
HYPOXIA AND THERAPEUTIC TARGETS
Anti-angiogenesis agents and cancer therapy
During the growth of a solid neoplasia, the tumor microenvironment (TM) undergoes biochemical changes that include depletion of glucose, bicarbonate, O2 (i.e. hypoxia and anoxia), high levels of lactate and adenosine and low pH values [1, 2]. As a response to hypoxia and nutrient deficiencies, cells activate angiogenesis, that is the development of new blood vessels sprouting from the existing ones [3-6]. However, neo-angiogenesis yields immature vessels with functional and structural alterations, leading to an irregular blood flow, which fails to provide enough oxygen to each cancer cell and the TM remains, essentially, hypoxic [7, 8]. It has been thought that blocking the oxygen supply could be a valid therapeutic strategy to stop cancer progression. Thus, scientific research on cancer treatment has made significant progresses in the development of new anti-angiogenic agents that have entered into clinical practice alone or in combination with the standard chemo-/radio- therapy [9]. Anti-VEGF compounds were developed, such as bevacizumab and ramucirumab as well as the VEGF antagonist aflibercept [7]. However, despite some promising results in clinical trials, the blockade of VEGF signaling yield positive results only in a fraction of solid tumors, such as colorectal, lung and breast HER2+/ER+/PR+ cancers [10, 11]. Moreover, compared to the previous standard of care, treatments based on angiogenesis inhibitors provide poor benefits in terms of progression-free (PFS) and overall survival (OS) [12]. VEGF blockade could exacerbate the oxygen deprivation in the TM leading to a further increased hypoxic state. In these conditions, in vivo studies have shown that tumor blood vessel pruning may even stimulate cancer cells to acquire pro-invasive and metastatic traits, a threatening form of tumor adaptation to a hypoxic TM (HTM) [7, 8, 12, 13]. As a matter of fact, it has been shown that glioblastoma patients treated with bevacizumab had a shorter OS [14].
Hypoxic tumor microenvironment, cancer progression and epithelial-mesenchymal transition
Following the observations reported above, it was made clear that one of the main problems in fighting cancer is the extreme adaptation of malignant cells to HTM, which promotes malignant progression, and confers increased resistance to chemo- and radio- therapies [15]. In fact, increased hypoxia was correlated to a high expression of markers of epithelial mesenchymal transition (EMT) [16-18] and hypoxic niches were shown to be enriched of cancer stem cells (CSCs) [19, 20]. In summary, cancer cells within a HTM undergo to dramatic changes, ultimately leading to a worsening of their malignant behavior. Thus, nowadays the research is devoted towards the identification of mechanisms involved in the adaptation of cancer cells to the HTM with the aim to detect targets for novel therapeutic agents. A key-molecule identified in mid-nineties is the HIF1A [21]. HIF1A is responsible for the formation of HIF complex that acts as a transcription factor in the activation of a broad spectrum of genes orchestrating large phenotypic changes and ultimately leading to EMT. Following EMT, cells lose their normal phenotype and acquire mesenchymal features [22, 23] including the acquisition of stemness characteristics [24], enhancement of invasiveness and metastasizing capabilities [25]. All these changes are associated with poor prognosis and chemo-/radio-resistance in different tumor models [26, 27]. EMT is characterized by loss of proteins involved in cell adhesion (e.g. E-cadherin) and the over-expression of mesenchymal-specific proteins such as SNAIL, the Vimentin, and TWIST [16, 18]. Phenotypically, these changes correspond to the acquisition of a spindle-like cell morphology, increased invasiveness, migration, and cell proliferation enabling cancer cells to invade surrounding tissues [16]. For this reason, this transition has been regarded as the possible first step in the complex process of generating distant metastases [28, 29] (Figure 1). In parallel with these considerations, it is not surprising that HIF1A over-expression was showed to be an independent predictor of poor prognosis in most of the solid tumors [30-34]. The hypoxia>HIF>EMT axis has been studied thoroughly in aggressive tumors [e.g. lung, triple negative breast (TNBC), gastric, ovarian cancer, melanoma, multiple myeloma, malignant pleural mesothelioma (MPM), pancreatic ductal adenocarcinoma (PDAC), hepatocellular and renal cell carcinoma (RCC)] and various important genes activated by HIF complex were identified (Figure 2). Among them, there are. Autophagy markers BECN1 and MAP1LC3 which are activated in lung cancer [35] and in PDAC [36]; the acidosis modulator CAIX, which is overexpressed in TNBC [37] and in RCC [38]; the epigenetic regulators [39] KLF8, CD24, JMJ2DB and the long-non coding RNA AK058003 which are overexpressed in gastric cancer [40-43], CD44 in TNBC [44], NANOG, OCT4, SRY, SOX2, SHH, SMO, GLI1 in PDAC [45, 46] and MPM [47]; the chemokines CXCR4 and CCL2 which are over-expressed in gastric cancer [48, 49] and multiple myeloma [50], CCR7 in ovarian cancer [51] whereas CX3CR1 in PDAC [52]; the cyclosporin CYPA which is over-expressed in PDAC [53]; the endothelin EDN1 which is activated in melanoma [54]; the fascin FSCN1 which is activated in PDAC [55]; the GTPase protein RND3 which is over-expressed in gastric cancer [56]; the growth factors related to insulin homeostasis IGF1, IGF1R, IGFBP3 which are over-expressed in lung cancer [57] and hepatocellular carcinoma [58]; the mucin MUC1 which is overexpressed in MPM [59], in PDAC [60], in RCC [61] and in lung cancer [62]; the matrix metalloproteinase MMP2 which is over-expressed in PDAC, lung cancer and ovarian cancer cell lines [63]; the protein kinase receptors TGFB/TGFBR1 which are up-regulated in gastric cancer [64], TNFAR in melanoma [65] and in RCC [66] and AXL in gastric cancer [67], TNBC [68], in lung cancer [69], and MPM [70]. Moreover, another important cell signaling is activated by HIF. It was shown that HGF, together with its ligand MET (HGF/MET), promotes cell proliferation, survival, migration, and angiogenesis in hepatocellular carcinoma [71]. Furthermore, ADM is overexpressed in RCC [72] and in PDAC [73]). ILK is activated by HIF1A and through a regulatory loop it is responsible for the increase of HIF1A expression, in a positive feed-back mechanism [74]. In addition, E-cadherin, which has always been defined as a tumor suppressor, showed an unexpected role in the regulation of genes involved in the response to hypoxia, suggesting a possible novel function in aggressive breast cancer [75]. These recent findings, taken together, prompted clinical researchers to focus on the identification of new compounds able to inhibit HIF1A or its targets. Hereon, we will focus on novel therapeutic agents developed for these purposes and for which the clinical trials are in an advanced stage (Table 1, Figure 3). These new molecules could open the way to effective therapies and increase the survival of patients suffering from these aggressive types of cancer.
Table 1: Summary of novel therapeutic approaches
Drugs |
Targets |
Phase |
Type of cancer |
Ref. study |
a. Targeting HIF1A |
||||
Digoxin/ DIG-HIF |
HIF1A |
2 |
Advanced breast cancer |
NCT01763931 |
Ganetespib |
HIF1A/Hsp90 |
3 |
Advanced NSCLC |
NCT01798485 |
b. Inhibiting HIF-targets involved in the regulation of acid TM |
||||
SLC-0111 |
CAIX |
1 |
Advanced solid tumors |
NCT02215850 |
DTP348 |
CAIX |
1 |
Advanced solid tumors |
NCT02216669 |
c. Inhibiting HIF-targets involved in the Epithelial Mesenchymal Transition |
||||
Rilotumumab |
MET/HGF/SF |
3 |
Advanced gastric cancer |
NCT01697072 |
Crizotinib Axitinib |
MET/ALK |
1b |
Advanced solid tumors |
NCT01999972 |
Cabozantinib |
MET/AXL RET/ROS1/NTRK1 |
2 |
Advanced NSCLC |
NCT01639508 |
d. Targeting mitochondrion dysfunction |
||||
CPI-613 |
Mitochondria |
1 |
Metastatic colorectal cancer |
NCT02232152 |
Fenofibrate |
Mitochondria |
2 |
Advanced multiple myeloma |
NCT01965834 |
Antibacterial agents |
Mitochondria |
2 |
Malignant diseases |
NCT02366884 |
Anti-bacterial pro-drug activated in hypoxia |
||||
TH302 |
Hypoxic TM |
3 |
Soft Tissue sarcoma |
NCT01440088 |
e. Targeting stromal-epithelial metabolic coupling |
||||
Metformin |
MCT4 |
0 |
Head and Neck squamous cells cancer |
NCT02083692 |
Topotecan/ Acetylcysteine |
MCT4/CAV1 |
2 |
Advanced ovarian cancer |
NCT02569957 |
Summary of clinical trial that are currently recruiting patients targeting: HIF1A, HIF-targets involved in the regulation of acid TM, HIF-targets involved in the epithelial mesenchymal transition, and metabolic changes such as mitochondrion dysfunction and stromal-epithelial metabolic coupling.
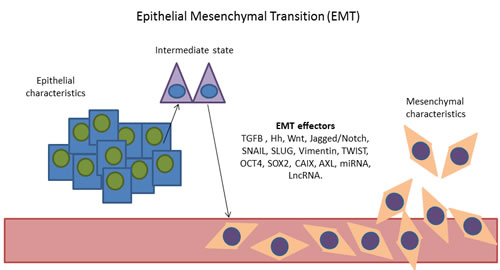
Figure 1: Representation of the epithelial mesenchymal transition. Cells with epithelial characteristics acquire mesenchymal characteristics through the activation of EMT effectors. [EMT effectors: TGFB, Hedgehog (Hh), Wnt, Jagged/Notch signalling pathways, SNAIL, Slug, Vimentin, TWIST, OCT4, SOX2, CAIX, AXL, MicroRNAs (miRNA) and long non-coding RNAs (lncRNA)].
THERAPEUTIC APPROACHES IN ADVANCED CLINICAL TRIALS TARGETING THE ADAPTATION TO HTM
Targeting HIF1A
Digoxin/ DIG-HIF
Digoxin, a cardiac glycoside, it has been shown to have anti-cancer activity due to the inhibition of HIF1A synthesis, in vitro and in vivo in several solid tumors [76-78]. Currently, Digoxin is involved in a phase 2 clinical trial (https://clinicaltrials.gov/ct2/show/NCT01763931) as a novel inhibitor of HIF1A in breast cancer. It will be administrated to breast cancer patients for 2 weeks prior to surgery. During the two weeks of daily oral digoxin therapy, researchers will measure, by immunohistochemistry, the expression of HIF1A protein in surgically resected breast cancer tissues. This clinical trial will be useful also to evaluate adverse events, to assess safety and tolerability of Digoxin in the pre-surgical breast cancer patients according to Common Terminology Criteria for Adverse Events, version 4.
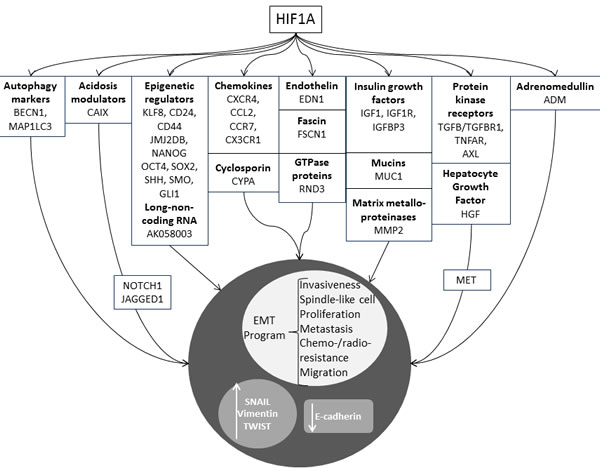
Figure 2: Genes whose expression was shown to be correlated to the activation status of HIF1A, leading to EMT. Autophagy markers: BECN1, MAP1LC3; acidosis modulators: CAIX; epigenetic regulators: KLF8, cell surface glycoproteins (CD24, CD44), lysine (K)-specific demethylase jumonji domain (JMJ2DB), nanog homeobox (NANOG), octamer-binding transcription factor 4 (OCT4), SRY sex determining region Y-box 2 (SOX2), sonic hedgehog (SHH), smoothened, frizzled class receptor (SMO), GLI family zinc finger 1 (GLI1); long non-coding RNA: AK058003; chemokines: CXCR4, CCL2, CCR7, CX3CR1; cyclospporins: cyclophilin A (CYPA); endothelins: endothelin1 (EDN1); fascins: fascin actin-bundling protein 1 (FSCN1); GTPase proteins: Rho family GTPase 3 (RND3); insulin growth factors: IGF1, IGF1R, IGFBP3; mucins: mucin 1, cell surface associated (MUC1); matrix metalloproteinase: MMP2, protein kinases receptors: TGFB/TGFBR1, TNFAR, AXL; HGF; adrenomedullin (ADM).
Ganetespib
The chaperone Hsp90 is involved in tumor growth, angiogenesis and cancer stem cells formation [79, 80]. Its pathway leads to the activation of several oncogenic proteins including HIF1A. It has been shown that Ganetespib (5-[2,4-dihydroxy-5-(1-methylethyl)phenyl]-4-(1-methyl-1H-indol-5-yl)-2,4-dihydro-3H- 1,2,4-triazol-3-one) is able to increase the proteasome-mediated degradation of Hsp90. By this mechanism Ganetespib can inhibit HIF1A in a TNBC mouse model [81]. This drug was already administered as mono-therapy in patients with genotypically defined advanced non-small cell lung cancer (NSCLC) in a multicenter phase 2 study and showed manageable side effects and an OS improvement of 11 months (https://www.clinicaltrials.gov/ct2/show/record/NCT01798485) [82]. Currently, Ganetespib is in a phase 3 study in combination with docetaxel in patients with advanced NSCLC (https://www.clinicaltrials.gov/ct2/show/NCT01798485). This clinical trial aims to the identification of a possible synergism between Ganetespib (150 mg/m2) and Docetaxel (75 mg/m2) to propose a more effective anti-cancer therapy than Docetaxel alone.
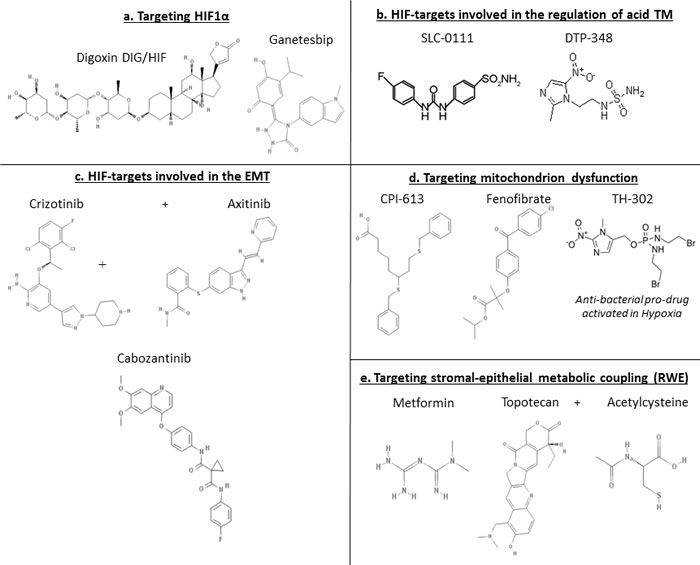
Figure 3: Chemical structures related to Table 1.
Inhibiting HIF-targets involved in the regulation of acid TM
CAIX is a hypoxia-inducible metal-enzyme that promotes cancer cell survival and invasion in HTM via HIF activation [83]. Its role is to catalyze the reversible hydration of carbon dioxide to bicarbonate and protons (H2O + CO2 to HCO3- + H+) regulating cellular pH. It is expressed selectively on the cell surface of tumor cells, particularly in the CSCs, [83] and it is one of the major factors contributing to cancer cell survival and metastasis. CAIX is highly expressed in breast cancer and PDAC and it has been shown as biomarker of poor prognosis for metastasis development and survival [83-85]. Further work has established a critical role of CAIX expression in the maintenance of EMT phenotype [84], “stem cell” function and tumor heterogeneity induced by hypoxia [86, 87].
SLC-0111
SLC-0111, as small molecule, it is able to reach the hypoxic niches and selectively bind and inhibit CAIX. Pre-clinical pharmacokinetic and toxicology analysis of SLC-0111 has been completed, demonstrating little toxicity and positive effects for the treatment of various tumors. Currently, SLC-0111 is involved in a phase I, Multi-center, Open-label, study (https://clinicaltrials.gov/ct2/show/NCT02215850). In this prospective single arm study, SLC-0111 will be given once daily in 28 day cycles, in subjects with advanced solid tumors. The aim is to investigate its safety, tolerability, and pharmacokinetic and to gain some information about its effectiveness in treating cancer.
DTP348
DTP348, namely 2-(2-methyl-5-nitro-1H-imidazol-1-yl) ethylsulfamide, was designed to target hypoxic tumor regions using low doses. Interestingly, this molecule is able to incorporate, in the same scaffold, different functionalities: the inhibition of CAIX, the acidification of the intracellular pH, and the radio-sensitization in HTM [88]. This oral dual CAIX inhibitor/ radio-sensitizer, is currently in a phase I multicenter, open-label, dose-escalation study (https://clinicaltrials.gov/ct2/show/NCT02216669). The clinical trial will investigate the effects of DTP348 alone and in combination with radiotherapy in patients with advanced solid tumors, to determine its recommended phase II Dose, safety and tolerability. Once defined these parameters, DTP348 will be administered every day for 7 weeks to evaluate its pharmacokinetic and anti-cancer activity.
Inhibiting HIF-targets involved in the Epithelial Mesenchymal Transition
HGF is the natural ligand of MET proto-oncogene, a receptor tyrosine kinase [89, 90]. It has been shown that the activation of the HGF/MET pathway induces EMT generating a mesenchymal population more tumorigenic and chemo-resistant than the parental one [91]. HGF/MET are induced by HIF1A [92] and it has been shown that HIF1A overexpression increases HGF’s mRNA stability [93].
Rilotumumab
Rilotumumab is a human monoclonal antibody immunoglobulin G type 2, that blocks the binding of HGF/SF to its receptor MET. Rilotumumab is currently in a phase 3, multicenter, randomized, double-blind, placebo controlled study, of epirubicin, cisplatin & capecitabine (ECX) or untreated advanced MET-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma (https://clinicaltrials.gov/ct2/show/NCT01697072). The aim of the study is to evaluate if the treatment of ECX in combination with Rilotumumab (15 mg/kg, IV every 21 days) leads to a better clinical outcome, evaluating OS, PFS, time to progression, objective response rate (ORR), disease control rate, time to response and immunogenicity in subjects with un-resettable locally advanced or metastatic MET positive gastric or GEJ cancer.
Crizotinib/axitinib
The hypothesis already discussed in the first part of this review that MET and HGF contribute to VEGF inhibitor resistance, is currently tested in a phase 1b, open label, dose escalation study combining Axitinib (N-methyl-2-[[3-[(E)-2-pyridin-2-ylethenyl]-1H-indazol-6-yl]sulfanyl]benzamide) with Crizotinib (3-[(\1R)-\1-(\2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine) in patients with advanced solid tumors (https://clinicaltrials.gov/ct2/show/NCT01999972). Axitinib is a potent and selective tyrosine kinase inhibitor of VEGF receptor (VEGFR) 1, 2, and 3 [94]. Crizotinib is a MET and ALK inhibitor, actually in use for patients with ALK-positive NSCLC [95]. This clinical trial is really innovative and complex, since this is the first time that Axitinib and Crizotinib are administered in combination. The first part of the study will be essential to evaluate if the oral administration of Axitinib (2-5 mg, twice daily for 28 days cycles) and Crizotinib (200-250 mg, twice daily for 28 days cycles) has an anti-cancer activity in advanced solid tumors. Moreover in this first part the effective dosage that will be administered, will be defined in advanced RCC to evaluate safety, pharmacokinetics and pharmacodynamics.
Cabozantinib
Cabozantinib (1-N-[4-(6,7-dimethoxyquinolin-4-yl)oxyphenyl]-1-N’-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide) is an oral medicine that inhibits MET, RET, ROS1, NTRK1, and AXL. The inhibiting activities of Cabozantinib towards MET and AXL is relevant. In fact, researchers found AXL, a member of receptor tyrosine kinase family [96] to increase EMT phenotype, commonly activated in HTM [97]. A key-role of this increased expression is due again by HIF that directly activates AXL gene transcription by binding to the hypoxia-response element within the AXL proximal promoter [98]. Recently, it has been demonstrated that increased AXL expression could contribute to drug resistance in cancer cell lines. In fact, AXL down-regulation inhibits cell survival and overcomes drug resistance in vitro. AXL could activate the downstream signals such as AKT and ERK1 to promote cell survival and growth. Therefore, AXL inhibitors could be effective in abrogating the growth of tumor cells [69]. Thus, through both MET and AXL inhibitions, Cabozantinib was shown to induce cancer shrinkage and cell growth reduction in medullary thyroid and prostate carcinoma. Currently, a phase 2 study is evaluating Cabozantinib in patients with RET fusion- (Group A) and those with ROS1 or NTRK1 fusions or increased MET or AXL activity (Group B) in advanced NSCLC (https://clinicaltrials.gov/ct2/show/NCT01639508). During the period of 28 days cycle, 60 mg of Cabozantinib will be administered to patients orally daily, to evaluate positive and negative effects in tumors with genes changes. Moreover, in group A and B ORR, PFS and OS will be evaluated.
METABOLIC CHANGES AND THERAPEUTIC TARGETS
Warburg effect, mitochondria changes and cancer stem cells
In parallel with the adaptation to a HTM, deep changes are observed also in the mitochondrial functions. A well-known phenomenon commonly found in cancer cells is the so-called Warburg effect (WE). While in normal tissues, glucose is bio-transformed to pyruvate and carried into the mitochondria for the oxidative phosphorylation (OXPHOS) (Figure 4a), in cancer, the glycolysis is 200-fold more active than the normal, either in the presence or absence of oxygen (aerobic glycolysis/WE) leading to high amount of lactate [99] (Figure 4b). Despite the first identification of the WE dates back to 1927, the mechanisms triggering WE are not fully understood, yet. It could be the consequence of damaged mitochondria in cancer cells, or of the adaptation of cancer cells to HTM. The up-regulation of the aerobic glycolysis seems related to oncogene activation. It appears that almost every major oncogene that drives oncogenesis also drives increased glucose metabolism. For example, activating mutations within BRAF, PTEN, and KRAS, were found to have effects in the transcriptional activation and repression of key metabolic enzymes and in the reprogramming of glutamine metabolism [100-102]. WE could be also triggered by the stabilization of HIF1A. In fact, once HIF1A is stabilized, it is observed that cancer cells increase glycolysis, where a good proportion of glucose is converted into lactate instead going through OXPHOS, therefore creating an acidic microenvironment and leading to an inefficient ATP production [103]. It is well-established that this change is important for survival in hypoxic conditions and it correlates also with CSCs formation. In fact, mitochondria show differences between CSCs and non-CSCs. For example, CSCs from lung cancer and leukemia showed low mitochondrial DNA content, OXPHOS, oxygen consumption rate, intracellular ATP and an enrichment of specific biomarkers (ALDH, CD24, CD44, CD133) responsible for their formation and survival [104]. However, these facts should not mislead regarding to the role of mitochondria in CSCs. In fact, it was also observed that the clonal expansion and survival of CSCs is, actually, dependent on mitochondrial biogenesis suggesting that the mitochondrial metabolism, although reduced, is crucial in CSC survival. In particular, CSCs have been shown to be dependent on OXPHOS. In fact, when an inhibitor of ATP-synthase activity is used, CSCs usually are killed [105]. Although, a reduced mitochondrion activity is observed in cancer, in oncocytoma a paradoxical effect is observed. These “oxyphilic” tumors (typically RCC, ovarian or thyroid cancer) have an impressively abnormal number of mitochondria in their cytoplasm [106-109]. A model was proposed where cancer genes could be responsible for a reduced mitochondria metabolism leading to an inefficient ATP production, and where the mitochondrial hyperplasia is a consequence of a compensatory effect in the attempt, unsuccessful, to restore the normal ATP production [110]. Among the players discovered to be involved in WE and related metabolic changes the Hsp90 chaperon has a role in maintaining energy production under nutrient deprivation [111]. Moreover, SLC2A1 and MCT4 were found frankly over-expressed in cancer cells in correlation to an improved glucose supply, and lactate disposal [112]. In addition, in gastric cancer also MACC1 was found up-regulated and it was found to contribute in triggering the WE through the up-regulation of glycolytic enzymes [113]. It has been hypothesized that the changes in mitochondrion activity could be a weak-point for CSCs [114]. Thus, novel therapeutic approaches were proposed based on the use of antibiotics targeting mitochondrion [105], in the reminiscence of its prokaryotic origins. Five classes of mitochondrial-targeted antibiotics including erythromycin, tetracycline, glycylcycline, and chloramphenicol effectively reduce CSCs formation [105, 115] and similar results were obtained with oligomycin A, an inhibitor of the mitochondrial ATP synthase [115]. When considering more in general specific mitochondrion inhibitors, a recent work showed that a specific Monocarboxylate Transporters (MCTs) inhibitor (AR-C155858), which blocks the cellular uptake of mitochondrial fuels, such as lactate, could effectively reduce the formation of CSCs [115]. Moreover, novel molecules, such as NV-128, an isoflavone-derivative, showed a significant activity in reducing the mitochondrial function (with decrease in ATP, COX1, and COX4 levels), leading to increased mitochondrial superoxide and hydrogen peroxide. This promoted a state of cellular starvation and oxidation that activated two independent pathways: (i) AMPKa1 pathway leading to MTOR inhibition; and (ii) mitochondrial MAPK/ERK kinase/extracellular signal pathway, leading to loss of mitochondrial membrane potential [116] with an inhibition of CSCs formation. It should be stressed that normal cells appeared more resistant to these inhibitors as compared to CSCs. This fact should provide a wide therapeutic window for reducing the side effects for cancer patients.
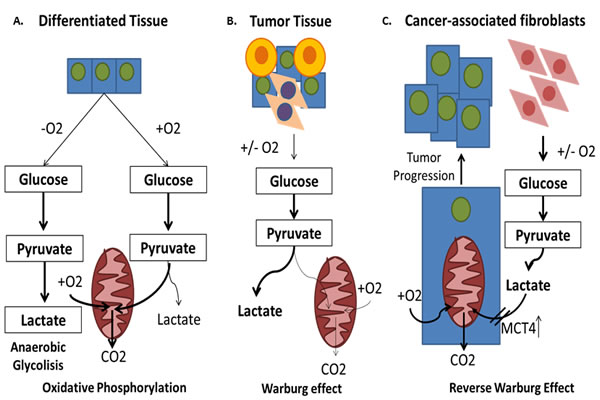
Figure 4: The Warburg effect and the reverse Warburg effect. A. In normal tissues, glucose is bio-transformed to pyruvate and carried into the mitochondria for the oxidative phosphorylation (OXPHOS). B. Most types of cancer engage themselves in glycolysis, irrespective to the presence of oxygen (aerobic glycolysis or Warburg Effect). C. Some cancer cells reprogram cancer associated fibroblasts (CAFs) to undergo aerobic glycolysis (WE) and to secrete energy-rich nutrients that feed into mitochondrial oxidative metabolism in cancer cells.
Reverse Warburg Effect and metabolic symbiosis
Once the research on cancer metabolism advances, it was found that WE is not a general rule. In fact, they are highly flexible in adapting to the micro-environmental conditions and they can easily reactivate the mitochondrial OXPHOS [117]. Differences in the metabolic phenotype can even occur within a single tumor mass [118]. Thus, for some tumors, cancer cells are found not completely dependent on accelerated glycolysis. Interestingly, it was discovered that they are “fed” through the activation of normal stromal cells surrounding cancer cells [119]. For example, it has been shown that the activation of HIF1A occurs in carcinoma-associated fibroblasts (CAFs) and it enhances aerobic glycolysis and lactate production, which is converted to pyruvate and utilized for mitochondrial OXPHOS in cancer cells [120, 121]. This phenomenon has been called ‘‘reverse WE’’ (RWE), which indicates increased aerobic glycolysis of stromal cells (CAFs) adjacent to cancer cells, which secrete energy-rich nutrients that feed cancer cells [122], leading to an acidic TM, increased tumor growth and malignant behaviors [123]. This host-parasite relationship is characterized also by the loss of caveolin-1 (CAV1), a molecule that has been shown to be responsible for HIF1A activation in stromal cells [120] and by the increase of MCTs and in particular MCT4, a transporter responsible for the regulation of both energetic supply and intracellular pH [124] (Figure 4c). Based on this consideration, the development of anticancer agents that target the enzymes involved in glycolysis appears to be promising.
NOVEL THERAPEUTIC APPROACHES TARGETING METABOLIC CHANGES
Targeting mitochondrion dysfunction
The mitochondrion changes discussed in the last two sections prompted clinical researchers to focus on the identification of new compounds able to inhibit some of the involved molecules. Hereon, we will describe novel therapeutic agents developed to these purposes (Table 1, Figure 3).
CPI-613
CPI-613 (6,8-bis(benzylsulfanyl)octanoic acid) is a molecule able to kill cancer cells through the shutdown of mitochondria. Consequently the deprivation of energy and other supplies, lead to the inhibition of cell growth. CPI-613 is currently involved in a pilot phase I trial (https://clinicaltrials.gov/ct2/show/NCT02232152) in combination with fluorouracil in patients with non-resettable metastatic colorectal cancer. The aim of the study is to evaluate if there is a synergism between a chemotherapy drug (fluorouracil, administered IV over 46 hours on days 2-4) and an inhibitor of mitochondria (CP-613, administered IV over 2 hours on days 1-4) to determine the Maximum Tolerated Dose, pharmacokinetic and safety in patients who have failed FOLFOX (leucovorin calcium, fluorouracil and oxaliplatin), or FOLFIRI (leucovorin calcium, fluorouracil, and irinotecan hydrochloride) therapies.
Fenofibrate
Fenofibrate (propan-2-yl 2-[4-(4-chlorobenzoyl)phenoxy]-2-methylpropanoate), that is currently in use to treat hyperlipoproteinemia (i.e high level of cholesterol and/or triglycerides) [125], has been shown to inhibit mitochondrial function by blocking ATP synthesis and inhibiting electron transport at NADH-ubiquinone (UQ) oxidoreductase. This inhibition leads to the induction of a stress signal known as the unfolded protein response and subsequently apoptosis, in particular in multiple myeloma. Thus fenofibrate is now studied in a phase II clinical trial (https://clinicaltrials.gov/ct2/show/NCT01965834) with the aim to determine response rate, safety and tolerability in multiple myeloma patients.
Anti-bacterial agents
A combination of two selected antibiotics from doxycycline, paramomycin, clarithromycin, clindamycin, dapsone, miltefosine, targeting mithocondria and with documented anti-cancer properties are currently employed in a phase 2 clinical trial (https://clinicaltrials.gov/ct2/show/study/NCT02366884) alone and/or in combination with anti-fungal and anti-protozoal agents. The aim of this study is to investigate the clinical efficacy of these agents in patients with malignant disease confirmed histologically that is considered untreatable, progressive and fatal within the next 16 months. Tumor regression is expected within 6 months of treatment and to avoid tumor recurrence the treatment will be continued until 10-12 months. This drug repositioning is interesting because we already know safety, tolerability and side effects.
Anti-bacterial pro-drug activated in hypoxia: TH-302
TH-302 is a prodrug that is activated only under hypoxic conditions; this characteristic prevents the production of a broad systemic toxicity, typical of chemotherapy. Within regions of tumor hypoxia, TH-302 releases a potent DNA alkylating agent called bromo-isophosphoramide mustard [126] that is responsible for blocking the replication and transcription of DNA, leading to cancer cell growth inhibition. Moreover, once TH-302 is activated in hypoxia, it can diffuse to surrounding regions inhibiting the cell growth also in oxygenated cancer tissues, via a “bystander effect” [127]. To date, TH-302 is involved in a randomized phase 3, multicenter, open-label study, in combination with Doxorubicin in patients with locally advanced un-resettable or metastatic soft tissue sarcoma (https://clinicaltrials.gov/ct2/show/NCT01440088). The clinical trial aims to evaluate if Doxorubicin that usually has poor tissue penetration and acts in regions of tumors that are located in proximity to the tumor vessels, in combination with TH-302 could increase the OS in patients with cancers where the presence of hypoxia is high, as shown in soft tissue sarcomas [128]. TH-302 will be administered by IV infusion on day 1 and 8 for 21-days. Doxorubicin will be administered on day 1, after the TH-302 infusion. The pharmacokinetics of each compound will be investigated.
Targeting stromal-epithelial metabolic coupling (RWE)
Metformin
Metformin (1,1-Dimethylbiguanide) is an antidiabetic medication used particularly for the treatment of type 2 diabetes, decreasing hyperglycemia primarily by suppressing gluconeogenesis. Recently metformin is used also for the polycystic ovary syndrome in patients with insulin resistance [129]. Currently metformin is involved in a phase 0 clinical trial (https://clinicaltrials.gov/ct2/show/study/NCT02083692 ) in patients with head and neck squamous cell cancer. The main purpose of the study is to evaluate if metformin is responsible for a decrease of MCT4 expression in fibroblast and its effect on tumor metabolism.
Topotecan/ acetylcysteine
Topotecan (9-Dimethylaminomethyl-10-hydroxycamptothecin) is an anticancer agent used in particular for the treatment of ovarian cancer. Acetylcysteine (N-Acetyl-L-cysteine) is used mainly as a mucolytic agent and it seems to block stromal induction of MCT4 [130]. Currently a phase 2 study (https://clinicaltrials.gov/ct2/show/study/NCT02569957) is recruiting patients with ovarian cancer. The aim of this study is to evaluate if the administration of acetylcysteine in combination with topotecan, can inhibit the cancer cell growth, due to changes in MCT4, CAV1 and HIF1A.
CONCLUSIONS
Tumor hypoxia is responsible for radio- and chemo-resistance as well as predisposing for increased CSCs formation and tumor metastases. Pro-angiogenic factors are synthesized and released from tumor, stromal, endothelial, and myeloid cells in response to HTM which are commonly found during cancer progression. The formation of an aberrant and heterogeneous vascular network is a key pathological event in the multistep process of tumor growth and metastasis. The use of VEGF inhibitors, such as avastin, gives some results, especially in colorectal, breast, lung cancer and glioblastoma where it is able to induce vascular normalization and reducing hypoxia (leading to improved efficacy of conventional therapies) [131] nevertheless anti-angiogenic drugs have been shown not to be always effective in the treatment of cancer, particularly for the highly aggressive ones [12]. These considerations have raised the questions that probably angiogenesis is not the main responsible of the tumor progression, shifting the focus on the metabolic changes associated with HTM. It has been thought that inhibiting the response to acidosis and hypoxia and blocking the proteins strictly correlated to HIF1A, EMT and mitochondria functions could be a promising way to treat this disease and worth to be further investigated. The clinical trials described in this review will provide in relatively short time new answers to the researchers of whether this approach will yield results worth to be fully undertaken.
AKNOWLEDGMENTS
This review was supported by the “Italian Group for the Study and Therapy of Malignant Mesothelioma” (GIMe:www.gime.it).
CONFLICTS OF INTEREST
All the authors declare no potential conflict of interest
REFERENCES
1. Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol. 2006; 70(5): 1469-80.
2. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995; 92(12): 5510-4.
3. Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007; 26(3-4): 489-502.
4. Ribatti D, Crivellato E. “Sprouting angiogenesis”, a reappraisal. Dev Biol. 2012; 372(2): 157-65.
5. Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003; 9(6): 677-84.
6. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989; 49(23): 6449-65.
7. Multhoff G, Radons J, Vaupel P. Critical role of aberrant angiogenesis in the development of tumor hypoxia and associated radioresistance. Cancers (Basel). 2014; 6(2): 813-28.
8. Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008; 8(6): 425-37.
9. Tournigand C, Chibaudel B, Samson B, Scheithauer W, Vernerey D, Mésange P, Lledo G, Viret F, Ramée JF, Tubiana-Mathieu N, Dauba J, Dupuis O, Rinaldi Y, et al. Bevacizumab with or without erlotinib as maintenance therapy in patients with metastatic colorectal cancer (GERCOR DREAM; OPTIMOX3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2015; 16(15): 1493-505.
10. Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006; 3(1): 24-40.
11. Kerbel RS. Reappraising antiangiogenic therapy for breast cancer. Breast. 2011; 20 Suppl 3: S56-60.
12. Choi HJ, Armaiz Pena GN, Pradeep S, Cho MS, Coleman RL, Sood AK. Anti-vascular therapies in ovarian cancer: moving beyond anti-VEGF approaches. Cancer Metastasis Rev. 2015; 34(1): 19-40.
13. Sennino B, McDonald DM. Controlling escape from angiogenesis inhibitors. Nat Rev Cancer. 2012; 12(10): 699-709.
14. Xu H, Rahimpour S, Nesvick CL, Zhang X, Ma J, Zhang M, Zhang G, Wang L, Yang C, Hong CS, Germanwala AV, Elder JB, Ray-Chaudhury A, et al. Activation of hypoxia signaling induces phenotypic transformation of glioma cells: implications for bevacizumab antiangiogenic therapy. Oncotarget. 2015; 6(14): 11882-93. doi: 10.18632/oncotarget.3592.
15. Cavazos DA, Brenner AJ. Hypoxia in astrocytic tumors and implications for therapy. Neurobiol Dis. 2015; pii: S0969-9961(15)00225-9.
16. Kogita A, Togashi Y, Hayashi H, Sogabe S, Terashima M, De Velasco MA, Sakai K, Fujita Y, Tomida S, Takeyama Y, Okuno K, Nakagawa K, Nishio K. Hypoxia induces resistance to ALK inhibitors in the H3122 non-small cell lung cancer cell line with an ALK rearrangement via epithelial-mesenchymal transition. Int J Oncol. 2014; 45(4): 1430-6.
17. Yang YJ, Na HJ, Suh MJ, Ban MJ, Byeon HK, Kim WS, Kim JW, Choi EC, Kwon HJ, Chang JW, Koh YW. Hypoxia Induces Epithelial-Mesenchymal Transition in Follicular Thyroid Cancer: Involvement of Regulation of Twist by Hypoxia Inducible Factor-1α. Yonsei Med J. 2015; 56(6): 1503-14.
18. Liu S, Kumar SM, Martin JS, Yang R, Xu X. Snail1 mediates hypoxia-induced melanoma progression. Am J Pathol. 2011; 179(6): 3020-31.
19. Peitzsch C, Perrin R, Hill RP, Dubrovska A, Kurth I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int J Radiat Biol. 2014; 90(8): 636-52.
20. Guo J, Wang B, Fu Z, Wei J, Lu W. Hypoxic Microenvironment Induces EMT and Upgrades Stem-Like Properties of Gastric Cancer Cells. Technol Cancer Res Treat. 2015; pii: 1533034614566413.
21. Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997; 272(36): 22642-7.
22. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119(6): 1420-8.
23. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010; 29(34): 4741-51.
24. Sutherland RM. Cell and environment interactions in tumor microregions: the multicell spheroid model. Science. 1988; 240(4849): 177-84.
25. Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004; 9 Suppl 5: 10-7.
26. Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008; 10(3): 295-305.
27. Chou CW, Wang CC, Wu CP, Lin YJ, Lee YC, Cheng YW, Hsieh CH. Tumor cycling hypoxia induces chemoresistance in glioblastoma multiforme by upregulating the expression and function of ABCB1. Neuro Oncol. 2012; 14(10): 1227-38.
28. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011; 331(6024): 1559-64.
29. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011; 147(2): 275-92.
30. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999; 15; 59(22): 5830-5.
31. Yang QC, Zeng BF, Dong Y, Shi ZM, Jiang ZM, Huang J. Overexpression of hypoxia-inducible factor-1alpha in human osteosarcoma: correlation with clinicopathological parameters and survival outcome. Jpn J Clin Oncol. 2007; 37(2): 127-34.
32. Hung JJ, Yang MH, Hsu HS, Hsu WH, Liu JS, Wu KJ. Prognostic significance of hypoxia-inducible factor-1alpha, TWIST1 and Snail expression in resectable non-small cell lung cancer. Thorax. 2009; 64(12): 1082-9.
33. Kaya AO, Gunel N, Benekli M, Akyurek N, Buyukberber S, Tatli H, Coskun U, Yildiz R, Yaman E, Ozturk B. Hypoxia inducible factor-1 alpha and carbonic anhydrase IX overexpression are associated with poor survival in breast cancer patients. J BUON. 2012; 17(4): 663-8.
34. Colbert LE, Fisher SB, Balci S, Saka B, Chen Z, Kim S, El-Rayes BF, Adsay NV, Maithel SK, Landry JC, Curran WJ Jr. High nuclear hypoxia-inducible factor 1 alpha expression is a predictor of distant recurrence in patients with resected pancreatic adenocarcinoma. Int J Radiat Oncol Biol Phys. 2015; 91(3): 631-9.
35. Zou YM, Hu GY, Zhao XQ, Lu T, Zhu F, Yu SY, Xiong H. Hypoxia-induced autophagy contributes to radioresistance via c-Jun-mediated Beclin1 expression in lung cancer cells. J Huazhong Univ Sci Technolog Med Sci. 2014; 34(5): 761-7.
36. Zhu H, Wang D, Zhang L, Xie X, Wu Y, Liu Y, Shao G, Su Z. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol Rep. 2014; 32(3): 935-42.
37. Tan EY, Yan M, Campo L, Han C, Takano E, Turley H, Candiloro I, Pezzella F, Gatter KC, Millar EK, O’Toole SA, McNeil CM, Crea P, et al. The key hypoxia regulated gene CAIX is upregulated in basal-like breast tumours and is associated with resistance to chemotherapy. Br J Cancer. 2009; 100(2): 405-11.
38. Grabmaier K, A de Weijert MC, Verhaegh GW, Schalken JA, Oosterwijk E. Strict regulation of CAIX(G250/MN) by HIF-1alpha in clear cell renal cell carcinoma. Oncogene. 2004; 23(33): 5624-31.
39. Wang JQ, Wu KJ. Epigenetic regulation of epithelial-mesenchymal transition by hypoxia in cancer: targets and therapy. Curr Pharm Des. 2015; 21(10): 1272-8.
40. Liu N, Wang Y, Zhou Y, Pang H, Zhou J, Qian P, Liu L, Zhang H. Krüppel-like factor 8 involved in hypoxia promotes the invasion and metastasis of gastric cancer via epithelial to mesenchymal transition. Oncol Rep. 2014; 32(6): 2397-404.
41. Zhang H, Sun L, Xiao X, Xie R, Liu C, Wang Y, Wei Y, Zhang H, Liu L. Krüppel-like factor 8 contributes to hypoxia-induced MDR in gastric cancer cells. Cancer Sci. 2014; 105(9): 1109-15.
42. Fujikuni N, Yamamoto H, Tanabe K, Naito Y, Sakamoto N, Tanaka Y, Yanagihara K, Oue N, Yasui W, Ohdan H. Hypoxia-mediated CD24 expression is correlated with gastric cancer aggressiveness by promoting cell migration and invasion. Cancer Sci. 2014; 105(11): 1411-20.
43. Wang Y, Liu X, Zhang H, Sun L, Zhou Y, Jin H, Zhang H, Zhang H, Liu J, Guo H, Nie Y, Wu K, et al. Hypoxia-inducible lncRNA-AK058003 promotes gastric cancer metastasis by targeting γ-synuclein. Neoplasia. 2014; 16(12): 1094-106.
44. Krishnamachary B, Penet MF, Nimmagadda S, Mironchik Y, Raman V, Solaiyappan M, Semenza GL, Pomper MG, Bhujwalla ZM. Hypoxia regulates CD44 and its variant isoforms through HIF-1α in triple negative breast cancer. PLoS One. 2012; 7(8): e44078.
45. Onishi H, Morifuji Y, Kai M, Suyama K, Iwasaki H, Katano M. Hedgehog inhibitor decreases chemosensitivity to 5-fluorouracil and gemcitabine under hypoxic conditions in pancreatic cancer. Cancer Sci. 2012; 103(7): 1272-9.
46. Onishi H, Morisaki T, Nakao F, Odate S, Morisaki T, Katano M. Protein-bound polysaccharide decreases invasiveness and proliferation in pancreatic cancer by inhibition of hedgehog signaling and HIF-1α pathways under hypoxia. Cancer Lett. 2013; 335(2): 289-98.
47. Goudarzi H, Hida Y, Takano H, Teramae H, Iizasa H, Hamada J. Hypoxia affects in vitro growth of newly established cell lines from patients with malignant pleural mesothelioma. Biomed Res. 2013; 34(1): 13-21.
48. Tao LL, Shi SJ, Chen LB, Huang GC. Expression of monocyte chemotactic protein-1/CCL2 in gastric cancer and its relationship with tumor hypoxia. World J Gastroenterol. 2014; 20(15): 4421-7.
49. Oh YS, Kim HY, Song IC, Yun HJ, Jo DY, Kim S, Lee HJ. Hypoxia induces CXCR4 expression and biological activity in gastric cancer cells through activation of hypoxia-inducible factor-1α. Oncol Rep. 2012; 28(6): 2239-46.
50. Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, Thompson B, Maiso P, Sun JD, Hart CP, Roccaro AM, Sacco A, Ngo HT, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012; 119(24): 5782-94.
51. Cheng S, Han L, Guo J, Yang Q, Zhou J, Yang X. The essential roles of CCR7 in epithelial-to-mesenchymal transition induced by hypoxia in epithelial ovarian carcinomas. Tumour Biol. 2014; 35(12): 12293-8.
52. Zhao T, Gao S, Wang X, Liu J, Duan Y, Yuan Z, Sheng J, Li S, Wang F, Yu M, Ren H, Hao J. Hypoxia-inducible factor-1α regulates chemotactic migration of pancreatic ductal adenocarcinoma cells through directly transactivating the CX3CR1 gene. PLoS One. 2012; 7(8): e43399.
53. Zhang H, Chen J, Liu F, Gao C, Wang X, Zhao T, Liu J, Gao S, Zhao X, Ren H, Hao J. CypA, a gene downstream of HIF-1α, promotes the development of PDAC. PLoS One. 2014; 9(3): e92824.
54. Spinella F, Caprara V, Cianfrocca R, Rosanò L, Di Castro V, Garrafa E, Natali PG, Bagnato A. The interplay between hypoxia, endothelial and melanoma cells regulates vascularization and cell motility through endothelin-1 and vascular endothelial growth factor. Carcinogenesis. 2014; 35(4): 840-8.
55. Zhao X, Gao S, Ren H, Sun W, Zhang H, Sun J, Yang S, Hao J. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014; 74(9): 2455-64.
56. Zhou J, Li K, Gu Y, Feng B, Ren G, Zhang L, Wang Y, Nie Y, Fan D. Transcriptional up-regulation of RhoE by hypoxia-inducible factor (HIF)-1 promotes epithelial to mesenchymal transition of gastric cancer cells during hypoxia. Biochem Biophys Res Commun. 2011; 415(2): 348-54.
57. Nurwidya F, Takahashi F, Kobayashi I, Murakami A, Kato M, Minakata K, Nara T, Hashimoto M, Yagishita S, Baskoro H, Hidayat M, Shimada N, Takahashi K. Treatment with insulin-like growth factor 1 receptor inhibitor reverses hypoxia-induced epithelial-mesenchymal transition in non-small cell lung cancer. Biochem Biophys Res Commun. 2014; 455(3-4): 332-8.
58. Liu Q, Xu Z, Mao S, Chen W, Zeng R, Zhou S, Liu J. Effect of hypoxia on hypoxia inducible factor-1α, insulin-like growth factor I and vascular endothelial growth factor expression in hepatocellular carcinoma HepG2 cells. Oncol Lett. 2015; 9(3): 1142-1148.
59. Goudarzi H, Iizasa H, Furuhashi M, Nakazawa S, Nakane R, Liang S, Hida Y, Yanagihara K, Kubo T, Nakagawa K, Kobayashi M, Irimura T, Hamada J. Enhancement of in vitro cell motility and invasiveness of human malignant pleural mesothelioma cells through the HIF-1α-MUC1 pathway. Cancer Lett. 2013; 339(1): 82-92.
60. Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X, Zhang B, Mehla K, Brown RB, Caffrey T, Yu F, Johnson KR, Powers R, et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci U S A. 2012; 109(34): 13787-92.
61. Aubert S, Fauquette V, Hémon B, Lepoivre R, Briez N, Bernard D, Van Seuningen I, Leroy X, Perrais M. MUC1, a new hypoxia inducible factor target gene, is an actor in clear renal cell carcinoma tumor progression. Cancer Res. 2009; 69(14): 5707-15.
62. Mikami Y, Hisatsune A, Tashiro T, Isohama Y, Katsuki H. Hypoxia enhances MUC1 expression in a lung adenocarcinoma cell line. Biochem Biophys Res Commun. 2009; 379(4): 1060-5.
63. Zhu S, Zhou Y, Wang L, Zhang J, Wu H, Xiong J, Zhang J, Tian Y, Wang C, Wu H. Transcriptional upregulation of MT2-MMP in response to hypoxia is promoted by HIF-1α in cancer cells. Mol Carcinog. 2011; 50(10): 770-80.
64. Matsuoka J, Yashiro M, Doi Y, Fuyuhiro Y, Kato Y, Shinto O, Noda S, Kashiwagi S, Aomatsu N, Hirakawa T, Hasegawa T, Shimizu K, Shimizu T, et al. Hypoxia stimulates the EMT of gastric cancer cells through autocrine TGFβ signaling. PLoS One. 2013; 8(5): e62310.
65. Marconi C, Peppicelli S, Bianchini F, Calorini L. TNFα receptor1 drives hypoxia-promoted invasiveness of human melanoma cells. Exp Oncol. 2013; 35(3): 187-91.
66. Chuang MJ, Sun KH, Tang SJ, Deng MW, Wu YH, Sung JS, Cha TL, Sun GH. Tumor-derived tumor necrosis factor-alpha promotes progression and epithelial-mesenchymal transition in renal cell carcinoma cells. Cancer Sci. 2008; 99(5): 905-13.
67. Sawabu T, Seno H, Kawashima T, Fukuda A, Uenoyama Y, Kawada M, Kanda N, Sekikawa A, Fukui H, Yanagita M, Yoshibayashi H, Satoh S, Sakai Y, et al. Growth arrest-specific gene 6 and Axl signaling enhances gastric cancer cell survival via Akt pathway. Mol Carcinog. 2007; 46(2): 155-64.
68. D’Alfonso TM, Hannah J, Chen Z, Liu Y, Zhou P, Shin SJ. Axl receptor tyrosine kinase expression in breast cancer. J Clin Pathol. 2014; 67(8): 690-6.
69. Wu F, Li J, Jang C, Wang J, Xiong J. The role of Axl in drug resistance and epithelial-to-mesenchymal transition of non-small cell lung carcinoma. Int J Clin Exp Pathol. 2014; 7(10): 6653-61.
70. Pinato DJ, Mauri FA, Lloyd T, Vaira V, Casadio C, Boldorini RL, Sharma R. The expression of Axl receptor tyrosine kinase influences the tumour phenotype and clinical outcome of patients with malignant pleural mesothelioma. Br J Cancer. 2013; 108(3): 621-8.
71. Goyal L, Muzumdar MD, Zhu AX. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res. 2013; 19(9): 2310-8.
72. Michelsen J, Thiesson H, Walter S, Ottosen PD, Skøtt O, Jensen BL. Tissue expression and plasma levels of adrenomedullin in renal cancer patients. Clin Sci (Lond). 2006; 111(1): 61-70.
73. Keleg S, Kayed H, Jiang X, Penzel R, Giese T, Büchler MW, Friess H, Kleeff J. Adrenomedullin is induced by hypoxia and enhances pancreatic cancer cell invasion. Int J Cancer. 2007; 121(1): 21-32.
74. Chou CC, Chuang HC, Salunke SB, Kulp SK, Chen CS. A novel HIF-1α-integrin-linked kinase regulatory loop that facilitates hypoxia-induced HIF-1α expression and epithelial-mesenchymal transition in cancer cells. Oncotarget. 2015; 6(10): 8271-85. doi: 10.18632/oncotarget.3186.
75. Chu K, Boley KM, Moraes R, Barsky SH, Robertson FM. The paradox of E-cadherin: role in response to hypoxia in the tumor microenvironment and regulation of energy metabolism. Oncotarget. 2013; 4(3): 446-62. doi: 10.18632/oncotarget.872.
76. Newman RA, Yang P, Pawlus AD, Block KI. Cardiac glycosides as novel cancer therapeutic agents. Mol Interv. 2008; 8(1): 36-49.
77. Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, Dang CV, Liu JO, Semenza GL. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008; 105(50): 19579-86.
78. Lin J, Denmeade S, Carducci MA. HIF-1alpha and calcium signaling as targets for treatment of prostate cancer by cardiac glycosides. Curr Cancer Drug Targets. 2009; 9(7): 881-7.
79. White PT, Subramanian C, Zhu Q, Zhang H, Zhao H, Gallagher R, Timmermann BN, Blagg BS, Cohen MS. Novel HSP90 inhibitors effectively target functions of thyroid cancer stem cell preventing migration and invasion. Surgery. 2015; pii: S0039-6060(15)00741-2.
80. Pillai RN, Ramalingam SS. Heat shock protein 90 inhibitors in non-small-cell lung cancer. Curr Opin Oncol. 2014; 26(2): 159-64.
81. Xiang L, Gilkes DM, Chaturvedi P, Luo W, Hu H, Takano N, Liang H, Semenza GL. Ganetespib blocks HIF-1 activity and inhibits tumor growth, vascularization, stem cell maintenance, invasion, and metastasis in orthotopic mouse models of triple-negative breast cancer. J Mol Med (Berl). 2014; 92(2): 151-64.
82. Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, Paschold E, Salgia R, West H, Sequist LV, Bonomi P, Brahmer J, Chen LC, et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res. 2013; 19(11): 3068-77.
83. Lock FE, McDonald PC, Lou Y, Serrano I, Chafe SC, Ostlund C, Aparicio S, Winum JY, Supuran CT, Dedhar S. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene. 2013; 32(44): 5210-9.
84. Touisni N, Maresca A, McDonald PC, Lou Y, Scozzafava A, Dedhar S, Winum JY, Supuran CT. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem. 2011; 54(24): 8271-7.
85. Couvelard A, O’Toole D, Leek R, Turley H, Sauvanet A, Degott C, Ruszniewski P, Belghiti J, Harris AL, Gatter K, Pezzella F. Expression of hypoxia-inducible factors is correlated with the presence of a fibrotic focus and angiogenesis in pancreatic ductal adenocarcinomas. Histopathology. 2005; 46(6): 668-76.
86. Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, Kyle A, Auf dem Keller U, Leung S, Huntsman D, Clarke B, Sutherland BW, Waterhouse D, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011; 71(9): 3364-76.
87. Ledaki I, McIntyre A, Wigfield S, Buffa F, McGowan S, Baban D, Li JL, Harris AL. Carbonic anhydrase IX induction defines a heterogeneous cancer cell response to hypoxia and mediates stem cell-like properties and sensitivity to HDAC inhibition. Oncotarget. 2015; 6(23): 19413-27. doi: 10.18632/oncotarget.4989.
88. Rami M, Dubois L, Parvathaneni NK, Alterio V, van Kuijk SJ, Monti SM, Lambin P, De Simone G, Supuran CT, Winum JY. Hypoxia-targeting carbonic anhydrase IX inhibitors by a new series of nitroimidazole-sulfonamides/sulfamides/sulfamates. J Med Chem. 2013; 56(21): 8512-20.
89. Ogunwobi OO, Liu C. Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via Akt and COX-2 pathways. Clin Exp Metastasis. 2011; 28(8): 721-31.
90. Grotegut S, von Schweinitz D, Christofori G, Lehembre F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006; 25(15): 3534-45.
91. Cañadas I, Rojo F, Taus Á, Arpí O, Arumí-Uría M, Pijuan L, Menéndez S, Zazo S, Dómine M, Salido M, Mojal S, García de Herreros A, Rovira A, et al. Targeting epithelial-to-mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin Cancer Res. 2014; 20(4): 938-50.
92. Yu F, Lin Y, Zhan T, Chen L, Guo S. HGF expression induced by HIF-1α promote the proliferation and tube formation of endothelial progenitor cells. Cell Biol Int. 2015; 39(3): 310-7.
93. Chu SH, Feng DF, Ma YB, Zhu ZA, Zhang H, Qiu JH. Stabilization of hepatocyte growth factor mRNA by hypoxia-inducible factor 1. Mol Biol Rep. 2009; 36(7): 1967-75.
94. Chen Y, Suzuki A, Tortorici MA, Garrett M, LaBadie RR, Umeyama Y, Pithavala YK. Axitinib plasma pharmacokinetics and ethnic differences. Invest New Drugs. 2015; 33(2): 521-32.
95. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010; 363(18): 1693-703.
96. Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000; 19(49): 5548-57.
97. Mishra A, Wang J, Shiozawa Y, McGee S, Kim J, Jung Y, Joseph J, Berry JE, Havens A, Pienta KJ, Taichman RS. Hypoxia stabilizes GAS6/Axl signaling in metastatic prostate cancer. Mol Cancer Res. 2012; 10(6): 703-12.
98. Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN, LaGory EL, Kariolis MS, Chan A, Lindgren D, Axelson H, Miao YR, Krieg AJ, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A. 2014; 111(37): 13373-8
99. Lu J, Tan M, Cai Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015; 356(2 Pt A): 156-64.
100. Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget. 2013; 4(4): 584-99. doi: 10.18632/oncotarget.965.
101. Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, Pearson RB, McGee SL, Long GV, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014; 4(4): 423-33.
102. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013; 496(7443): 101-5.
103. Muñoz-Pinedo C, El Mjiyad N, Ricci JE. Cancer metabolism: current perspectives and future directions. Cell Death Dis. 2012; 3: e248.
104. Farnie G, Sotgia F, Lisanti MP. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget. 2015; 6(31): 30472-86. doi: 10.18632/oncotarget.5401.
105. Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget. 2015; 6(7): 4569-84. doi: 10.18632/oncotarget.3174.
106. Tickoo SK, Lee MW, Eble JN, Amin M, Christopherson T, Zarbo RJ, Amin MB. Ultrastructural observations on mitochondria and microvesicles in renal oncocytoma, chromophobe renal cell carcinoma, and eosinophilic variant of conventional (clear cell) renal cell carcinoma. Am J Surg Pathol. 2000; 24(9): 1247-56.
107. Ebner D, Rödel G, Pavenstaedt I, Haferkamp O. Functional and molecular analysis of mitochondria in thyroid oncocytoma. Virchows Arch B Cell Pathol Incl Mol Pathol. 1991; 60(2): 139-44.
108. Goebel HH, Schulz F, Rama B. Ultrastructurally abnormal mitochondria in the pituitary oncocytoma. Acta Neurochir (Wien). 1980; 51(3-4): 195-201.
109. Gasparre G, Romeo G, Rugolo M, Porcelli AM. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim Biophys Acta. 2011; 1807(6): 633-42.
110. Savagner F, Franc B, Guyetant S, Rodien P, Reynier P, Malthiery Y. Defective mitochondrial ATP synthesis in oxyphilic thyroid tumors. J Clin Endocrinol Metab. 2001; 86(10): 4920-5.
111. Caino MC, Chae YC, Vaira V, Ferrero S, Nosotti M, Martin NM, Weeraratna A, O’Connell M, Jernigan D, Fatatis A, Languino LR, Bosari S, Altieri DC. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J Clin Invest. 2013; 123(7): 2907-20.
112. Choi J, Kim do H, Jung WH, Koo JS. Metabolic interaction between cancer cells and stromal cells according to breast cancer molecular subtype. Breast Cancer Res. 2013; 15(5): R78.
113. Lin L, Huang H, Liao W, Ma H, Liu J, Wang L, Huang N, Liao Y, Liao W. MACC1 supports human gastric cancer growth under metabolic stress by enhancing the Warburg effect. Oncogene. 2015; 34(21): 2700-10
114. Song IS, Jeong JY, Jeong SH, Kim HK, Ko KS, Rhee BD, Kim N, Han J. Mitochondria as therapeutic targets for cancer stem cells. World J Stem Cells. 2015; 7(2): 418-27.
115. Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 2014; 5(22): 11029-37. doi: 10.18632/oncotarget.2789.
116. Alvero AB, Montagna MK, Holmberg JC, Craveiro V, Brown D, Mor G. Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells. Mol Cancer Ther. 2011; 10(8): 1385-93.
117. Kucharzewska P, Christianson HC, Belting M. Global profiling of metabolic adaptation to hypoxic stress in human glioblastoma cells. PLoS One. 2015; 10(1): e0116740.
118. Miccheli A, Tomassini A, Puccetti C, Valerio M, Peluso G, Tuccillo F, Calvani M, Manetti C, Conti F. Metabolic profiling by 13C-NMR spectroscopy: [1,2-13C2]glucose reveals a heterogeneous metabolism in human leukemia T cells. Biochimie. 2006; 88(5): 437-48.
119. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010; 9(17): 3506-14.
120. Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, Tanowitz HB, Casimiro MC, Wang C, Pestell RG, Grieshaber P, Caro J, Sotgia F, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010; 9(17): 3534-51.
121. Chiavarina B, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Tanowitz HB, Pestell RG, Sotgia F, Lisanti MP. Metabolic reprogramming and two-compartment tumor metabolism: opposing role(s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle. 2012; 11(17): 3280-9.
122. Morandi A, Chiarugi P. Metabolic implication of tumor: stroma crosstalk in breast cancer. J Mol Med (Berl). 2014; 92(2): 117-26.
123. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009; 8(23): 3984-4001.
124. Sanità P, Capulli M, Teti A, Galatioto GP, Vicentini C, Chiarugi P, Bologna M, Angelucci A. Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC Cancer. 2014; 14: 154.
125. Harvengt C, Heller F, Desager JP. Hypolipidemic and hypouricemic action of fenofibrate in various types of hyperlipoproteinemias. Artery. 1980; 7(1): 73-82.
126. Duan JX, Jiao H, Kaizerman J, Stanton T, Evans JW, Lan L, Lorente G, Banica M, Jung D, Wang J, Ma H, Li X, Yang Z, et al. Potent and highly selective hypoxia-activated achiral phosphoramidate mustards as anticancer drugs. J Med Chem. 2008; 51(8): 2412-20.
127. Sun JD, Liu Q, Wang J, Ahluwalia D, Ferraro D, Wang Y, Duan JX, Ammons WS, Curd JG, Matteucci MD, Hart CP. Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin Cancer Res. 2012; 18(3): 758-70.
128. Yoon C, Lee HJ, Park DJ, Lee YJ, Tap WD, Eisinger-Mathason TS, Hart CP, Choy E, Simon MC, Yoon SS. Hypoxia-activated chemotherapeutic TH-302 enhances the effects of VEGF-A inhibition and radiation on sarcomas. Br J Cancer. 2015; 113(1): 46-56.
129. Victor VM, Rovira-Llopis S, Bañuls C, Diaz-Morales N, Castelló R, Falcón R, Gómez M, Rocha M, Hernández-Mijares A. Effects of metformin on mitochondrial function of leukocytes from polycystic ovary syndrome patients with insulin resistance. Eur J Endocrinol. 2015; 173(5): 683-91.
130. Martinez-Outschoorn UE, Curry JM, Ko YH, Lin Z, Tuluc M, Cognetti D, Birbe RC, Pribitkin E, Bombonati A, Pestell RG, Howell A, Sotgia F, Lisanti MP. Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: RAS and NFκB target stromal MCT4. Cell Cycle. 2013; 12(16): 2580-97.
131. Blagosklonny MV. How Avastin potentiates chemotherapeutic drugs: action and reaction in antiangiogenic therapy. Cancer Biol Ther. 2005; 4(12): 1307-10.