future studies employing this platform may facilitate more rapid analyses of the contributions of other cooperating events to MPNST pathogenesis, the role of the tumor microenvironment in MPNST growth, and the development of radiologic and molecular biomarkers of malignant transformation.
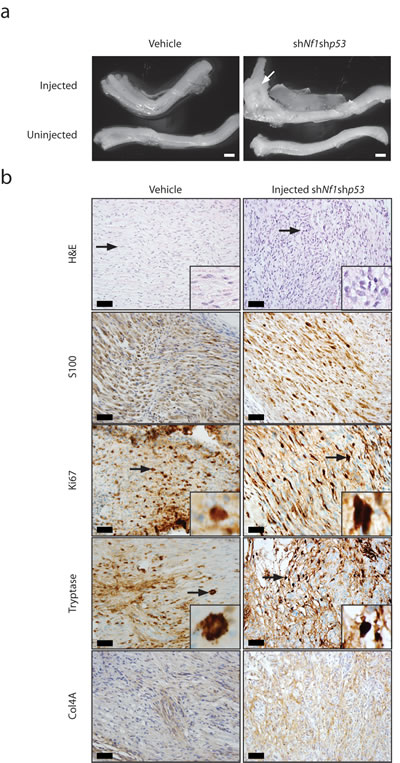
Figure 4: MPNST formation is accelerated in Nf1+/- mice harboring embryonic Nf1 loss and somatic Trp53 reduction. a. Gross images of the sciatic nerves from GFAP-Cre; Nf1flox/null mice injected with vehicle or pTomo-shNf1;shp53 virus. Low-grade MPNSTs were only observed in the sciatic nerves of GFAP-Cre; Nf1flox/null mice injected with pTomo-shNf1;shp53 virus. Scale bar, 1000µm. b. Sciatic nerve sections from GFAP-Cre; Nf1flox/null mice injected with pTomo-shNf1;shp53 virus demonstrate increased cellularity and nuclear pleiomorphism, consistent with low-grade MPNST (H&E). Induced MPNSTs exhibit reduced S100 β-staining, increased Ki67 labeling, increased mast cell infiltration (tryptase staining), and collagen-4A (Col4A) basement membrane immunoreactivity. Scale bar, 40µm.
MATERIALS AND METHODS
Mice
GFAP-Cre (FVB-Tg(GFAP-cre)25Mes/J) and Rosa-Green reporter mice were purchased from the Jackson Laboratories [24, 25]. Periostin-Cre [22] and Nf1flox/flox [47] mice have previously been described. All mice were maintained on a C57Bl/6 background and used in accordance with approved animal studies protocols at the Washington University School of Medicine.
Lentivirus generation
The pTomo-shNf1;shp53 lentivirus construct used has been previously described [29]. Viral particles were produced by the Viral Vectors Core Facility of the Hope Center for Neurological Diseases at the Washington University School of Medicine.
Sciatic nerve injections
The right flanks of 6-8 week old mice were shaved and prepped with betadine. The right sciatic nerves were surgically isolated while mice were under anesthesia. 10μl of pTomo-shNf1;shp53 lentivirus (5x106 IU) or 10μl vehicle was injected into the right sciatic nerves of mice using a10μl Hamilton syringe equipped with a 33-gauge needle. The surgical incision was closed with Vetbond and sutured. Mice were followed and examined three times per week until right-sided limping was observed. At this time, mice were euthanized with carbon dioxide and the sciatic nerves isolated for gross and histologic analysis.
Tissue preparation
For eight-week-old GFAP-Cre; Rosa Green and Periostin-Cre; Rosa Green mouse nerves and bones were Formalin-fixed and paraffin-embedded. 5 micron-thick sections were generated for analysis. Tissues were subsequently dehydrated and coverslipped with Vectashield mounting media containing DAPI (Vector Laboratories).
Immunohistochemistry
Nerves were processed as described above using primary (Supplemental Table 2) and horseradish peroxidase-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA, USA) in combination with Vectastain Elite ABC development.
Microscopy
For eight-week-old GFAP-Cre; Rosa Green and Periostin-Cre; Rosa Green mouse nerves and bones, images were acquired at 100x magnification on a Nikon Eclipse TE300 fluorescence inverted microscope equipped with an optical camera (Leica DFC 3000G) and analyzed using Leica Application Suite Advanced Fluorescence 3.20.9652. Seven day post-injection images were acquired at 200x magnification on a Nikon Eclipse TE300 fluorescence inverted microscope equipped with an optical camera (Leica DFC 3000G). Images from tumor bearing mice were acquired at 400x magnification using an Olympus BX51 camera.
Western blotting
Cell pellets, sciatic nerves, or tumors dissected from sciatic nerves were lysed in buffer containing 1% NP-40 (nonylphenoxypolyethoxylethanol) supplemented with protease inhibitors. Western blotting was performed as previously described [48]. Antibodies used included neurofibromin (Santa Cruz Biotechnology; dilution 1:100), p53 (Cell Signaling; dilution 1:1000), and α-tubulin (Sigma; dilution 1:10000). Densitometry was measured using Life Science Software from UVP VisionWorks LS Version 8.1.1 Image Acquisition and Analysis Software system. α-tubulin serves as an internal protein loading control.
ACKNOWLEDGEMENTS
We thank Suellen Greco, Luethia Hinkle, Crystal Idleburg, and Nada Husic for technical assistance and Drs. Anthony J. Apicelli and Brian A. Van Tine for helpful discussions.
CONFLICTs OF INTEREST
The authors have no conflicts to disclose.
FUNDING
This work was partly funded by a generous gift from Schnuck Markets Inc. (to D.H.G) and The St. Louis Men’s Group Against Cancer. A.C.H. is supported on the T32 HL007088. Lentivirus was generated by the Washington University Hope Center Viral Vectors Core which is supported by a Neuroscience Blueprint Core grant NIH P30 NS057105.
References
1. Hruban RH, Shiu MH, Senie RT and Woodruff JM. Malignant peripheral nerve sheath tumors of the buttock and lower extremity. A study of 43 cases. Cancer. 1990; 66:1253-1265.
2. Kourea HP, Bilsky MH, Leung DH, Lewis JJ and Woodruff JM. Subdiaphragmatic and intrathoracic paraspinal malignant peripheral nerve sheath tumors: a clinicopathologic study of 25 patients and 26 tumors. Cancer. 1998; 82:2191-2203.
3. Wong WW, Hirose T, Scheithauer BW, Schild SE and Gunderson LL. Malignant peripheral nerve sheath tumor: analysis of treatment outcome. International journal of radiation oncology, biology, physics. 1998; 42:351-360.
4. Arun D and Gutmann DH. Recent advances in neurofibromatosis type 1. Current opinion in neurology. 2004; 17:101-105.
5. Rasmussen SA, Overman J, Thomson SA, Colman SD, Abernathy CR, Trimpert RE, Moose R, Virdi G, Roux K, Bauer M, Rojiani AM, Maria BL, Muir D and Wallace MR. Chromosome 17 loss-of-heterozygosity studies in benign and malignant tumors in neurofibromatosis type 1. Genes, chromosomes & cancer. 2000; 28:425-431.
6. Perry A, Roth KA, Banerjee R, Fuller CE and Gutmann DH. NF1 deletions in S-100 protein-positive and negative cells of sporadic and neurofibromatosis 1 (NF1)-associated plexiform neurofibromas and malignant peripheral nerve sheath tumors. The American journal of pathology. 2001; 159:57-61.
7. Rahrmann EP, Moriarity BS, Otto GM, Watson AL, Choi K, Collins MH, Wallace M, Webber BR, Forster CL, Rizzardi AE, Schmechel SC, Ratner N and Largaespada DA. Trp53 haploinsufficiency modifies EGFR-driven peripheral nerve sheath tumorigenesis. The American journal of pathology. 2014; 184:2082-2098.
8. Riddle ND, Gorden L, Rojiani MV, Hakam A and Rojiani AM. CD44 and p53 immunoexpression patterns in NF1 neoplasms - indicators of malignancy and infiltration. International journal of clinical and experimental pathology. 2010; 3:515-521.
9. Lothe RA, Smith-Sorensen B, Hektoen M, Stenwig AE, Mandahl N, Saeter G and Mertens F. Biallelic inactivation of TP53 rarely contributes to the development of malignant peripheral nerve sheath tumors. Genes, chromosomes & cancer. 2001; 30:202-206.
10. Brossier NM and Carroll SL. Genetically engineered mouse models shed new light on the pathogenesis of neurofibromatosis type I-related neoplasms of the peripheral nervous system. Brain research bulletin. 2012; 88:58-71.
11. Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, Stemmer-Rachamimov AO, Cancelas JA and Ratner N. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer cell. 2008; 13:105-116.
12. Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, Zhang S, Yang Y, Vakili ST, Yu M, Burns D, Robertson K, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/— and c-kit-dependent bone marrow. Cell. 2008; 135:437-448.
13. Zheng H, Chang L, Patel N, Yang J, Lowe L, Burns DK and Zhu Y. Induction of abnormal proliferation by nonmyelinating schwann cells triggers neurofibroma formation. Cancer cell. 2008; 13:117-128.
14. Zhu Y, Ghosh P, Charnay P, Burns DK and Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002; 296:920-922.
15. Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, Bronson RT and Jacks T. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999; 286:2172-2176.
16. Vogel KS, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ and Parada LF. Mouse tumor model for neurofibromatosis type 1. Science. 1999; 286:2176-2179.
17. Wu J, Patmore DM, Jousma E, Eaves DW, Breving K, Patel AV, Schwartz EB, Fuchs JR, Cripe TP, Stemmer-Rachamimov AO and Ratner N. EGFR-STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene. 2014; 33:173-180.
18. Gregorian C, Nakashima J, Dry SM, Nghiemphu PL, Smith KB, Ao Y, Dang J, Lawson G, Mellinghoff IK, Mischel PS, Phelps M, Parada LF, Liu X, Sofroniew MV, Eilber FC and Wu H. PTEN dosage is essential for neurofibroma development and malignant transformation. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:19479-19484.
19. Dodd RD, Mito JK, Eward WC, Chitalia R, Sachdeva M, Ma Y, Barretina J, Dodd L and Kirsch DG. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Molecular cancer therapeutics. 2013.
20. King D, Yang G, Thompson MA and Hiebert SW. Loss of neurofibromatosis-1 and p19(ARF) cooperate to induce a multiple tumor phenotype. Oncogene. 2002; 21:4978-4982.
21. Hirbe A, Dahiya SM, Miller C, Li T, Fulton R, Zhang X, McDonald S, DeSchryver K, Duncavage E, Walrath J, Reilly KM, Abel HJ, Pekmezci M, Perry A, Ley TJ and Gutmann DH. Whole exome sequencing reveals the order of genetic changes during malignant transformation and metastasis in a single patient with NF1-plexiform neurofibroma. Clinical cancer research. 2015.
22. Lindsley A, Snider P, Zhou H, Rogers R, Wang J, Olaopa M, Kruzynska-Frejtag A, Koushik SV, Lilly B, Burch JB, Firulli AB and Conway SJ. Identification and characterization of a novel Schwann and outflow tract endocardial cushion lineage-restricted periostin enhancer. Developmental biology. 2007; 307:340-355.
23. Joseph NM, Mosher JT, Buchstaller J, Snider P, McKeever PE, Lim M, Conway SJ, Parada LF, Zhu Y and Morrison SJ. The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer cell. 2008; 13:129-140.
24. Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K and Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001; 31:85-94.
25. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES and Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience. 2010; 13:133-140.
26. Ryan JJ, Klein KA, Neuberger TJ, Leftwich JA, Westin EH, Kauma S, Fletcher JA, DeVries GH and Huff TF. Role for the stem cell factor/KIT complex in Schwann cell neoplasia and mast cell proliferation associated with neurofibromatosis. Journal of neuroscience research. 1994; 37:415-432.
27. Yang FC, Ingram DA, Chen S, Hingtgen CM, Ratner N, Monk KR, Clegg T, White H, Mead L, Wenning MJ, Williams DA, Kapur R, Atkinson SJ and Clapp DW. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/- mast cells. The Journal of clinical investigation. 2003; 112:1851-1861.
28. Yang FC, Chen S, Clegg T, Li X, Morgan T, Estwick SA, Yuan J, Khalaf W, Burgin S, Travers J, Parada LF, Ingram DA and Clapp DW. Nf1+/- mast cells induce neurofibroma like phenotypes through secreted TGF-beta signaling. Human molecular genetics. 2006; 15:2421-2437.
29. Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, Ellisman MH and Verma IM. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012; 338:1080-1084.
30. Gehlhausen JR, Park SJ, Hickox AE, Shew M, Staser K, Rhodes SD, Menon K, Lajiness JD, Mwanthi M, Yang X, Yuan J, Territo P, Hutchins G, Nalepa G, Yang FC, Conway SJ, et al. A murine model of neurofibromatosis type 2 that accurately phenocopies human schwannoma formation. Human molecular genetics. 2015; 24:1-8.
31. Holzel M, Landsberg J, Glodde N, Bald T, Rogava M, Riesenberg S, Becker AJ, Jonsson G and Tuting T. A preclinical model of malignant peripheral nerve sheath tumor-like melanoma is characterized by infiltrating mast cells. Cancer research. 2015.
32. Stemmer-Rachamimov AO, Louis DN, Nielsen GP, Antonescu CR, Borowsky AD, Bronson RT, Burns DK, Cervera P, McLaughlin ME, Reifenberger G, Schmale MC, MacCollin M, Chao RC, Cichowski K, Kalamarides M, Messerli SM, et al. Comparative pathology of nerve sheath tumors in mouse models and humans. Cancer research. 2004; 64:3718-3724.
33. Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR and Gutmann DH. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer research. 2003; 63:8573-8577.
34. Daginakatte GC and Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Human molecular genetics. 2007; 16:1098-1112.
35. Pong WW, Higer SB, Gianino SM, Emnett RJ and Gutmann DH. Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Annals of neurology. 2013; 73:303-308.
36. Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F and Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer cell. 2014; 26:288-300.
37. Mo W, Chen J, Patel A, Zhang L, Chau V, Li Y, Cho W, Lim K, Xu J, Lazar AJ, Creighton CJ, Bolshakov S, McKay RM, Lev D, Le LQ and Parada LF. CXCR4/CXCL12 mediate autocrine cell- cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell. 2013; 152:1077-1090.
38. Le LQ, Shipman T, Burns DK and Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009; 4:453-463.
39. Le LQ, Liu C, Shipman T, Chen Z, Suter U and Parada LF. Susceptible stages in Schwann cells for NF1-associated plexiform neurofibroma development. Cancer research. 2011; 71:4686-4695.
40. Chen Z, Liu C, Patel AJ, Liao CP, Wang Y and Le LQ. Cells of origin in the embryonic nerve roots for NF1-associated plexiform neurofibroma. Cancer cell. 2014; 26:695-706.
41. Upadhyaya M, Spurlock G, Monem B, Thomas N, Friedrich RE, Kluwe L and Mautner V. Germline and somatic NF1 gene mutations in plexiform neurofibromas. Human mutation. 2008; 29:E103-111.
42. Reuss DE, Habel A, Hagenlocher C, Mucha J, Ackermann U, Tessmer C, Meyer J, Capper D, Moldenhauer G, Mautner V, Frappart PO, Schittenhelm J, Hartmann C, Hagel C, Katenkamp K, Petersen I, et al. Neurofibromin specific antibody differentiates malignant peripheral nerve sheath tumors (MPNST) from other spindle cell neoplasms. Acta neuropathologica. 2014; 127:565-572.
43. Huijbregts RP, Roth KA, Schmidt RE and Carroll SL. Hypertrophic neuropathies and malignant peripheral nerve sheath tumors in transgenic mice overexpressing glial growth factor beta3 in myelinating Schwann cells. The Journal of neuroscience. 2003; 23:7269-7280.
44. Brosius SN, Turk AN, Byer SJ, Brossier NM, Kohli L, Whitmire A, Mikhail FM, Roth KA and Carroll SL. Neuregulin-1 overexpression and Trp53 haploinsufficiency cooperatively promote de novo malignant peripheral nerve sheath tumor pathogenesis. Acta neuropathologica. 2014; 127:573-591.
45. Kazmi SJ, Byer SJ, Eckert JM, Turk AN, Huijbregts RP, Brossier NM, Grizzle WE, Mikhail FM, Roth KA and Carroll SL. Transgenic mice overexpressing neuregulin-1 model neurofibroma-malignant peripheral nerve sheath tumor progression and implicate specific chromosomal copy number variations in tumorigenesis. The American journal of pathology. 2013; 182:646-667.
46. Rahrmann EP, Watson AL, Keng VW, Choi K, Moriarity BS, Beckmann DA, Wolf NK, Sarver A, Collins MH, Moertel CL, Wallace MR, Gel B, Serra E, Ratner N and Largaespada DA. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nature genetics. 2013; 45:756-766.
47. Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD and Parada LF. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes & development. 2001; 15:859-876.
48. Uhlmann EJ, Li W, Scheidenhelm DK, Gau CL, Tamanoi F and Gutmann DH. Loss of tuberous sclerosis complex 1 (Tsc1) expression results in increased Rheb/S6K pathway signaling important for astrocyte cell size regulation. Glia. 2004; 47:180-188.