
INTRODUCTION
Amyloid precursor protein (APP) is an evolutionarily conserved protein with two homologues in mammals: amyloid precursor-like protein 1 (APLP1) and amyloid precursor-like protein 2 (APLP2) [1, 2]. APLP1 was identified as the first homologue of APP [3, 4], and APLP2 (also known as YWK-II) was subsequently identified as an APP homologue [5-8]. All of these three proteins share high sequence homology and conserved domain structure. Each has an extracellular domain, a transmembrane-spanning domain and a ~50 amino acid-long cytoplasmic tail domain (shown for APP and APLP2 in Figure 1A, 1B) [9,10]. APP and APLP2 are broadly expressed, while APLP1 expression is restricted to neural tissue [5, 6, 11, 12]. As described in detail below, APP and APLP2 are overexpressed in many cancers. APP and/or APLP2 have been described as having notable functions in many cancers, such as cancers of the prostate, breast, colon, thyroid, lung, nasopharynx, and gastrointestinal tract (for a more complete list, see Table 1). Both APP and APLP2 have been linked to characteristics of cancer cells such as abnormal growth, migration, and invasion (Table 1).
Table 1: Expression and role of APP and APLP2 in multiple cancer cell types
Cancer |
APP Expression and Effects |
APLP2 Expression and Effects |
||
Acute myeloid leukemia |
↑ |
APP increases cell migration, as well as extramedullary infiltration due to matrix metalloproteinase-2 [67]. |
- |
- |
|
Breast |
↑ |
APP increases cell growth, motility, survival, and phosphorylation of AKT pathway proteins [63, 64]. |
- |
APLP2 is differentially spliced in breast cancer cell lines and human mammary epithelial cells [47]. |
|
Colon |
↑ |
APP increases phosphorylation of ERK pathway proteins and increases proliferation [62, 68]. |
↑ |
APLP2 increases proliferation [69]. |
EBV-negative Burkitt’s lymphoma |
↑ |
APP causes rapid proliferation of Epstein-Barr virus-negative Burkitt’s lymphoma cells [70]. |
- |
- |
|
Ewing’s sarcoma |
- |
- |
↑ |
APLP2 interferes with radiation-induced apoptosis and reduces MHC class I expression; APLP2 is increased in immune-evasive Ewing’s sarcoma cells [71, 72]. |
Gastrointes-tinal neuro- endocrine |
↑ |
APP is expressed in intestinal carcinoids, and is colocalized partly with markers of microvesicles and early endosomes [73]. |
↑ |
APLP2 is expressed in intestinal carcinoids, and is colocalized partly with markers of microvesicles and early endosomes [73]. |
|
Lung |
↑ |
APP increases proliferation and causes cell size abnormalities [74]. |
↓ |
APLP2 expression is decreased in lung neuroendocrine tumors, though the consequences are not well understood [73]. |
|
Melanoma |
↑ |
Perinuclear APP staining and soluble APP secretion are increased. APP facilitates proliferation, and its knockdown induces differentiation [75]. |
- |
APLP2 decreases HLA class I surface expression on MDA-MB435S cells (formerly classified as breast cancer cells but recently classified as melanoma cells) [76]. |
|
Naso-pharynx |
↑ |
APP increases cell growth and migration, and there is EGFR-mediated upregulation of soluble APP production [77]. |
- |
- |
Oral |
↑ |
Upregulation of AP2α and positive correlation between APP and AP2α in tumor tissue [78]. |
- |
- |
Pancreas |
↑ |
↑ |
APLP2 increases migration, proliferation, invasion, and metastasis [42, 61]. |
|
Prostate |
↑ |
APP increases proliferation and migration, modulates levels of metalloproteinase and EMT-related proteins, and downregulates MAP kinase phosphatase and Notch signaling pathways [65]. |
- |
- |
Testicular germ cell |
↑ |
APP expression was detected in ~39% of nonseminomatous germ cell tumors (NSGC), and APP was associated with venous invasion [79]. |
↑ |
APLP2 is expressed in testicular germ cell tumor tissue [80]. |
Thyroid |
↑ |
APP staining is increased in tumor tissue, and is associated with bigger tumor size, extracapsular invasion, and spread to the lymph nodes [81]. |
- |
- |
APP AND APLP2 GENES AND PROTEIN STRUCTURES
The APP family members are encoded by separate multi-exon genes located on three different chromosomes [13-15]. The human APP gene is positioned at chromosome 21 (specifically, at band 21q21), and the human APLP2 gene is found at 11q24 [16, 17]. Although APP and APLP2 are related genetically, it has been observed that they are transcriptionally divergent, and there are unique sequence motifs in each gene that suggest specialized, non-overlapping functions [18].
The extracellular domain of APP contains two disulfide knots (three overlapping disulfide bonds) that resemble the disulfide knots within growth factors [19]. The cysteine residues used to form the disulfide knots of APP are conserved in APLP2, which by similarity is proposed to have two disulfide knots as well (Figure 1A, 1B). In the extracellular portion of APP and APLP2 are several smaller domains, including a bovine pancreatic trypsin inhibitor (BPTI)/Kunitz protease inhibitor domain, and a domain in which aspartic and glutamic acid residues are very abundant (called the Asp-Glu-rich domain) (Figure 1A, 1B) [9, 20].
The Kunitz protease inhibitor domain in the extracellular region of APP and APLP2 (Figure 1A, 1B) inhibits multiple proteases (such as trypsin, chymotrypsin, plasmin, plasmin, and kallikrein enzymes) with varying efficiencies [21]. Another protease inhibited by the Kunitz protease inhibitor domain (as part of the cleaved, secreted APP ectodomain) is coagulation factor XIa [22]. Notably, mesotrypsin produced by tumor cells acts to cleave the Kunitz protease inhibitor domain of the soluble, secreted APP ectodomain, which likely contributes to the procoagulant character of the tumor microenvironment [23].
At the positions indicated in Figure 1, the extracellular domains of APLP2 and APP bind copper or zinc ions [9, 24-27]. Recently, it was shown that the presence of copper increases the expression of APP in prostate cancer cells [28]. Furthermore, this same study revealed that the copper-binding region of the APP 770 splice variant (in conjunction with tyrosine residues in the APP intracellular domain) reduces copper-mediated inhibition of prostate cancer cell growth [28].
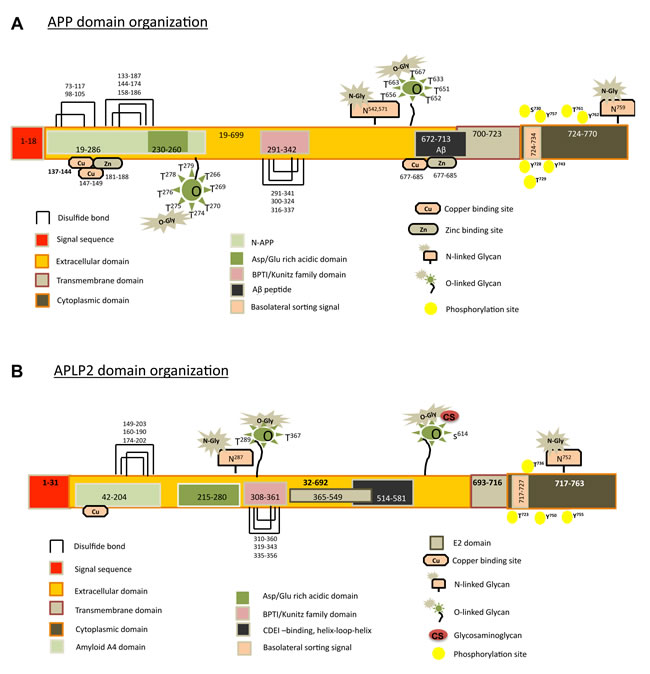
Figure 1: Graphical representations of the domains and sub-domains for APP and APLP2 are shown, along with disulfide bonds and predicted post-translational modifications. A. For APP, the 770 amino acid isoform (UniProt accession number P05067) is displayed. B. For APLP2, the 763 amino acid isoform (UniProt accession number Q06481) is shown. N-glycan prediction was done with the NetNGlyc 1.0 Server (http://www.cbs.dtu.dk/services/NetNGlyc/). O-glycan prediction was performed by NetOGlyc 3.1 Server (http://www.cbs.dtu.dk/ services/NetOGlyc/). Phosphorylated residues were predicted with PhosphoSite (http://www.phosphosite.org/ homeAction.do). For some isoforms of APLP2, but not of APP, chondroitin sulfate (CS) glycosaminoglycan modification occurs at Serine 614.
CLEAVAGE OF APP FAMILY MEMBERS
Of the three APP family members, APP is the most studied, notorious for its cleavage generating the β amyloid peptide (Figure 1A) that contributes to Alzheimer’s disease [29-31]. Each of the APP family members (APP, APLP1, and APLP2) undergoes cleavage by secretases that release a large extracellular domain and produce smaller C-terminal fragments (Figure 2) [32-33]. APP and APLP2 cleavage is regulated by the hormone insulin-like growth factor 1 (IGF-1) [34-37]. IGF-1 has a known role in cancer progression, and multiple studies have explored the impact of inhibiting IGF-1 receptor signaling as a potential therapeutic approach for cancers, including neuroblastoma [38-41]. Some reports suggest that IGF-1 increases α-secretase cleavage of APP and APLP2, which would presumably result in down-regulation of β amyloid production [34, 36]. However, other reports indicate that IGF-1 actually promotes the generation of β amyloid [35], and therefore additional studies are needed to fully discern the effect of IGF-1 on APP and APLP2 processing and function in cancer.
In our studies, we have examined the effect of blocking beta-secretase activity on the viability of pancreatic cancer cells [42]. Many chemical inhibitors of beta-secretase have been developed, and some have already shown safety and efficacy in clinical trials for Alzheimer’s disease patients [43-46]. We incubated pancreatic cancer cells with inhibitors of the beta-secretase enzyme, and observed a reduction in APLP2 C-terminal fragments in the cells. Furthermore, we demonstrated that treatment of the pancreatic cancer cells with beta-secretase inhibitors decreased the growth and viability of the cells. A non-transformed pancreatic cell line was included as a control, and the beta-secretase inhibitors did not diminish the growth or survival of the non-transformed cell line. Our results suggest that although no chemical inhibitors have been designed with the specific goal of targeting APLP2, existing beta-secretase inhibitors that have been made to target APP for the treatment of Alzheimer’s disease may potentially be repurposed to target APLP2.
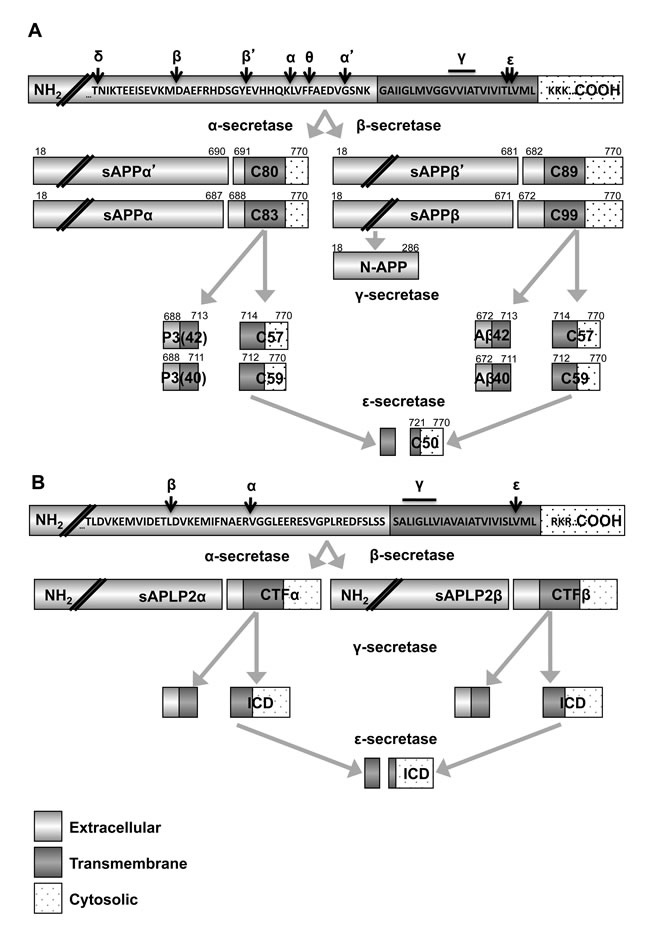
Figure 2: Secretase processing of APP and APLP2 generates many fragments. Fragments of APP A. and APLP2 B. generated following cleavage by several secretase enzymes are shown. Amino acids in canonical APP and APLP2 are provided. Double forward slashes are used to denote truncated sequences. The N-terminal ends are indicated with NH2 and the carboxyl ends are indicated with COOH. A. Beta secretase 1 and 2 (BACE1 and BACE2) can cleave at site β, while the alternate cleavage site for BACE1 is site β’ and the alternative cleavage site for BACE2 is site θ. Two α-secretase cleavage sites have been described, α and α’. Sites γ and ε are cleaved by γ-secretase enzymes. Fragments of APP generated by various cleavage sites are provided with their nomenclature (text inside rectangles) and the residues forming the various fragments (superscript text above rectangles). The C-terminal fragments (CTF) of APP are C99, C89, C83 and C80 and the intracellular domains (ICDs) of APP are C59, C57 and C50. B. The BACE1 (β), ADAM10 (α) and γ-secretase cleavage sites (γ) have been determined for APLP2. APLP2 C-terminal fragments are distinguished by the secretase responsible for their formation. Intracellular domains of APLP2 are denoted as ICDs.
APLP2 SPLICING
In APLP2 (and in APP, as will be described below), splicing leads to diversity and to specialized functions influential in cancer. Several isoforms of APLP2 arise from splicing (Figure 3) [14, 20, 21]. The canonical isoform of human APLP2 is 763 residues long and ~110 kDa in molecular mass, and it utilizes all 18 exons. Alternative APLP2 isoforms of 751, 707, 695, and 522 amino acids in length have been reported, and the first four of these isoforms arise from exclusion of exons 7 and/or 14. Omission of exon 7 removes the Kunitz protease inhibitor domain, and the frequency with which this APLP2 exon is excluded was found to vary in a comparison of two human breast cancer lines (MCF7 and MDA-MD-231) and human mammary epithelial cells [47].
Only APLP2 isoforms that exclude exon 14 are modified by chondroitin sulfate glycosaminoglycan modification on Ser614 (Figures 1 and 3) [48, 49]. Among the pancreatic cancer cell lines that we examined, 4 out of 5 expressed high levels of endogenous chondroitin sulfate glycosaminoglycan-modified APLP2 [42], which suggests that there may be preferential expression in pancreatic cancer cells of APLP2 isoforms lacking exon 14. Since transfected cells expressing various APLP2 isoforms had an increased migratory response when the isoform that was overexpressed was APLP2-751 (which bears the chondroitin sulfate glycosaminoglycan modification) [50], it is possible that the presence of the chondroitin sulfate glycosaminoglycan modification may also increase the migratory tendencies of pancreatic cancer cells.
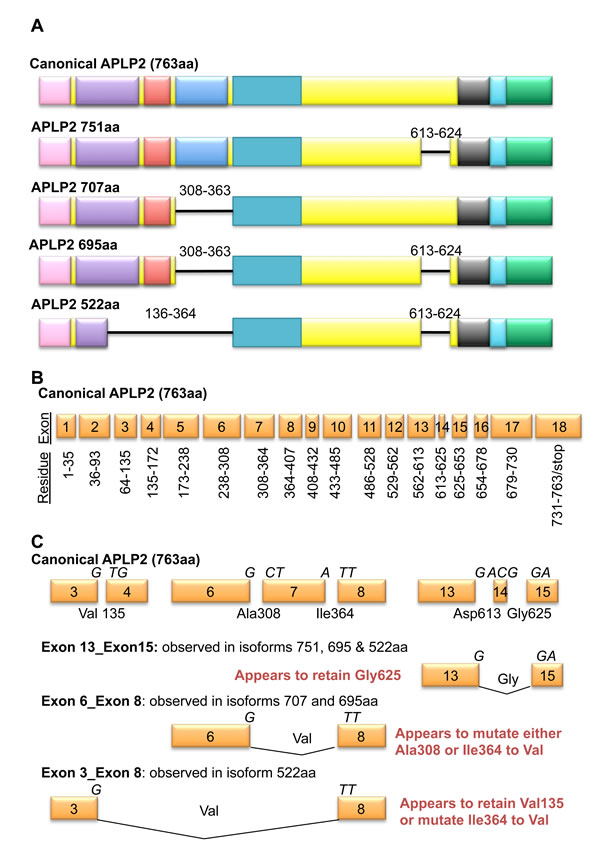
Figure 3: Isoforms of APLP2 arise through alternate splicing. A. Reported isoforms of APLP2 are shown with numbers denoting residues not encoded in smaller isoforms. B. The 18 exons of APLP2 are displayed alongside the residues that they encode. C. Exon junctions in the canonical APLP2 sequence (top row) and known splicing sites (bottom three rows) are depicted. Letters above exons indicate the nucleic acid codes, and residues and their location within the canonical APLP2 isoform are provided.
APP SPLICING
As is the case with APLP2, splicing also leads to generation of APP variants that differ in molecular mass. Alternative splicing of APP RNA gives rise to at least 9 translated isoforms, with variable expression across normal and disease states. The largest isoform, which contains the sequences encoded by all 18 exons, is 770 amino acids in length (Figure 4A). Aside from the canonical APP-770, the other primary APP isoforms contain 751 or 695 amino acid residues (Figure 4B). Some APP splice variants are restricted in localization. For example, APP-751 and APP-695 are abundantly present in mammalian brain [51-53], and APP 752, 733, 696, and 677 are found in astrocytes and leukocytes [54]. Upon mitogenic stimulation with phytohemagglutinin, T cells secrete APP-733 [55]. In the various APP isoforms, the spliced regions include a group of N-terminal residues near a heparin-binding domain (exon 2), the Kunitz-type protease inhibitor domain (exon 7), an OX-2 antigen domain (exon 8), and the amyloid beta processing sequence (exon 15) (Figure 4C).
Several studies have demonstrated that in cancer there is upregulation of the 751-amino acid APP isoform, which lacks the OX-2 antigenic domain encoded by exon 8 [56-58]. High throughput reverse transcriptase-polymerase chain reaction screens that profiled the expression of >600 cancer-related genes revealed the APP-751 splice variant in primary epithelial ovarian tumors and primary breast tumors, but not in corresponding tissues from normal donors, suggesting that the production of APP-751 is cancer-specific rather than simply characteristic of the tissue of origin [56-58]. The high degree of APP exon 8 excision in breast tumor versus normal tissue (p<10-5) prompted its inclusion in a 12-marker APP splice variant panel that correctly identified 33 of 35 (96%) of tumor samples in a blinded validation assay. Likewise, quantitative microarray followed by reverse transcriptase-polymerase chain reaction showed a >10% decrease in APP mRNAs containing exon 8 in non-squamous cell lung carcinoma, breast cancer, and colon cancer (as compared to patient-matched controls), indicating a predilection towards APP-751 expression in each of these cancers [58].
The role of the OX-2 domain that is omitted in APP-751 is currently unclear. The OX-2 domain was named for its homology to a region of the OX-2 antigen (an immunoglobulin superfamily member found on neuronal and lymphoid cells), but the function of this specific sequence within the OX-2 protein is not known [59]. Hence, on a mechanistic basis, what effect is exerted by the presence versus absence of exon 8 in cancer cells is not yet apparent. An alternative perspective (proposed by Misquitta-Ali et al. [58]) is that exclusion of the OX-2 domain could alter the production of specific APP cleavage products, and thus the ability of the APP-751 isoform to facilitate tumorigenesis is actually a consequence of the different levels of these products.
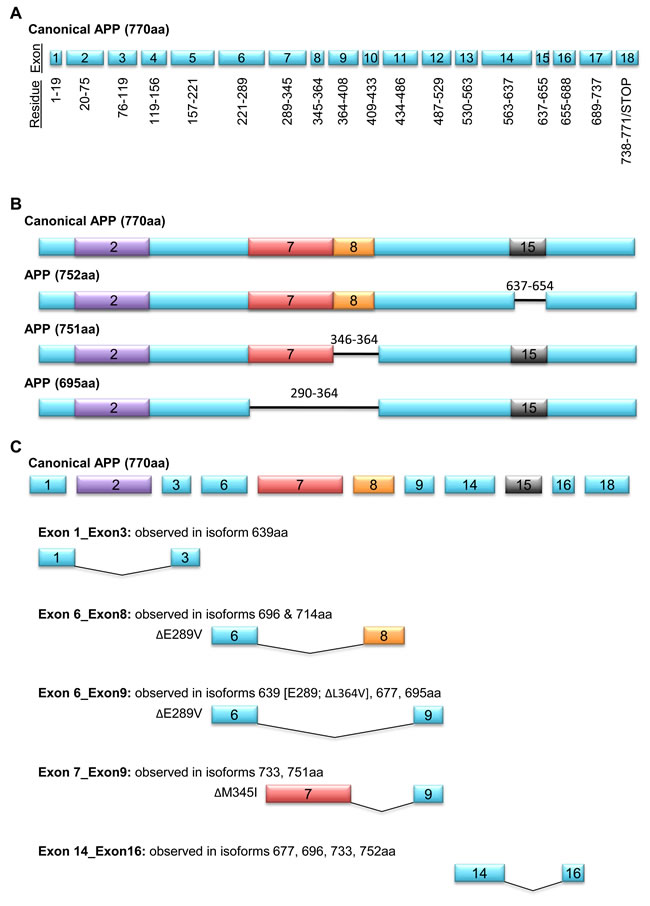
Figure 4: APP has alternatively spliced isoforms. A. 18 exons encode the canonical 770-amino acid APP uncleaved glycoprotein, with several overlapping residues resulting from ligated mRNA of different exons forming a single codon. B. Diagrams of primary isoforms APP 751, APP 695, and leukocyte (L)-APP 752, with excised residues denoted by black horizontal lines. Excised exons are differentially colored: heparin-binding domain in exon 2 (purple), Kunitz-type protease inhibitor in exon 7 (red), OX-2 antigen sequence in exon 8 (orange), and amyloid beta processing sequence (grey). The deletion of the amyloid beta processing sequence enables the attachment of chondroitin sulfate glycosaminoglycan. C. Alternative splicing events and amino acid substitutions in 8 APP isovariants.
EXPRESSION OF APP FAMILY MEMBERS IN CANCER
APP and/or APLP2 expression is aberrantly altered in many types of cancers (Table 1), such as pancreatic [42, 60, 61], colon [62], breast [63, 64], prostate [65], lung [66], and other cancers [43, 67-81]. Furthermore, APP and APLP2 have been shown to have a range of roles in cancer cells, including both pro-growth and pro-invasion functions. In this review, we will now focus on the expression and functions of APP and APLP2 in specific types of cancer.
APP family members in germ cell cancers
APLP2 is expressed in normal mouse germ cells, is present on the plasma membrane on mature spermatozoa [82], and has been implicated in sperm survival [83]. Notably, recent studies done by Venkataramani et al. [80], reported that APP expression positively correlates with pluripotency-linked gene expression in testicular germ cell cancers but APLP2 expression does not, indicating that APP (and not APLP2) is a biomarker of pluripotent stem cell transformation. APP is widely expressed at increased levels in several subsets of testicular germ cell tumors, such as seminomas, choriocarcinomas, yolk sac tumors and teratomas [80].
APP family members in Ewing’s sarcoma
In cell lines derived from the pediatric cancer Ewing’s sarcoma, APLP2 is typically overexpressed [71]. Upon radiation treatment, increased expression of APLP2 reduces the proportion of Ewing’s sarcoma cells in the sub-G1 stage (i.e., the apoptotic subset) [71]. Consistent with APLP2’s ability to increase Ewing’s sarcoma cell survival following radiation treatment, higher APLP2 overexpression was found in Ewing’s sarcoma cells that had resisted lysis by lymphokine-actived killer cells (which destroy target cells by an apoptotic mechanism) [71]. APLP2 overexpression also reduces the level of MHC class I molecules at the plasma membrane of Ewing’s sarcoma cell lines [72]. This effect of APLP2 on MHC class I has also been observed with HeLa cells and other cancer cell lines [76, 84-87], suggesting that APLP2 may generally assist in cancer immune escape from T cell killing.
APP family members in breast cancer
APP expression is increased in breast cancer cell lines that exhibit greater metastatic tendencies, such as motility and proliferation [64]. When APP in a variety of breast cancer cell lines with increasing metastatic potential was knocked down, the cell lines had reduced cell growth and underwent G1 arrest due to induction of the cell cycle inhibitor p27kip1 [64]. Additionally, induction of apoptosis markers such as cleaved caspase 3 and the PARP cleavage product were also noted in the breast cancer cell lines in which APLP2 expression had been knocked down, especially in those cell lines that had been identified as having higher metastatic potential [64]. The APP-knockdown breast cancer cells also showed decreased tumor growth in both a 3D in vitro cell culture and in an in vivo mouse model [64]. APP was also shown to induce migration in breast cancer cells, especially in the presence of IGF-1 [64]. Another study demonstrated that APP was positively correlated with a higher risk of recurrence in ER-positive breast cancer cases, as compared to ER-negative cases, suggesting that increased APP is associated with a worse prognosis [63].
APP family members in prostate cancer
Studies done by Takayama et al. [88] showed APP induces androgen-mediated signaling pathways contributing to the growth and proliferation of prostate cancer. Also, immunohistology revealed a lack of APP in normal human prostate cells, while in tumor samples from a group of prostate cancer patients with a 50% survival rate there was intense cytoplasmic staining of APP [88]. The importance of APP in prostate cancer was further validated by an in vivo animal model in which knock-down of APP repressed tumor growth [88]. In addition, APP has been demonstrated to be involved in the migration and proliferation of prostate cancer cells via mechanisms involving metalloproteinases and epithelial-to-mesenchymal transition-related pathways [65].
APP family members in lung cancer
APP, especially the secreted form of APP, is upregulated in lung cancers [66]. APP’s role in this type of cancer was further verified by a recent study by Sobol et al. [74]. Using APP-specific siRNA transfection of non-small cell lung cancer (NSCLC) cells, this group showed that upon APP downregulation there was destabilization of cyclin C, leading to G0/G1 cell cycle arrest, as well as to decreased phosphorylation of pRb, abnormalities in cell size, and necrosis due to membrane permeabilization [74]. In regard to other APP family members, according to the ONCOMINE database (Compendia Bioscience, Ann Arbor, MI), significant upregulation and downregulation of APLP1 and APLP2 (respectively) was observed in neuroendocrine lung tumors [73]. More work remains to be done, not only to examine the role of APP in lung cancer, but also to investigate the role that APLP1 and APLP2 might play in this particular type of cancer.
APP family members in melanoma
Metastatic melanoma has a very poor prognosis due to its frequent resistance to traditional chemotherapies and radiation treatments. Botelho et al. [75] showed by immunohistochemistry and immunofluorescence that there is differential expression of transmembrane and secreted APP in the vertical and metastatic growth phase of melanomas, as compared to earlier stages of the disease. Transient knock-down of APP in advanced melanoma cell lines reduced proliferation and increased the expression of melanocyte pigmentation/differentiation markers such as human tyrosinase, tyrosinase-related protein-1, and microphthalmia-associated transcription factor, indicating that the loss of APP leads to a more differentiated phenotype [75]. It was also observed upon APP downregulation in melanoma cells that there was lower expression of ABCB5 (doxorubicin-resistant transporter), which has been implicated in chemoresistance [75]. Consistent with the observed reduction in ABCB5, upon APP downregulation aggressive melanoma cell lines became sensitive to chemotherapeutic drugs to which they were not previously sensitive [75].
APP family members in pancreatic cancer
Hansel et al. [60] demonstrated that secreted APP enhances cell proliferation in pancreatic cancer cells, as well as thyroid epithelial cells and fibroblasts, by acting as an autocrine growth factor. Other investigators have likewise shown that a secreted form of APP that is produced by α-secretase cleavage aids in cell survival and migration [89]. Pancreatic cancer cell proliferation was significantly reduced upon treatment of the cells with batimastat (which inhibits α-secretase cleavage of APP) along with gemcitabine, as compared to gemcitabine alone [90]. When cells that had been incubated with batimastat were treated with a recombinant form of the secreted APP fragment, growth capacity was restored, confirming that α-secretase-mediated secretion of APP contributes to pancreatic cancer cell growth [90].
Our research group has demonstrated that in addition to overexpression of APP in pancreatic cancer cell lines, there is overexpression of APLP2 (both full-length and cleaved forms) in pancreatic cancer cell lines, and by immunohistochemistry we have demonstrated overexpression of APLP2 in human pancreatic tumor samples [42, 61]. Transient knock-down of APLP2 or APP reduced pancreatic cancer cell growth and viability [42]. We found that a series of cell lines derived from human ductal epithelial cells by transfection with hTERT plus an increasing number of oncogenes had escalating levels of full-length and cleaved APLP2, which suggests that APLP2 may increase gradually during the process of pancreatic cancer development [42].
In an orthotopic mouse model of pancreatic cancer, we demonstrated that down-regulation of APLP2 expression resulted in decreased tumor weight and limited metastasis. We also investigated the expression of APLP2 in human pancreatic cancer metastases, and found that APLP2 is increased in metastatic lesions at many sites, particularly the intestine and the diaphragm [61]. Furthermore, we found positive APLP2 expression in a large proportion (38%) of paired primary tumor and liver metastasis samples from the same patients [61].
APP family members in colon cancer
Colon cancer also exhibits overexpressed APP and APLP2 [68, 69]. Both in vitro and in vivo studies have shown that APP promotes growth and proliferation of colon cancer [62]. The gene that encodes APP is part of a genetic signature for increased likelihood of metastasis in patients with early stage mismatch-repair proficient sporadic colon cancer [91].
Consistent with the findings obtained in studies of APP, knock-down of APLP2 reduced proliferation of the Caco2 colon cancer cell line [69]. In colon cancer cells, APLP2 expression is positively correlated with expression of human leukocyte antigen-B-associated transcript 3 (Bat3). Bat3 associates with APLP2 and inhibits its ubiquitylation, thereby blocking its degradation by the proteasome [92]. These findings suggest that Bat3 facilitates the ability of APLP2 to increase colon cancer cell growth by stabilizing APLP2.
The regulation of APP and APLP2
Since it is becoming increasingly clear that APP and APLP2 have pro-cancer functions, how APP and APLP2 expression and function are regulated is also evidently relevant to their roles in cancer. In regard to transcriptional control, there are distinctions between APP and APLP2. The APP promoter has putative recognition sites that are predicted to allow NFĸB/Rel and activator protein-1 (AP-1) to regulate APP expression [93, 94]. The promoter for the APP gene contains transcription factor sites that are absent in the promoter for the APLP2 gene, including a possible heat shock transcription element [95, 96]. Retinoic acid and interleukin-1 have been associated with increased transcription of APP, and retinoic acid also upregulates APLP2 expression [97-99].
The processing of APP and APLP2 is regulated by phosphorylation events. For example, the Pin 1 protein binds to the phosphorylated form of APP at Thr668 [100], and this binding regulates the processing of APP [101]. In addition, the processing of APLP2 is influenced by epidermal growth factor, phorbol 12-myristate 13-acetate (PMA), IGF-1, and retinoic acid [36, 102]. Epidermal growth factor and PMA activate protein kinase C-ε/δ, leading to cleavage and processing of APLP2 via the mitogen-activated protein kinase pathway in corneal epithelial cells [102]. Phosphorylation of protein kinase C by IGF-1 initiates intracellular events leading to the cleavage and shedding of APLP2 from the cell [36]. In cells of the nervous system, the processing and secretion of APP is increased by the presence of okadaic acid, estrogen, or testosterone [103-105].
CONCLUSIONS
The APP family members are highly conserved, but despite structural similarities, APP and APLP2 have frequently been observed to have divergent and unique functions. However, both APP and APLP2 are typically upregulated with advancement of cancer progression, and each has been implicated in several phenotypes related to cancer (Table 1; Figure 5). We are also examining the levels within cancer cells of the enzymes (such as beta-secretases) that cleave APP and APLP2, since expression levels of these enzymes regulate the biological influences of both APP and APLP2 [32, 33]. At present, the signaling pathways by which APP and APLP2 wield their effects in cancer cells (which may be multiple pathways) are not well understood, though there are a few clues that the pathways leading to the transcription coactivator YAP (known to be an important factor in cancer growth and migration) might potentially be involved [106-112]. Much remains to be discovered in cancer models about the impact of APP/APLP2 post-translational modifications, such as phosphorylation and glycosylation (Figure 1) [93, 113-125]. Deciphering the specific pathways in which APP and APLP2 function in cancer cells to increase malignancy (and, with APLP2, also to increase cancer immune evasion [76, 84-86]) will be necessary to fully comprehend the roles of these proteins in cancer progression and to develop new therapeutic regimens for cancer that are based on targeting APP and/or APLP2.
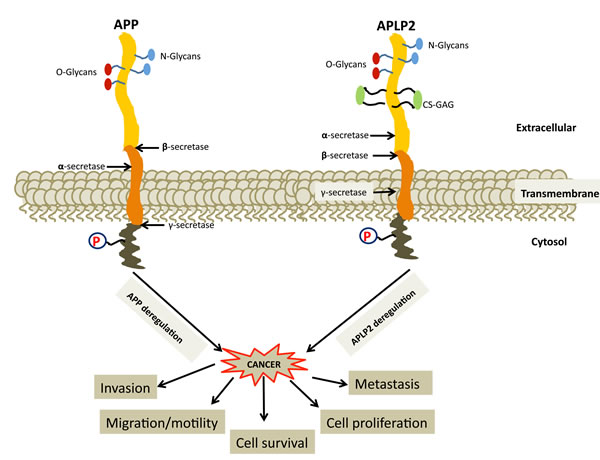
Figure 5: Deregulation of APP and APLP2 causes cancer progression and metastasis, but the roles in cancer of most of the protein interactions involving APP and APLP2 are not well understood. Illustrations of transmembrane APP and APLP2 are displayed with cleavage sites indicated. Proteolytic α-, β-. and γ-secretases cleave at various sites on APP and APLP2, generating protein fragments. Interactions between APP and APLP2 with various interacting partners are mediated by glycosylation and phosphorylation.
ACKNOWLEDGMENTS
We thank Alexandra Moffitt for her reading of this manuscript and her comments. We gratefully acknowledge funding from the Nebraska Research Initiative, the Fred & Pamela Buffett Cancer Center’s NIH Cancer Center Support Grant (P30CA036727), the Nebraska Center for Cellular Signaling (NIH Grant P30GM106397), the State of Nebraska through the Pediatric Cancer Research Group, NIH R03CA169953, NIH R03CA176557, and NIH SPORE P50CA127297. Support for this work was also provided by University of Nebraska Medical Center Graduate Studies Office Emley and Regents Tuition Fellowships (H.L.P.) and by NIH Training Grant T32CA009476. A.T. is a recipient of the Wellcome Trust/DBT India Alliance Intermediate Fellowship Award and acknowledges financial support from the Wellcome Trust/DBT Alliance and CSIR-IMTECH intramural funding.
CONFLICTs OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Zheng H, Koo EH. The amyloid precursor protein: beyond amyloid. Mol. Neurodegener. 2006; 1: 5.
2. Jacobsen KT, Iverfeldt K. Amyloid precursor protein and its homologues: a family of proteolysis-dependent receptors. Cell Mol Life Sciences. 2009; 66: 2299-2318.
3. Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T. Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci USA. 1988; 85: 7341-7345.
4. Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F. Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc Natl Acad Sci USA. 1992; 89: 10758-10762.
5. Sprecher CA, Grant FJ, Grimm G, O’Hara PJ, Norris F, Norris K, Foster DC. Molecular cloning of the cDNA for a human amyloid precursor protein homolog: evidence for a multigene family. Biochemistry. 1993; 32: 4481-4486.
6. Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE. Isolation and characterization of APLP2 encoding a homologue of the Alzheimer’s associated amyloid beta protein precursor. Nat Genet. 1993; 5: 95-100.
7. Huang P, Miao S, Fan H, Sheng Q, Yan Y, Wang L, Koide SS. Expression and characterization of the human YWK-II gene, encoding a sperm membrane protein related to the alzheimer betaA4-amyloid precursor protein. Mol Hum Reprod. 2000; 6: 1069-1078.
8. Yin X, Ouyang S, Xu W, Zhang X, Fok KL, Wong HY, Zhang J, Qiu X, Miao S, Chan HC, Wang L. YWK-II protein as a novel G(o)-coupled receptor for Mullerian inhibiting substance in cell survival. J Cell Sci. 2007; 120: 1521-1528.
9. Dyrks T, Weidemann A, Multhaup G, Salbaum JM, Lemaire HG, Kang J, Muller-Hill B, Masters CL, Beyreuther K. Identification, transmembrane orientation and biogenesis of the amyloid A4 precursor of Alzheimer’s disease. EMBO J. 1988; 7: 949-957.
10. Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987; 325: 733-736.
11. McNamara MJ, Ruff CT, Wasco W, Tanzi RE, Thinakaran G, Hyman BT. Immunohistochemical and in situ analysis of amyloid precursor-like protein-1 and amyloid precursor-like protein-2 expression in Alzheimer disease and aged control brains. Brain Res. 1998; 804: 45-51.
12. Slunt HH, Thinakaran G, Von Koch C, Lo AC, Tanzi RE, Sisodia SS. Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP). J Biol Chem. 1994; 269: 2637-2644.
13. Yoshikai S, Sasaki H, Doh-ura K, Furuya H, Sakaki Y. Genomic organization of the human amyloid beta-protein precursor gene. Gene. 1990; 87: 257-263.
14. von der Kammer H, Loffler C, Hanes J, Klaudiny J, Scheit KH, Hansmann I. The gene for the amyloid precursor-like protein APLP2 is assigned to human chromosome 11q23-q25. Genomics. 1994; 20: 308-311.
15. Yang Y, Martin L, Cuzin F, Mattei MG, Rassoulzadegan M. Genomic structure and chromosomal localization of the mouse CDEI-binding protein CDEBP (APLP2) gene and promoter sequences. Genomics. 1996; 35: 24-29.
16. Robakis NK, Wisniewski HM, Jenkins EC, Devine-Gage EA, Houck GE, Yao XL, Ramakrishna N, Wolfe G, Silverman WP, Brown WT. Chromosome 21q21 sublocalisation of gene encoding beta-amyloid peptide in cerebral vessels and neuritic (senile) plaques of people with Alzheimer disease and Down syndrome. Lancet. 1987; 1: 384-385.
17. Leach R, Ko M, Krawetz SA. Assignment of amyloid-precursor-like protein 2 gene (APLP2) to 11q24 by fluorescent in situ hybridization. Cytogenet Cell Genet. 1999; 87: 215-216.
18. Shariati SA, De Strooper B. Redundancy and divergence in the amyloid precursor protein family. FEBS Lett. 2013; 587: 2036-2045.
19. Reinhard C, Hebert SS, De Strooper B. The amyloid-beta precursor protein: integrating structure with biological function. EMBO J. 2005; 24: 3996-4006.
20. Sandbrink R, Masters CL, Beyreuther K. Similar alternative splicing of a non-homologous domain in betaA4-amyloid protein precursor-like proteins. J Biol Chem. 1994; 269: 14227-14234.
21. Petersen LC, Bjorn SE, Norris F, Norris K, Sprecher C, Foster DC. Expression, purification and characterization of a Kunitz-type protease inhibitor domain from human amyloid precursor protein homolog. FEBS Lett. 1994; 338: 53-57.
22. Badellino KO, Walsh PN. Localization of a heparin binding site in the catalytic domain of factor XIa. Biochemistry. 2001; 39: 4769-4777.
23. Salameh MA, Robinson JL, Navaneetham D, Sinha D, Madden BJ, Walsh PN, Radisky ES. The amyloid precursor protein/protease nexin 2 Kunitz inhibitor domain is a highly specific substrate of mesotrypsin. J Biol Chem. 2010; 285: 1939-1949.
24. Bush AI, Pettingell WH Jr, de Paradis M, Tanzi RE, Wasco W. (1994). The amyloid beta-protein precursor and its mammalian homologues. Evidence for a zinc-modulated heparin-binding superfamily. J Biol Chem. 1994; 269: 26618-26621.
25. Simons A, Ruppert T, Schmidt C, Schlicksupp A, Pipkorn R, Reed J, Masters CL, White AR, Cappai R, Beyreuther K, Bayer TA, Multhaup G. Evidence for a copper-binding superfamily of the amyloid precursor protein. Biochemistry. 2002; 41: 9310-9320.
26. White AR, Reyes R, Mercer JF, Camakaris J, Zheng H, Bush AI, Multhaup G, Beyreuther K, Masters CL, Cappai R. Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 1999; 84: 439-444.
27. Cappai R, Cheng F, Ciccotosto GD, Needham BE, Masters CL, Multhaup G, Fransson LA, Mani K. The amyloid precursor protein (APP) of Alzheimer disease and its paralog, APLP2, modulate the Cu/Zn-Nitric Oxide-catalyzed degradation of glypican-1 heparan sulfate in vivo. J Biol Chem. 2005; 280: 13913-13920.
28. Gough M, Blanthorn-Hazell S, Delury C, Parkin E. The E1 copper binding domain of full-length amyloid precursor protein mitigates copper-induced growth inhibition in brain metastatic prostate cancer DU145 cells. Biochem Biophys Res Commun. 2014; 453: 741-747.
29. Goedert M. Neuronal localization of amyloid beta protein precursor mRNA in normal human brain and in Alzheimer’s disease. EMBO J. 1987; 6: 3627-3632.
30. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002; 297: 353-356.
31. Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010; 330: 1774.
32. Eggert S, Paliga K, Soba P, Evin G, Masters CL, Weidemann A, Beyreuther K. The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves α-, β-, γ-, and ε-like cleavages. J Biol Chem. 2004; 279: 18146-18156.
33. Vassar R, Kovacs DM, Yan R, Wong PC. The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J. Neurosci. 2009; 29: 12787-12794.
34. Adlerz L, Holback S, Multhaup G, Iverfeldt K. IGF-1-induced processing of the amyloid precursor protein family is mediated by different signaling pathways. J Biol Chem. 2007; 282: 10203-10209.
35. Araki W, Kume H, Oda A, Tamaoka A, Kametani F. IGF-1 promotes beta-amyloid production by a secretase-independent mechanism. Biochem Biophys Res Commun. 2009; 380: 111-114.
36. Jacobsen KT, Adlerz L, Multhaup G, Iverfeldt K. Insulin-like growth factor-1 (IGF-1)-induced processing of amyloid-beta precursor protein (APP) and APP-like protein 2 is mediated by different metalloproteinases. J Biol Chem. 2010; 285: 10223-10231.
37. Zhang H, Gao Y, Dai Z, Meng T, Tu S, Yan Y. IGF-1 reduces BACE-1 expression in PC12 cells via activation of PI3-K/Akt and MAPK/ERK1/2 signaling pathways. Neurochem Res. 2011; 36: 49-57.
38. Coulter DW, Blatt J, D’Ercole AJ, Moats-Staats BM. IGF-1 receptor inhibition combined with rapamycin or temsirolimus inhibits neuroblastoma cell growth. Anticancer Res. 2008; 28: 1509-1516.
39. Coulter DW, Wilkie MB, Moats-Staats BM. Inhibition of IGF-1 receptor signaling in combination with rapamycin or temsirolimus increases MYC-N phosphorylation. Anticancer Res. 2009; 29: 1943-1949.
40. Seccareccia E, Brodt P. The role of the insulin-like growth factor-I receptor in malignancy: an update. Growth Hormone & IGF Res. 2012; 22: 193-199.
41. Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, Krailo M, Villaluna D, Adamson PC, Blaney SM. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2014; 61: 452-456.
42. Peters HL, Tuli A, Wang X, Liu C, Pan Z, Ouellette MM, Hollingsworth MA, MacDonald RG, Solheim JC. Relevance of amyloid precursor-like protein 2 C-terminal fragments in pancreatic cancer cells. Int J Oncol. 2012; 41: 1464-1474.
43. Luo X, Yan R. Inhibition of BACE1 for therapeutic use in Alzheimer’s disease. Int J Clin Exp Pathol. 2010; 3: 618-628.
44. Chang W-P, Huang X, Downs D, Cirrito JR, Koelsch G, Holtzman DM, Ghosh AK, Tang J. Beta-secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 2011; 25: 775-784.
45. Tang J, Ghosh A. Treating transgenic Alzheimer mice with a beta-secretase inhibitor, what have we learned? Aging (Albany NY). 2011; 3: 14-16. doi: 10.18632/aging.100267.
46. Ghosh AK, Tang J. Prospects of beta-secretase inhibitors for the treatment of Alzheimer’s disease. ChemMedChem. 2015; 10: 1463-1466.
47. Li C, Kato M, Shiue L, Shively JE, Ares M Jr, Lin R-J. Cell type and culture condition-dependent alternative splicing in human breast cancer cells revealed by splicing-sensitive microarrays. Cancer Res. 2006; 66: 1990-1999.
48. Thinakaran G, Sisodia SS. Amyloid precursor-like protein 2 (APLP2) is modified by the addition of chondroitin sulfate glycosaminoglycan at a single site. J Biol Chem. 1994; 269: 22099-22104.
49. Thinakaran G, Slunt HH, Sisodia SS. Novel regulation of chondroitin sulfate glycosaminoglycan modification of amyloid precursor protein and its homologue, APLP2. J Biol Chem. 1995; 270: 16522-16525.
50. Li XF, Thinakaran G, Sisodia SS, Yu FS. Amyloid precursor-like protein 2 promotes cell migration toward fibronectin and collagen IV. J Biol Chem. 1999; 274: 27249-27256.
51. Tanaka S, Shiojiri, S, Takahashi Y, Kitaguchi, N, Ito H, Kameyama M, Kimura J, Nakamura S, Ueda, K. Tissue-specific expression of three types of beta-protein precursor mRNA: enhancement of protease inhibitor-harboring types in Alzheimer’s disease brain. Biochem Biophys Res Commun. 1989; 165: 1406-1414.
52. Kang J, Muller-Hill B. Differential splicing of Alzheimer’s disease amyloid A4 precursor RNA in rat tissues: PreA4(695) mRNA is predominantly produced in rat and human brain. Biochem Biophys Res Commun. 1990; 166: 1192-1200.
53. Moir RD, Lynch T, Bush AI, Whyte S, Henry A, Portbury S, Multhaup G, Small DH, Tanzi, RE, Beyreuther K, Masters CL. Relative increase in Alzheimer’s disease of soluble forms of cerebral Abeta amyloid protein precursor containing the Kunitz protease inhibitory domain. J Biol Chem. 1998; 273: 5013-5019.
54. König G, Mönning U, Czech C, Prior R, Banati R, Schreiter-Gasser U, Bauer J, Masters CL, Beyreuther K. Identification and differential expression of a novel alternative splice isoform of the beta A4 amyloid precursor protein (APP) mRNA in leukocytes and brain microglial cells. J Biol Chem. 1992; 267: 10804-10809.
55. Mönning U, König G, Banati RB, Mechler H, Czech C, Gehrmann J, Schreiter-Gasser U, Masters CL, Beyreuther K. Alzheimer beta A4-amyloid protein precursor in immunocompetent cells. J Biol Chem. 1992; 267: 23950-23956.
56. Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Gervais-Bird J, Madden R, Paquet ER, Koh C, Venables JP, Prinos P, Jilaveanu-Pelmus M, Wellinger R, Rancourt C, Chabot B, Elela SA. Multiple alternative splicing markers for ovarian cancer. Cancer Res. 2008; 68: 657-663.
57. Venables JP, Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Koh C, Gervais-Bird J, Lapointe E, Froehlich U, Durand M, Gendron D, Brosseau JP, Thibault P, Lucier JF, Tremblay K, Prinos P, Wellinger RJ, Chabot B, Rancourt C, Elela SA. Identification of alternative splicing markers for breast cancer. Cancer Res. 2008; 68: 9525-9531.
58. Misquitta-Ali CM, Cheng E, O’Hanlon D, Liu N, McGlade CJ, Tsao MS, Blencowe BJ. Global profiling and molecular characterization of alternative splicing events misregulated in lung cancer. Mol Cell Biol. 2011; 31: 138-150.
59. Dawkins E, Small DH. Insights into the physiological function of the beta-amyloid precursor protein: beyond Alzheimer’s disease. J Neurochem. 2014; 129: 756-769.
60. Hansel DE, Rahman A, Wehner S, Herzog V, Yeo CJ, Maitra A. Increased expression and processing of the Alzheimer amyloid precursor protein in pancreatic cancer may influence cellular proliferation. Cancer Res. 2003; 63: 7032-7037.
61. Pandey P, Rachagani S, Das S, Seshacharyulu P, Sheinin Y, Naslavsky N, Pan Z, Smith BL, Peters HL, Radhakrishnan P, McKenna NR, Giridharan SS, Haridas D, Kaur S, Hollingsworth MA, MacDonald RG, Meza JL, Caplan S, Batra SK, Solheim JC. (2015). Amyloid precursor-like protein 2 (APLP2) affects the actin cytoskeleton and increases pancreatic cancer growth and metastasis. Oncotarget. 2015; 6: 2064-2075. doi: 10.18632/oncotarget.2990.
62. Meng JY, Kataoka H, Itoh H, Koono M. Amyloid beta protein precursor is involved in the growth of human colon carcinoma cell in vitro and in vivo. Int. J Cancer. 2001; 92, 31-39.
63. Takagi K, Ito S, Miyazaki T, Miki Y, Shibahara Y, Ishida T, Watanabe M, Inoue S, Sasano, H, Suzuki T. Amyloid precursor protein in human breast cancer: an androgen-induced gene associated with cell proliferation. Cancer Sci. 2013; 104: 1532-1538.
64. Lim S, Yoo BK, Kim HS, Gilmore HL, Lee Y, Lee HP, Kim SJ, Letterio J, Lee HG. Amyloid-beta precursor protein promotes cell proliferation and motility of advanced breast cancer. BMC Cancer. 2014; 14: 928-2407-14-928.
65. Miyazaki T, Ikeda K, Horie-Inoue K, Inoue S. Amyloid precursor protein regulates migration and metalloproteinase gene expression in prostate cancer cells. Biochem Biophys Res Commun. 2014; 452: 828-833.
66. Itoh H, Kataoka H, Koita H, Nabeshima K, Inoue T, Kangawa K, Koono M. Establishment of a new human cancer cell line secreting protease nexin-II/amyloid beta protein precursor derived from squamous-cell carcinoma of lung. Int J Cancer. 1991; 49: 436-443.
67. Jiang L, Yu G, Meng W, Wang Z, Meng F, Ma W. Overexpression of amyloid precursor protein in acute myeloid leukemia enhances extramedullary infiltration by MMP-2. Tumour Biol. 2013; 34: 629-636.
68. Venkataramani V, Rossner C, Iffland L, Schweyer S, Tambol IY, Walter J, Wirths O, Bayer TA. Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the Alzheimer amyloid precursor protein. J Biol Chem. 2010; 285: 10678-10689.
69. Moss AC, Doran PP, Macmathuna P. In silico promoter analysis can predict genes of functional relevance in cell proliferation: validation in a colon cancer model. Transl Oncogenomics. 2007; 2: 1-16.
70. Maesako Y, Uchiyama T, Ohno H. Comparison of gene expression profiles of lymphoma cell lines from transformed follicular lymphoma, Burkitt’s lymphoma and de novo diffuse large B-cell lymphoma. Cancer Sci. 2003; 94: 774-781.
71. Peters HL, Yan Y, Nordgren TM, Cutucache CE, Joshi SS, Solheim JC. Amyloid precursor-like protein 2 suppresses irradiation-induced apoptosis in Ewing sarcoma cells and is elevated in immune-evasive Ewing sarcoma cells. Cancer Biol Ther. 2013; 14: 752-760.
72. Peters HL, Yan Y, Solheim JC. APLP2 regulates the expression of MHC class I molecules on irradiated Ewing’s sarcoma cells. Oncoimmunology. 2013; 2: e26293.
73. Arvidsson Y, Andersson E, Bergstrom A, Andersson MK, Altiparmak G, Illerskog AC, Ahlman H, Lamazhapova D, Nilsson O. Amyloid precursor-like protein 1 is differentially upregulated in neuroendocrine tumours of the gastrointestinal tract. Endocr Relat Cancer. 2008; 15, 569-581.
74. Sobol A, Galluzzo P, Weber MJ, Alani S, Bocchetta, M. Depletion of Amyloid Precursor Protein (APP) causes G0 arrest in non-small cell lung cancer (NSCLC) cells. J Cell Physiol. 2015; 230:1332-1341.
75. Botelho MG, Wang X, Arndt-Jovin DJ, Becker D, Jovin TM. Induction of terminal differentiation in melanoma cells on downregulation of beta-amyloid precursor protein. J Invest Dermatol. 2010; 130: 1400-1410.
76. Tuli A, Sharma M, Wang X, Simone LC, Capek HL, Cate S, Hildebrand WH, Naslavsky N, Caplan S, Solheim JC. Amyloid precursor-like protein 2 association with HLA class I molecules. Cancer Immunol Immunother. 2009; 58: 1419-1431.
77. Tang CE, Guan YJ, Yi B, Li XH, Liang K, Zou HY, Yi H, Li MY, Zhang PF, Li C, Peng F, Chen ZC, Yao KT, Xiao ZQ. Identification of the amyloid beta-protein precursor and cystatin C as novel epidermal growth factor receptor regulated secretory proteins in nasopharyngeal carcinoma by proteomics. J Proteome Res. 2010; 9: 6101-6111.
78. Provenzano MJ, Yu L, Hitchler MJ, Fitzgerald MP, Robinson RA, Wayne S, Ver Meer M, Domann FE. (2007). AP-2 participates in the transcriptional control of the amyloid precursor protein (APP) gene in oral squamous cell carcinoma. Exp Mol Pathol. 2007; 83: 277-282.
79. Yamada Y, Fujimura T, Takahashi S, Takayama K, Urano T, Murata T, Obinata D, Ouchi Y, Homma Y, Inoue S. Clinical significance of amyloid precursor protein in patients with testicular germ cell tumor. Adv Urol. 2013; 2013: 348438.
80. Venkataramani V, Thiele K, Behnes CL, Wulf GG, Thelen P, Opitz L, Salinas-Riester G, Wirths O, Bayer TA, Schweyer S. Amyloid precursor protein is a biomarker for transformed human pluripotent stem cells. Am J Pathol. 2012; 180: 1636-1652.
81. Yang Z, Fan Y, Deng Z, Wu B, Zheng Q. Amyloid precursor protein as a potential marker of malignancy and prognosis in papillary thyroid carcinoma. Oncol Lett. 2012; 3: 1227-1230.
82. Zhuang D, Qiao Y, Zhang X, Miao S, Koide SS, Wang L. YWK-II protein/APLP2 in mouse gametes: potential role in fertilization. Mol Reprod Dev. 2006; 73: 61-67.
83. Fok KL, Chen H, Ruan YC, Chan HC. Novel regulators of spermatogenesis. Seminars Cell & Dev Biol. 2014; 29: 31-42.
84. Tuli A, Sharma M, McIlhaney MM, Talmadge JE, Naslavsky N, Caplan S, Solheim JC. Amyloid precursor-like protein 2 increases the endocytosis, instability, and turnover of the H2-Kd MHC class I molecule. J Immunol. 2008; 181: 1978-1987.
85. Tuli A, Sharma M, Naslavsky N, Caplan S, Solheim JC. Specificity of amyloid precursor-like protein 2 interactions with MHC class I molecules. Immunogenetics. 2008; 60: 303-313.
86. Tuli A, Sharma M, Capek HL, Naslavsky N, Caplan S, Solheim JC. Mechanism for amyloid precursor-like protein 2 enhancement of major histocompatibility complex class I molecule degradation. J Biol Chem. 2009; 284: 34296-34307.
87. Peters HL, Tuli A, Sharma M, Naslavsky N, Caplan S, MacDonald RG, Solheim JC. Regulation of major histocompatibility complex class I molecule expression on cancer cells by amyloid precursor-like protein 2. Immunol Res. 2011; 51: 39-44.
88. Takayama K, Tsutsumi, S, Suzuki T, Horie-Inoue K, Ikeda K, Kaneshiro K, Fujimura T, Kumagai J, Urano T, Sakaki Y, Shirahige K, Sasano H, Takahashi S, Kitamura T, Ouchi Y, Aburatani H, Inoue S. Amyloid precursor protein is a primary androgen target gene that promotes prostate cancer growth. Cancer Res. 2009; 69: 137-142.
89. Schmitz A, Tikkanen R, Kirfel G, Herzog V. The biological role of the Alzheimer amyloid precursor protein in epithelial cells. Histochem Cell Biol. 2002; 117: 171-180.
90. Woods NK, Padmanabhan, J. (2013). Inhibition of amyloid precursor protein processing enhances gemcitabine-mediated cytotoxicity in pancreatic cancer cells. J Biol Chem. 2013; 288: 30114-30124.
91. Hong Y, Downey T, Eu KW, Koh PK, Cheah PY. A ‘metastasis-prone’ signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin Exp Metastasis. 2010; 27: 83-90.
92. Wu W, Song W, Li S, Ouyang S, Fok KL, Diao R, Miao S, Chan HC, Wang L. Regulation of apoptosis by Bat3-enhanced YWK-II/APLP2 protein stability. J Cell Sci. 2012; 125: 4219-4229.
93. Trejo J, Massamiri T, Deng T, Dewji NN, Bayney RM, Brown JH. A direct role for protein kinase C and the transcription factor Jun/AP-1 in the regulation of the Alzheimer’s beta-amyloid precursor protein gene. J Biol Chem. 1994; 269: 21682-21690.
94. Grilli M, Goffi F, Memo M, Spano P. Interleukin-1beta and glutamate activate the NF-kappaB/Rel binding site from the regulatory region of the amyloid precursor protein gene in primary neuronal cultures. J Biol Chem. 1996; 271: 15002-15007.
95. Salbaum JM, Weidemann A, Lemaire HG, Masters CL, Beyreuther K. The promoter of Alzheimer’s disease amyloid A4 precursor gene. EMBO J. 1988; 7: 2807-2813.
96. von Koch CS, Lahiri DK, Mammen AL, Copeland NG, Gilbert DJ, Jenkins NA, Sisodia SS. The mouse APLP2 gene chromosomal localization and promoter characterization. J Biol Chem. 1995; 270:25475-25480.
97. Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, Vitek MP, Gajdusek DC. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci USA 1989; 86: 7606-7610.
98. Lahiri DK, Nall C. Promoter activity of the gene encoding the beta-amyloid precursor protein is up-regulated by growth factors, phorbol ester, retinoic acid and interleukin-1. Brain Res Mol Brain Res. 1995; 32: 233-240.
99. Beckman M, Iverfeldt K. Increased gene expression of beta-amyloid protein and its homologues APLP1 and APLP2 in human neuroblastoma cells in response to retinoic acid. Neurosci Lett. 1997; 221:73-76.
100. Balastik M, Lim J, Pastorino L, Lu KP. Pin1 in Alzheimer’s disease: multiple substrates, one regulatory mechanism? Biochim Biophys Acta 2007; 1772, 422-429.
101. Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 2006; 440: 528-534.
102. Xu KP, Zoukhri D, Zieske JD, Dartt DA, Sergheraert C, Loing E, Yu FS. A role for MAP kinase in regulating ectodomain shedding of APLP2 in corneal epithelial cells. Am J Physiol Cell Physiol. 2001; 281: C603-14.
103. Holzer M, Brückner MK, Beck M, Bigl V, Arendt T. Modulation of APP processing and secretion by okadaic acid in primary guinea pig neurons. J Neural Transm. (Vienna) 2000; 107: 451-461.
104. Goodenough S, Engert S, Behl C. Testosterone stimulates rapid secretory amyloid precursor protein release from rat hypothalamic cells via the activation of the mitogen-activated protein kinase pathway. Neurosci Lett. 2000; 296: 49-52.
105. Manthey D, Heck S, Engert S, Behl C. Estrogen induces a rapid secretion of amyloid beta precursor protein via the mitogen-activated protein kinase pathway. Eur J Biochem. 2001; 268: 4285-4291.
106. Swistowski A, Zhang Q, Orcholski ME, Crippen D, Vitelli C, Kurakin A, Bredesen DE. Novel mediators of amyloid precursor protein signaling. J Neurosci. 2009; 29: 15703-15712.
107. Orcholski ME, Zhang Q, Bredesen DE. Signaling via amyloid precursor-like proteins APLP1 and APLP2. J Alzheimers Dis. 2011; 23: 689-699.
108. Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, Dong J. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res. 2013; 73: 6722-6733.
109. Zhang L, Yang S, Chen X, Stauffer S, Yu F, Lele SM, Fu K, Datta K, Palermo N, Chen Y, Dong, J. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol Cell Biol. 2015; 35: 1350-1362.
110. Yang S, Zhang L, Purohit V, Shukla SK, Chen X, Yu F, Fu K, Chen Y, Solheim J, Singh PK, Dong J. Active YAP promotes pancreatic cancer cell motility, invasion, and tumorigenesis in a mitotic phosphorylation-dependent manner through LPAR3. Oncotarget. 2015; 6:36019-31. doi: 10.18632/oncotarget.5935.
111. Fu D, Lv X, Hua G, He C, Dong J, Lele SM, Li DW, Zhai Q, Davis JS, Wang C. YAP regulates cell proliferation, migration, and steroidogenesis in adult granulosa cell tumors. Endocr Relat Cancer 2014; 21: 297-310.
112. Steinhardt AA, Gayyed MF, Klein AP, Dong J, Maitra A, Pan D, Montgomery EA, Anders RA. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008; 39: 1582-1589.
113. Acevedo KM, Opazo CM, Norrish D, Challis LM, Li QX, White AR, Bush AI, Camakaris J. Phosphorylation of amyloid precursor protein at threonine 668 is essential for its copper-responsive trafficking in SH-SY5Y neuroblastoma cells. J Biol Chem. 2014; 289: 11007-11019.
114. Barbagallo AP, Wang Z, Zheng H, D’Adamio L. A single tyrosine residue in the amyloid precursor protein intracellular domain is essential for developmental function. J Biol Chem. 2011; 286: 8717-8721.
115. Gandy S, Czernik AJ, Greengard P. Phosphorylation of Alzheimer disease amyloid precursor peptide by protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Proc Natl Acad Sci USA. 1988; 85: 6218-6221.
116. Griffith LS, Mathes M, Schmitz, B. Beta-amyloid precursor protein is modified with O-linked N-acetylglucosamine. J Neurosci Res. 1995; 41: 270-278.
117. Jacobsen KT, Iverfeldt K. O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-beta precursor protein (APP). Biochem Biophys Res Commun. 2011; 404: 882-886.
118. Kitazume S, Tachida Y, Kato M, Yamaguchi Y, Honda T, Hashimoto Y, Wada Y, Saito T, Iwata N, Saido T, Taniguchi N. Brain endothelial cells produce amyloid beta from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J Biol Chem. 2010; 285, 40097-40103.
119. Matrone C, Barbagallo AP, La Rosa LR, Florenzano F, Ciotti MT, Mercanti D, Chao MV, Calissano P, D’Adamio L. APP is phosphorylated by TrkA and regulates NGF/TrkA signaling. J Neurosci. 2011; 31: 11756-11761.
120. Perdivara I, Petrovich R, Allinquant B, Deterding LJ, Tomer KB, Przybylski M. Elucidation of O-glycosylation structures of the beta-amyloid precursor protein by liquid chromatography-mass spectrometry using electron transfer dissociation and collision induced dissociation. J Proteome Res. 2009; 8: 631-642.
121. Suzuki T, Nairn AC, Gandy SE, Greengard P. Phosphorylation of Alzheimer amyloid precursor protein by protein kinase C. Neuroscience. 1992; 48: 755-761.
122. Suzuki T, Oishi M, Marshak DR, Czernik AJ, Nairn AC, Greengard P. Cell cycle-dependent regulation of the phosphorylation and metabolism of the Alzheimer amyloid precursor protein. EMBO J. 1994; 13: 1114-1122.
123. Suzuki T, Ando K, Isohara T, Oishi M, Lim GS, Satoh Y, Wasco W, Tanzi RE, Nairn AC, Greengard P, Gandy SE, Kirino Y. Phosphorylation of Alzheimer beta-amyloid precursor-like proteins. Biochemistry. 1997; 36: 4643-4649.
124. Tarr PE, Contursi C, Roncarati R, Noviello C, Ghersi E, Scheinfeld MH, Zambrano N, Russo T, D’Adamio L. Evidence for a role of the nerve growth factor receptor TrkA in tyrosine phosphorylation and processing of beta-APP. Biochem Biophys Res Commun. 2002; 29: 324-329.
125. Taru H, Suzuki T. Facilitation of stress-induced phosphorylation of beta-amyloid precursor protein family members by X11-like/Mint2 protein. J Biol Chem. 2004; 279: 21628-21636.