
INTRODUCTION
Ovarian cancer is the fifth leading cause of cancer deaths among women worldwide with an estimated 225,500 new cases annually [1]. Although ovarian cancer is among the most chemo-sensitive of solid tumors and generally shows a good initial response to platinum/taxane treatment and optimal debulking surgery, the disease will recur in 60-80% of women with advanced disease within five years [2, 3]. Considerable effort has been focused on identifying predictors of outcome at the somatic level, but less emphasis has been placed on the identification of germline predictors of outcome. We and others have used the candidate gene approach to identify ATP-binding cassette family members that might be associated with PFS [4, 5]. However, these findings have not been convincingly validated [5, 6].
Genome-wide association studies (GWAS) have been extremely successful at finding susceptibility loci for many different complex diseases [7], including multiple cancers [8]. The successful identification of loci associated with response to treatment could have profound clinical implications for individualizing anti-cancer treatment but there have been very few successful GWAS identifying loci associated with outcome for any cancer [9-16]. One factor that might explain this is that for most cancers there is considerable heterogeneity in the chemotherapeutic regimens used, which is likely to contribute to heterogeneity in treatment response [17-19]. In addition, it has been difficult to compile germline DNA and detailed treatment and clinical follow-up information on a sufficiently large number of patients to provide enough statistical power to detect loci associated with PFS or overall survival (OS).
In this study, we aimed to identify germline polymorphisms that influence response to first-line chemotherapy in patients with EOC. Based on previous ex vivo studies in lymphoblastoid cell lines derived from related family members that have shown moderately high heritability (0.21 to 0.7, depending on dose) for sensitivity to docetaxel [20] and cisplatin-induced cytotoxicity [21], we hypothesized that inter-patient variability in response to these drugs may be in part be explained by genetic variation that could be identified if we used a cohort of patients who had been uniformly treated. Therefore, we conducted the GWAS of PFS in ovarian cancer patients treated with carboplatin and paclitaxel, with the initial GWAS on 385 patients with high-grade, serous cancer (HGSC) and follow-up phases on serous EOC patients from ten studies from the Ovarian Cancer Association Consortium (OCAC).
We identified two rare SNPs that fall within a regulatory element within intron 2 of TTC39B. Chromatin conformation assays showed that the targets of the regulatory element are PSIP1, CCDC171 and an alternative promoter of TTC39B. DNA-protein analyses indicated that the likely functional SNP is rs7874043, which alters Sp1 transcription factor binding, a factor that is critical for chromatin looping between the PRE and the PSIP1 promoter. Furthermore, we show that silencing of PSIP1 significantly impaired DNA damage-induced homologous recombination function in ovarian cancer cell lines. According to KM-plotter (an online database linking expression to ovarian outcome in publicly available data), high expression of PSIP1 is associated with poor PFS in ovarian cancer suggesting that altered PSIP1 expression may be driving the association between the associated SNPs and outcome in EOC patients [22].
RESULTS
Four-Phase GWAS
We carried out a four-phase genome-wide association study of PFS in a total of 1,244 serous ovarian cancer patients who had debulking surgery and were uniformly treated with only carboplatin and paclitaxel as first-line therapy (Figure 1).
In Phase 1, we conducted a genome-wide scan on germline DNA from 385 patients from the Australian Ovarian Cancer Study (AOCS, n = 183), the Mayo Clinic (MAYO, n = 68) and The Cancer Genome Atlas (TCGA, n = 134) and performed a meta-analysis summarizing results from these cohorts (refer to Methods for details of genotyping and imputation). The Manhattan plot showing SNP association with PFS is presented in Supplementary Figure 1. We then prioritized 190 SNPs primarily ranked by P-value in Phase 1 for validation and further replication (Supplementary Table 1). We also included 10 SNPs in the gene YAP1, in light of its association with response to platinum-based chemotherapy in small-cell lung cancer patients [14], to test whether this finding would replicate in our study. These SNPs were also genotyped on Phase 1 AOCS and MAYO samples (DNA samples were not available for TCGA) so that we could replace imputed data with actual genotypes for these samples in subsequent analyses.
In Phase 2, we genotyped these 200 SNPs in 706 patients from 8 studies participating in OCAC. Following further data cleaning, we updated treatment details and other clinical information prior to Phase 2 analysis, which revealed 106 patients included in the Phase 1 GWAS who were no longer eligible for inclusion mainly due to the fact that these patients received additional agents or did not meet the dose requirement (Methods). Therefore, we performed a pooled analysis of 985 patients from both phases with these 106 patients excluded. This analysis identified two uncommon SNPs in strong linkage disequilibrium (LD), rs72700653 and rs7874043 (minor allele frequency (MAF) ~ 1.85% and imputation r2 = 0.9) in intron 2 of TTC39B, most strongly associated with PFS in serous ovarian cancer patients (P = 3.5x10-7 and 3.6x10-7 for rs72700653 and rs7874043 respectively; Supplementary Table 1). Both SNPs were imputed with high quality (imputation quality score r2 = 0.81 in MACH [23]). None of the 10 tag SNPs in the YAP1 gene were associated with PFS in these 985 patients (P > 0.05, Supplementary Table 1).
In Phase 3, we genotyped 38 tagSNPs, in addition to rs72700653 and rs7874043, in 985 OCAC samples to perform fine-mapping of the TTC39B locus. rs7874043 and rs72700653 remained the SNPs most associated with PFS at this locus, and the variants in moderate LD with rs7874043 showed consistent association with PFS (Supplementary Table 2). In Phase 4, we sought further replication of the association between these two variants and PFS in two additional cohorts, MAC (n = 26) and the clinical trial, ICON7 (n = 124) and additional samples from OCAC (n = 109). As there were only a small number of eligible cases in MAC, and both MAC and MAYO studies were recruited at the Mayo Clinic, we combined these two sets for analysis.
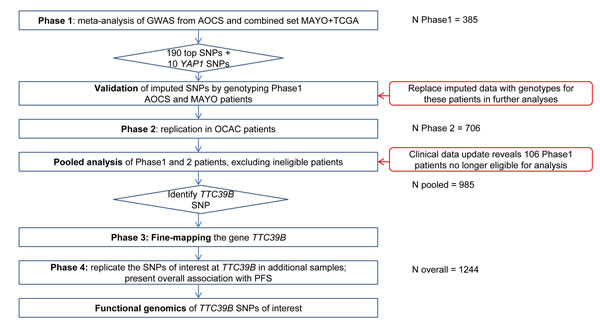
Figure 1: Study Design. Overview of the study design for the identification of TTC39B SNPs using a four-phase GWAS of PFS in serous EOC patients.
To get an overall estimate of the hazard ratio, we pooled all available data from Phase 1, 2 (again excluding the ineligible patients) and 4 (n = 1244). Details of all the OCAC sites contributing to this study are given in Supplementary Table 3. This analysis showed that the minor allele of rs7874043 was associated with significantly worse PFS (HR = 1.90, 95% CI = 1.38 to 2.61, P = 7.3x10-5; Figure 2a). The median PFS in patients homozygous for the common allele of rs7874043 was 16.0 months (95% CI = 15.0 to 17.1), compared to 11.5 months (95% CI = 9.5 to 15.4) in heterozygous patients, without adjustment for covariates (log-rank test P = 0.0098); while the difference was 17.2 months (95% CI = 16 to 18.1) versus 11.5 months (95% CI = 9.6 to 14.7) when we assumed all prognostic factors at their mean values (Figure 2b, Supplementary Figure 2). The result of association between this SNP and PFS was similar when restricted to the high-grade serous patients at advanced disease stage (n = 1061, HR = 1.86, 95% CI = 1.33 to 2.6, P = 2.6x10-4). The median PFS was 14.8 months (95% CI = 14 to 15.8) for these patients with homozygous genotypes versus 11.0 months (95% CI = 9.3 to 14.1) for heterozygote patients, assuming mean covariates. The other SNP in high LD, rs72700653, despite a similar HR, had weaker association than rs7874043 due to more missing genotypes (HR = 1.91, 95% CI = 1.36 to 2.69, P = 2.2x10-4). We found attenuated associations with PFS in OCAC patients who were selected with no regard to chemotherapy (HR = 1.31, 95% CI = 1.04 to 1.66, P = 0.02; Supplementary Figure 3),
We also determined whether TTC39B SNPs were associated with OS. rs7874043 showed a significant, but weaker association with OS. The minor allele was also associated with worse OS (HR = 1.56, 95% CI = 1.09 to 2.23, P = 0.015). The median OS differences unadjusted for covariates were 46.3 months (95% CI = 43.2 to 49.9) for patients with homozygous genotypes versus 37 months (95% CI = 29.8 to 53.8) for heterozygous patients (log-rank test P = 0.048), and 48.7 months (95% CI = 45.4 to 55.3) versus 38.9 months (95% CI = 29.8 to 55.5) assuming mean covariates (Supplementary Figure 4).
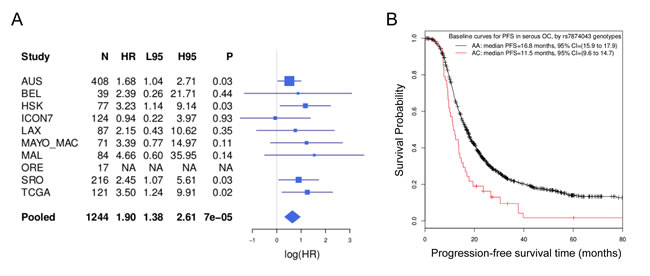
Figure 2: Association with PFS in serous EOC patients. A. Associations between the TTC39B SNP rs7874043 and PFS in individual studies (rows denoted by study names), and the overall association pooling all samples together while stratifying for studies (the row denoted by “Pooled”). “HR” indicates the point estimates of hazard ratio. “L95” and “H95” represents its lower and upper 95% confidence intervals. “NA” indicates no minor allele was found in the eligible cases. The forest plot on the right is on the log scale. B. Baseline survival curves of the two genotypes (AA vs AC) of rs7874043 in a stratified Cox regression, assuming all other prognostic factors at mean values. Patients with CC genotypes were not observed due to the low minor allele frequency of rs7874043. The survival curves were truncated at 80 months as only a few events occurred after that.
PFS-associated SNPs fall within a distal regulatory element of PSIP1, CCDC171 and an alternative promoter of TTC39B
Regulatory elements such as promoters and transcriptional enhancers/silencers can be identified by distinct chromatin marks. Tri-methylation of histone 3 lysine 4 (H3K4Me3) marks promoters, while mono-methylation (H3K3Me1) marks promoters and enhancers. ENCODE ChIP-seq data for H3K4Me1 from eight different cell lines covering the TTC39B locus revealed that both rs7874043 and rs72700653 fall within a putative regulatory element (PRE) marked by H3K4Me1 within intron 2 of TTC39B (Figure 3a). Transcription factor binding prediction indicated potential SNP-altered binding of Sp1, FOXA1 and AP1 (Supplementary Figure 5; [24]). We performed electrophoretic mobility shift assays (EMSAs) to assess binding of these transcription factors to the common and minor alleles of each of these variants and showed allele-specific protein binding for rs7874043 (Figure 3b; lanes 1, 2 and 8, 9). EMSAs using an Sp1 consensus oligonucleotide as competitor suggested that a strong higher mobility band and a weaker lower mobility band (in JAMs only) is likely to be Sp1 (Figure 3b, Supplementary Figure 6a). Using chromatin immunoprecipitation experiments we have shown that Sp1 is able to bind this site in JAM and A2780 ovarian cancer cell lines in vivo (Figure 3c). We also observed protein interaction at rs72700653, but found no difference in binding between alleles (Supplementary Figure 6b).
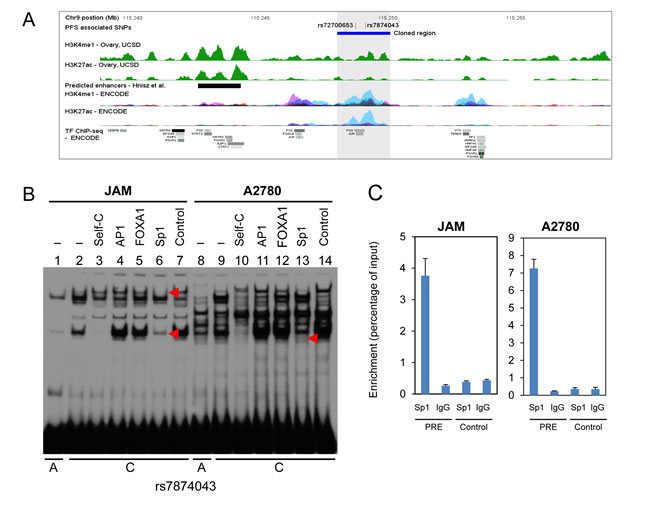
Figure 3: Chromatin structure and DNA-protein interactions surrounding the 9p22 PFS-associated SNPs. A. Colored histograms denote histone modification ChIP-seq data from UCSD and ENCODE. Epigenetic marks for H3K4me1 and H3K27ac in ovary from UCSD and 7 cell types from ENCODE, and transcription factor ChIP-seq data from ENCODE are shown. The grey shaded region denotes the PRE containing SNPs rs72700653 and rs7874043. B. EMSA for oligonucleotides containing SNP rs7874043 with the A = common allele and C = minor allele as indicated below the panel, assayed using JAM and A2780 nuclear extracts. Labels above each lane indicate inclusion of competitor oligonucleotides at 30-fold molar excess: (-) no competitor (Lanes 1,2,8,9); Self-C allele (Lanes 3,10), AP1 (Lanes 4,11), FOXA1 (Lanes 5,12), Sp1 (Lanes 6,13) and a control sequence (Lanes 7,14; containing binding site for ATF, a TF not predicted to bind). The Sp1-containing complexes are indicated with red arrowheads. C. ChIP-qPCR on the PRE in JAM and A2780 cell lines. ChIP assays were performed with Sp1 antibodies or non-immune IgG, with a region 2.3kb upstream of the predicted Sp1-binding site (Control) used as a control for nonspecific binding. Graphs represent two biological replicates. Error bars denote SD.
To determine the likely target genes of the PRE, we performed chromosome conformation capture (3C) using an anchor primer within the restriction fragment encompassing the PRE and a series of primers within restriction fragments spanning all protein coding gene promoters within two megabases of the PRE (Figure 4a). The results showed that the PRE frequently interacts with an alternative (1B) promoter of TTC39B in both A2780 and JAM cells (Figure 4b). The PRE also frequently interacted with the PSIP1 (also known as LEDGF) promoter, located approximately 260kb away, in JAM but not A2780 cells, and the CCDC171 promoter, approximately 300kb away, in both JAM and A2780 cells (Figure 4c and 4d; Supplementary Figure 7). All chromatin interactions were confirmed by performing 3C with an independent restriction enzyme using anchor primers in the relevant gene promoters and a series of primers spanning the PRE (Supplementary Figure 8). No significant interactions were detected between the PRE and other flanking genes including NFIB, ZDHHC21, CER1, FREM1 or SNAPC3 (Supplementary Figure 9).
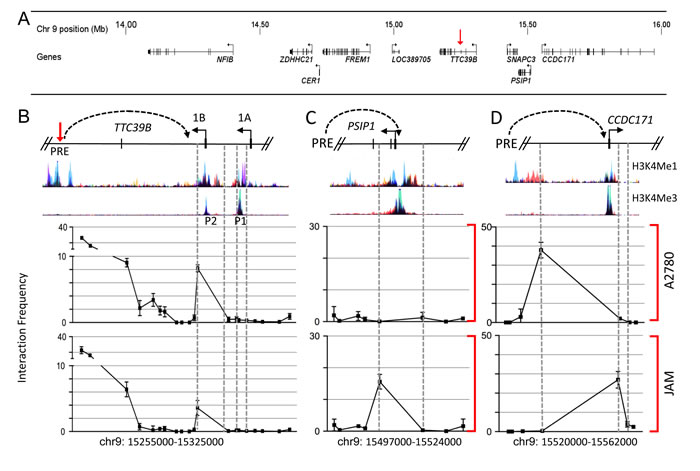
Figure 4: Chromatin interactions at 9p22 in ovarian cancer cell lines. A. Physical map spanning 2Mb of the 9p22 region showing the position of all annotated genes assessed by 3C. The red arrowhead denotes TTC39B. B.-D. 3C interaction profiles between the PRE (containing rs72700653 and rs7874043) and (B) TTC39B, (C) PSIP1 and (D) CCDC171 promoter regions. 3C libraries were generated with either HindIII (B) or EcoRI (C and D), with the anchor point set at the PRE. A physical map of the region interrogated by 3C and relevant ENCODE histone modification data is shown above. A representative graph of three biological replicates is shown. Error bars denote SD.
Using luciferase reporter assays we demonstrated that the PRE acts as a strong transcriptional enhancer on the PSIP1 and CCDC171 promoters (Figure 5). Interestingly, the PRE had no significant effect on the TTC39B 1B promoter in A2780 cells and acted as a silencer in JAM cells suggesting that, depending on the cellular context, the PRE can act as an enhancer or silencer. To examine the effect of the SNPs on the activity of the PRE, we generated reporter constructs containing the minor alleles of both rs7874043 and rs72700653 (Figure 5, TTC PRE HAP). In A2780 cells, inclusion of the PRE-minor alleles significantly increased TTC39B 1B promoter activity and in JAM cells the minor alleles ablated the PREs silencer activity. In both cell lines, inclusion of the PRE-minor alleles had no additional effect on PSIP1 and CCDC171 promoter activity. While this appears to rule out a direct effect of these SNPs on transactivation of these promoters, Sp1 is reported to regulate chromatin looping and therefore the SNPs may be influencing the physical interactions between the PRE and target genes [25]. To address this question, we performed 3C analysis on JAM cells after Sp1 siRNA-mediated silencing and showed that the chromatin interaction between the PRE and PSIP1 but not CCDC171 was ablated (Figure 5b and 5c; Supplementary Figure 10). Consistent with this, the expression of PSIP1, but not CCDC171, decreased with Sp1 knockdown (Figure 5d).
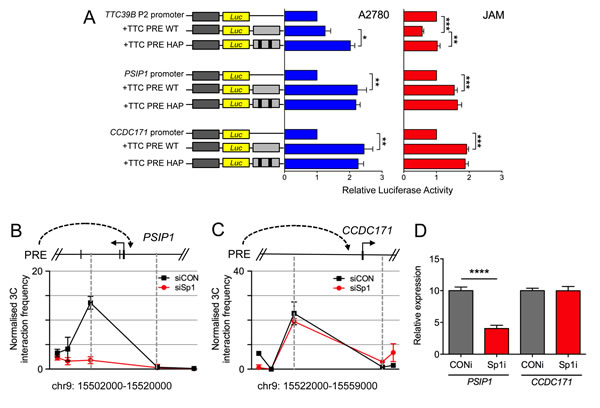
Figure 5: Evaluation of the function of rs72700653 and rs7874043 in ovarian cancer cell lines. A. Luciferase assays comparing effect of minor alleles on the function of TTC39B, PSIP1 and CCDC171 promoters. The PRE was cloned upstream of TTC39B, PSIP1 or CCDC171 promoter-driven luciferase reporter constructs with rs72700653 and rs7874043 (PRE HAP) or without (PRE WT). A2780 or JAM ovarian cancer cells were transiently transfected with each construct and assayed for luciferase activity after 24h. Error bars denote SEM (N = 3). P values were determined with a two-tailed t test. *p < 0.05, **p < 0.01,***p < 0.001. Effect of siRNA knock-down of SP1 on 3-C interactions between the PRE with B. PSIP1 and C. CCDC171 promoter regions in JAM cells. 3C libraries were generated with EcoRI, with the anchor point set at the PRE. A physical map of the region interrogated by 3C data is shown above. A representative graph of three biological replicates is shown. Error bars denote SD. D. Effect of siRNA knock-down on gene expression levels of PSIP and CCDC171 in JAM cells. JAM cells were transiently transfected using Sp1 (siSp1) RNAi smartpools or nontargeting control (siCON) and assayed after 48 hours. Gene expression was measured by TaqMan and is given relative to B-glucuronidase. Error bars denote SEM (N = 3). P values were determined with a two-tailed t test. ****p < 0.0001.
High expression of PSIP1 and CCDC171 is associated with PFS
We used Rapid Amplification of cDNA ends (RACE) to identify the transcript initiated from the 1B promoter of TTC39B, as there was none described. We identified a novel first exon of TTC39B, located ~13 kb downstream of the canonical exon 1, with a successive exon structure similar to the published TTC39B-202 (Supplementary Figure 11a). Negligible expression of this novel transcript was found in 18 ovarian cancer cell lines, whilst in 149 serous ovarian epithelial tumors from the AOCS low or minimal expression was observed (Supplementary Figure 11b and 11c). There was no association between expression of this transcript and PFS (HR = 0.7 (95% CI 0.38 - 1.25); P = 0.21 for the upper decile vs remaining patients); nor were expression levels associated with rs7874043 genotype (P = 0.22), but there were only six heterozygous carriers among the 142 tumors analyzed (Supplementary Figure 11c). However, analysis of 1171 epithelial ovarian tumors in KM-plotter [22], the online tool for survival-associated biomarkers, showed a strong association between high PSIP1 expression and shorter PFS (Figure 6; HR = 1.44 (95% CI 1.23 - 1.68; P = 6.6 x 10-6 for comparison above and below median PSIP1 expression). Since there were no data available in KM-plotter for CCDC171, we used the more limited TCGA serous ovarian cancer dataset and found that among the 68 patients with nil residual disease, high levels of CCDC171 were associated with PFS (Supplementary Figure 12; HR 5.04 (95% CI 1.99 - 12.79); P = 0.001 for the upper decile vs remaining patients). However, this was not evident among 374 patients with any debulking status (HR 1.25 (95% CI 0.84 - 1.86); P = 0.266 for the upper decile vs remaining patients). In the TCGA dataset, expression of PSIP1 was positively correlated with that of CCDC171 and many other genes on the short arm of chromosome 9 (Supplementary Table 4). This appears to be largely because of co-amplification, as the correlations dropped noticeably upon correction for copy number.
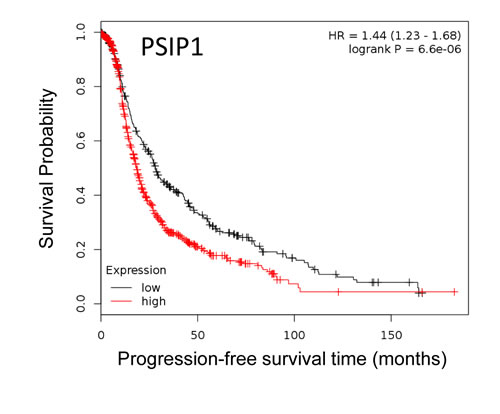
Figure 6: Kaplan-Meier curves of association between expression of PSIP1 with PFS in EOC. Expression of PSIP1 (Affymetrix probe 205961_s_at; log rank P = 6.6 x 10-6) and PFS in 1171 patients with serous and endometrioid EOC using the online tool KM-plotter [22]. High and low expression are defined as above and below the median.
PSIP1 is required for RAD51 foci formation after DNA damage in ovarian cancer cell lines
PSIP1 is known to facilitate the resection step during homologous recombination mediated-repair and is required for RAD51 foci formation after DNA damage in a number of cancer cell lines [26]. Therefore, to assess this function in ovarian cancer we silenced PSIP1 using siRNA in two high grade serous ovarian cancer cell lines, OVCAR3 and FUOV1 [27], which express relatively high levels of PSIP1 (Supplementary Figure 13a and 13b). We observed a significant reduction in DNA damage-induced Rad51 foci formation in both cell lines. Representative images and quantification are shown for OVCAR3 using two independent siRNA sequences (Figure 7a and 7b). Furthermore, exposure of OVCAR3 and FUOV1 cells to carboplatin and paclitaxel caused a moderate increase in PSIP1 levels consistent with PSIP1 being responsive to cellular stress, and in the case of carboplatin, potentially responsive to DNA damage (Supplementary Figure 13c). In long-term cell viability assays we also found that depletion of PSIP1 itself has significant effect on cell viability (Figure 7c) of ovarian cancer cell lines suggesting some level of dependency on PSIP1 levels for cell survival.
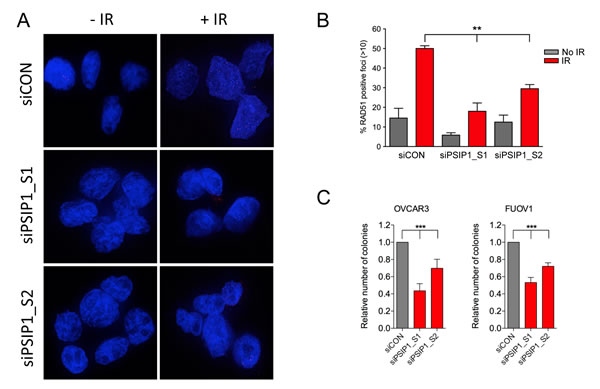
Figure 7: PSIP1 inhibition impaired DNA damage-induced homologous recombination function in ovarian cancer cell lines. A. Representative images of the OVCAR3 cell line transfected either with nontargeting scramble control (siCON) or PSIP1 (siPSIP1) RNAi for 48h, irradiated (IR) with 6 Gy and immunostained with anti-RAD51 (red) and DAPI (blue). B. Quantification of RAD51 positive foci after PSIP1 depletion alone and with 6 Gy IR. The percentage of cells with > 10 RAD51 foci were calculated. Error bars denote SEM (N = 2 with more than 50 cells were counted for each experiment). C. Effect of PSIP1 silencing on long-term colony formation in OVCAR3 and FUOV1 determined using crystal violet staining. P values were determined with a two-tailed t test. **p < 0.01,***p < 0.001.
DISCUSSION
We have carried out the first GWAS of PFS in European women diagnosed with EOC. We identified two SNPs in strong linkage disequilibrium (LD) in an intron of the TTC39B gene that were associated with worse PFS in patients with serous EOC. PFS in patients carrying the rare allele of rs7874043 is ~5 months shorter than in patients carrying the common allele. In particular for patients with high-grade serous EOC at advanced stage who often have poor survival, the SNP is also associated with ~4 months difference in PFS. Despite the large differences in PFS, these associations did not reach genome-wide significance (P < 5x10-8). We estimate that to reach genome-wide significance for the allele frequency of rs7874043, we would need twice as many serous EOC patients, depending on the true HR, with germline DNA samples and detailed clinical follow up data. We found attenuated associations with PFS in ovarian cancer patients who were selected with no regard to chemotherapy, reflecting either chance variation or that the effects of this SNP are specific to the treatment response. Furthermore, we found these SNPs had a significant, albeit weaker association with OS.
We have shown that the regulatory element, in which rs7874043 and rs72700653 lies, acts as a transcriptional enhancer on the PSIP1 and CCDC171 promoters, and that haplotype carrying the minor alleles of these SNPs enhances expression of the non-canonical TTC39B promoter. All three genes are located at 9p22.3, a region of chromosomal gain previously identified by comparative genomic hybridization in ovarian tumors as being associated with resistance to paclitaxel/carboplatin therapy [28]. The best characterized of the affected genes is PSIP1 (PC4 And SFRS1 Interacting Protein 1), also known as LEDGF (Lens Epithelial Derived Growth Factor), which is a epigenetic reader recognizing H3K36 marks that preferentially associate with the internal coding areas of actively transcribed genes [29, 30]. PSIP1 is known to have oncogenic activity that controls a caspase-independent lysosomal cell death pathway [31], and can protect against cell death induced by many different stimuli including etopside, anthracyclines, docetaxel and oxidative stress [32]. PSIP1 is overexpressed in chemoresistant acute myelogenic leukemia and protects leukemic cells from apoptosis in vitro [33]. PSIP1 is thought to protect cells from stress-induced apoptosis by transcriptionally activating protective genes such as HSP27 and CRYAB (alphaB-crystallin) [34]. More recently, PSIP1/LEDGF has been shown to regulate homologous recombination DNA repair pathway by guiding the tethering of Retinoblastoma binding protein 8 (RBBP8; also known as CtBP-Interacting Protein) to specific areas of chromatin (H3K36me3) in a DNA damage-dependent manner, providing a mechanism for its ability to protect cancer cells from DNA damage [35]. PSIP1 is also involved in HIV integration and is therefore a promising candidate for anti-retroviral therapy [36, 37].
Almost nothing is known about the function of CCDC171 (Coiled-Coil Domain Containing 171) except that it has been identified as an NRF1 target gene based on a Chip-Seq screen [38]. NRF1 plays a crucial role in maintenance of mitochondrial function and oxidative stress response. TTC39B (c9orf52) encodes a potential transmembrane protein and two GWAS for lipid levels have identified common SNPs in this locus associated with high density lipoprotein cholesterol (HDL-C) levels [39, 40]. This association was confirmed in a mouse model which showed that knockdown of TTC39B resulted in elevated levels of HDL-C [39]. These common SNPs are not strongly correlated with the rare SNPs we found to be associated with PFS (r2 < 0.1), and it is unknown whether they regulate the expression of the same novel TTC39B isoform that we identified. We cannot predict the function of this novel isoform but we have shown that it is not expressed at detectable levels in most ovarian cell lines, nor in approximately one-third of serous ovarian tumors.
Our data implicate Sp1 as a potential mediator of target gene(s) expression. Sp1 binds GC-rich DNA elements and regulates target genes by recruiting and complexing with transcription-associated proteins to activate or repress gene expression [41]. Notably, Sp1 is also able to mediate long-range activation of transcription through chromatin looping [30], and the PSIP1 promoter CpG island contains Sp1-responsive sites [42]. We showed the minor C allele of rs7874043 preferentially binds Sp1 in vitro, and that Sp1 binds to a region encompassing rs7874043, in vivo. Consistent with the known function of Sp1 in chromatin looping, we showed that Sp1 silencing ablated chromatin looping with the PSIP1 promoter and reduced PSIP1 expression. We therefore suggest that the minor allele of rs7874043 enhances chromatin looping between the PRE and the PSIP1 promoter to increase PSIP1 expression. Unfortunately, we were unable to identify any heterozygous cell lines for rs7874043, and therefore confirmation of these findings by evaluation of additional allele-specific effects was not possible in this study.
Although the absolute differences in PFS for carriers of the rare TTC39B alleles, compared to the common alleles, are quite large, the alleles are too rare to be responsible for much of the observed variation in PFS between affected women. Amongst the strengths of our study is the inclusion of only cases who had received standard first line treatment with carboplatin and paclitaxel, and the focus on clinically measurable PFS following first-line treatment, rather than on OS following exposure to multiple different drugs. Although we were able to obtain sufficient data in the population-based OCAC sites to conduct this study, it is much easier to do these studies in the context of clinical trials. Our study therefore emphasizes the importance of collecting germline DNA in clinical trials, and using them to detect biomarkers of response.
In conclusion, through a GWAS we have identified a SNP, rs7874043, as a very strong candidate for having a direct causal effect on PFS in ovarian cancer patients following first-line chemotherapy. We provide evidence that this SNP falls within a distal regulatory element that regulates several genes, including PSIP1, and show that high expression of PSIP1 is associated with poor PFS in ovarian cancer patients. We observed a significant reduction in cell viability following PSIP1 inhibition, suggesting that PSIP1 is a potential target for therapeutic intervention in ovarian cancer as previously suggested for other cancers [31]. Moreover, like other cancer cell lines, transient silencing of PSIP1 in an ovarian cancer cell line significantly impaired DNA damage-induced RAD51 foci formation suggesting involvement of PSIP1 in the regulation of homologous recombination-mediated DNA repair. PSIP1 is involved in HIV integration, and so there is already some interest in developing specific inhibitors. Successful inhibition of PSIP1 may provide a novel approach to target ovarian cancer.
MATERIALS AND METHODS
Patient selection criteria
All participating studies received approval from the respective Institutional Ethics Boards. In order to study a homogeneous group of patients in Phase 1, we selected patients from AOCS, MAYO and TCGA based on the following criteria: the primary cancer sites described as ovarian, fallopian tube or primary peritoneal, invasive serous histology, collection of primary treatment response data completed at the time of patient selection, high grade (grade 2 or 3), FIGO stage III or IV. Similar patient selection criteria were applied to OCAC studies in the following phases, except that the patients with low grade and low stage were also included. To improve cost efficiency, we preferentially genotyped AOCS patients with extreme phenotypes in Phase 1 (Supplementary Methods).
We observed substantial heterogeneity among treatments that patients in OCAC received, with more than 80 different chemotherapy drug combinations, dosage levels and schedules used for first-line chemotherapy, so we further selected patients who were treated with only three weekly paclitaxel and carboplatin as first-line therapy. Among these patients, the majority received > 4 cycles of paclitaxel at a dose of 135 to 175 mg/m2 and carboplatin at AUC 5 to 7), while dosage for some patients was unknown.
Prior to commencing data analysis in Phase 2, we obtained updated clinical data, which revealed that 91 AOCS and MAYO patients included in Phase 1 no longer met the criteria we applied because they had received agents in addition to carboplatin/paclitaxel, or < 4 cycles of paclitaxel and carboplatin. Other data updates on residual disease and other clinical features meant that an additional 15 cases were no longer eligible for the analysis. We therefore excluded these cases from further analyses. All the following analyses were based on the most up-to-date clinical data. To summarize, we analyzed a total of 1244 cases who had received standard chemotherapy plus an additional 1346 cases who had received non-standard chemotherapy for the analyses selected with no regard to chemotherapy. The details of their treatment is provided in the Supplementary Table 5.
Genotyping and Imputation
AOCS, MAYO and TCGA patients were genotyped using Illumina HumanOmni1-Quad arrays, HumanHap 610 arrays and Human1M arrays, respectively. We applied the following quality control steps for all three sets of GWAS data separately: 1) removing samples with > 10% missing genotypes; 2) excluding any SNP with less than 1% minor allele frequency (MAF); 3) excluding any SNP that failed the Hardy-Weinberg Equilibrium (HWE) test at the significance level of 5e-6; 4) excluding the SNPs with MAF > 5% when per SNP no-call rate > 5%, and those with MAF < 5% when per SNP no-call rate > 1%. We then assessed the cryptic relationship between the sample pairs using the ‘—genome’ command in PLINK [43]. The proportion of identical by descent (IBD) was estimated from the cleaned whole genome data, and then either of the paired IDs showing high levels of IBD sharing (PI_HAT > 0.2) was dropped. Patients who have been recruited in both MAYO and TCGA were identified using this approach and were included in the analyses only once. Following the check of cryptic relationships, we assessed potential population stratification using a Principal Components Analysis (PCA) algorithm EIGENSTRAT [44]. The HapMap 3 and GenomeEUTwin [45] individuals were used as a reference panel in the calculation of the principal components, and the current samples were projected into the background of reference population. We used 6 standard deviations (SDs) as the cut-off to identify ancestral outliers, which were removed from further association analysis. After these data cleaning, 183 AOCS patients (766,728 SNPs) and combined 68 MAYO and 134 TCGA patients (525,792 SNPs) were included in Phase 1 analysis.
To improve array comparison, we inferred the missing genotypes for the initial GWAS samples with the reference of the CEU samples from the 1000 Genomes pilot 1 data (June, 2010 release) using MACH 1.0 [23]. We had 385 samples on a common set of 297,906 SNPs without strand ambiguity (i.e. A|T or C|G genotypes) for imputation. We have imputed up to 6.86 million SNPs, with 88.7% (~6 million) SNPs obtaining reasonable imputation quality (imputed R2 > 0.3).
We used the Sequenom MassARRAY iPLEX platform for genotyping in Phase 2-4, using previously described methods and quality control measures [46]. ‘Tag SNP Picker’ option at the HapMap web site was used to pick 45 SNPs which captured 187 TTC39B variants (MAF > 5%, in r2 > 0.5 with tag SNPs) from HapMap CEU set. 40 SNPs among this tag-SNP list were successfully genotyped and passed QC.
Progression-free survival and overall survival
PFS was defined as the time interval between the date of histologic diagnosis and the first confirmed sign of disease recurrence, or progression (Supplementary Table 3). As a related survival trait, OS was defined as the time interval from date of diagnosis to time of last follow-up or time to death from any cause. To control for ascertainment bias, prevalent cases (with an interval > 12 months between the date of histological diagnosis and DNA collection) were excluded from analysis. There were a small number of cases who died without any reported evidence of progression (N = 17), and for them we applied right censoring to PFS at the time of last assessment.
Statistical analysis
The allelic association with PFS or OS was assessed in a Cox Proportional Hazards (CPH) model, adjusting for potential site differences and prognostic factors of grade (low vs high), stage (4 levels), residual disease (nil vs any) and age of diagnosis (specific to the analysis of OS). We tested the proportional hazards assumption for the adjusted variables and stratified by those that violated the assumption. In most analyses, we found that study site was the major variable violating the assumption; therefore, we fitted sites as strata in the model. By fitting strata, we assumed that there were baseline level differences in PFS between the patients from different sites but no difference in the hazard ratio conferred by the SNP being tested.
The data from Phase 1 MAYO and TCGA sets were combined for analysis while stratifying for site differences, because, unlike those from AOCS, they were not selected for extreme phenotypes. To increase the statistical power in Phase 1, we performed a meta-analysis of the results from the AOCS and the combined MAYO and TCGA set using an inverse-variance weighting approach. When performing pooled analyses in Phase 2-4, we pooled data from all studies while stratifying by study sites.
Cell lines
Human ovarian carcinoma cell lines A2780, JAM, OVCAR3 and FUOV1 were grown either in RPMI medium or DMEM/F12 with 10-20% FCS and antibiotics. Cell lines were maintained under standard conditions, routinely tested for Mycoplasma and identity profiled with short tandem repeat markers.
Electrophoretic mobility shift assays (EMSAs)
EMSAs were carried out as previously described [47], except that oligonucleotides were detected using a Chemiluminescent Nucleic Acid Detection Module kit (Cat no 89880, Thermo Scientific). Oligonucleotide sequences used in the assays are listed in Supplementary Table 6. Competitor oligonucleotides were used at 30-fold molar excess.
In silico prediction of transcription factor binding sites
Prediction of transcription factor binding sites was performed using the AliBaba 2.1 program (http://www.generegulation.com/pub/programs/alibaba2/index.html) [24].
Chromatin immunoprecipitation (ChIP) qPCR
Sp1 ChIP-qPCR (Sp1; D4C3 rabbit monoclonal, Cell Signalling) assays were conducted as described previously [48] with a sheared fragment size of 300 bp to 1 kb. For qPCR, 1 µl from 30 µl of DNA extract was used. Primers are listed in Supplementary Table 6.
Chromatin conformation capture (3C)
3C libraries were generated using HindIII, EcoRI or BglII as described previously [49]. 3C interactions were quantitated by real-time PCR (qPCR) using primers designed within restriction fragments (Supplementary Table 6). All qPCRs were performed on a RotorGene 6000 using MyTaq HS DNA polymerase with the addition of 5 mM of Syto9, annealing temperature of 66oC and extension of 30sec. 3C analyses were performed in three independent experiments with each experiment quantified in duplicate. BAC clones (RP11-746M21, RP11-940C5, RP11-356J15, RP11-728G24) covering the 9p22 region were used to create artificial libraries of ligation products in order to normalize for PCR efficiency. Data were normalized to the signal from the BAC clone library and, between cell lines, by reference to a region within GAPDH. All qPCR products were electrophoresed on 2% agarose gels, gel purified and sequenced to verify the 3C product.
Plasmid construction and luciferase assays
The TTC39B, PSIP1 and CCDC171 promoter-driven luciferase reporter constructs were generated by inserting PCR-generated promoter fragments into the multiple cloning site (MCS) of pGL3-Basic. A 2.2 kb fragment containing the PRE was inserted into the BamHI and SalI sites downstream of luciferase. The minor alleles of rs72700653 and rs7874043 were introduced into promoter and PRE constructs by overlap extension PCR. All constructs were sequenced to confirm variant incorporation (AGRF, Australia). Primers used to generate all constructs are listed in Supplementary Table 6. A2780 and JAM ovarian cancer cells were transfected with equimolar amounts of luciferase reporter plasmids and 50ng of pRLTK using Lipofectamine 2000. The total amount of transfected DNA was kept constant per experiment by adding carrier plasmid (pUC19). Luciferase activity was measured 24 hours post-transfection using the Dual-Glo Luciferase Assay System on a Beckman-Coulter DTX-880 plate reader. To correct for any differences in transfection efficiency or cell lysate preparation, Firefly luciferase activity was normalized to Renilla luciferase. The activity of each test construct was calculated relative to promoter constructs, the activity of which was arbitrarily defined as 1.
siRNA silencing
Sp1 (L-026959-00) and non-targeting (D-001810-10-20) ON-TARGETplus SMARTpool, PSIP1 (J-015209-05-0005 and J-015209-06-0005) and non-targeting (D-001810-01-05) siRNAs for were purchased from Thermo Scientific. For siRNA silencing, JAM cells were transfected with 25 nM of either Sp1 or non-targeting siRNAs using Lipofectamine 2000 and lysates prepared for 3C after 48 hours.
Taqman expression assays
JAM total RNA was extracted using Trizol (Life Technologies). Residual DNA contaminants were removed by DNAse treatment (Ambion) and complementary DNA was synthesized using random primers as per manufacturers’ instructions (Life Technologies). All qPCRs were performed on a RotorGene 6000 (Corbett Research) with TaqMan Gene Expression assays (Hs01045711_g1 for Sp1, Hs00916521_m1 for PSIP1 and Hs00411735_m1 for CCDC171) and TaqMan Universal PCR master mix. All reactions were normalized against β-glucuronidase (Catalogue No. 4326320E).
RAD51 foci formation
OVCAR3 cells were reverse transfected with 10nM of siRNAs targeting PSIP1 (siPSIP1) described above for 24 h and later seeded on the 0.1% poly-l-lysine coated coverslips followed by second reverse transfection for additional 24 h. To determine RAD51 foci accumulation, cells were irradiated (IR) with 6 Gy (137Cesium) and analyzed 6 h after irradiation as described previously [50].
Colony formation assays
48 h after siRNA transfection, 10,000-20,000 cells were seeded in 24 well plates and incubated for additional 7 days to determine colony viability. The colonies were fixed with 0.05% crystal violet for 30 minutes, washed and quantified for crystal violet intensity after destaining using Sorenson’s buffer (0.1 M sodium citrate in 50% Ethanol, pH 4.2) at 590 nM absorbance using PowerWave HT Microplate Spectrophotometer (BioTeK, USA).
ACKNOWLEDGMENTS
Manuscript writing group: JDF, SEJ, YL, GCT, AdeF, SLE, SM. Locus SNP selection: YL, SM. Phenotypic data cleaning: BG and SJ. Genotyping, calling and QC: YL, XC and JB. Imputation, Statistical analyses and programming: YL. Functional analysis and bioinformatics: JDF, JB, BG, MJH, AJR, MK, KMH, SK, HS, JSL, SK, CW, DK. All other authors provided participant samples and phenotype information, and read and approved the manuscript. The authors also acknowledge the cooperation of all participating institutions and the contributions of the women who participated in this study. The full AOCS Study Group is available at http://www.aocstudy.org/. The results published here are in part based upon data generated by The Cancer Genome Atlas Pilot Project established by the National Cancer Institute and National Human Genome Research Institute. Information about TCGA can be found at http://cancergenome.nih.gov/. Children’s Cancer Institute Australia is affiliated with University of New South Wales and Sydney Children’s Hospital (Randwick).
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
GRANT SUPPORT
This project has been supported by a grant from Cancer Australia. The Mayo Clinic GWAS was supported by R01CA114343 (Haplotype-based genome screen for ovarian cancer loci). The Ovarian Cancer Association Consortium is supported by a grant from the Ovarian Cancer Research Fund thanks to donations by the family and friends of Kathryn Sladek Smith. The AOCS was supported by the U.S. Army Medical Research and Materiel Command under DAMD17-01-1-0729, the National Health and Medical Research Council (NHMRC) of Australia (grants 400281, 400413), Cancer Council Victoria, Cancer Council Queensland, Cancer Council New South Wales, Cancer Council South Australia, The Cancer Foundation of Western Australia, and Cancer Council Tasmania. G. Chenevix-Trench is a Senior Principal Research fellow of the NHMRC. Y. Lu is funded by NHMRC grant 496675, S. MacGregor is supported by an NHMRC career development award, S. Edwards and J. French are supported by Fellowships from the National Breast Cancer Foundation (NBCF) Australia. The QIMR Berghofer groups were supported by NHMRC project grants (1051698 to SM and 1058415 to SLE and JDF) and a Weekend to End Women’s Cancer Research Grant (to SLE). A deFazio is funded by the University of Sydney Cancer Research Fund and A deFazio and PR Harnett are funded by the Cancer Institute NSW through the Sydney-West Translational Cancer Research Centre. B. Gao is supported by NHMRC and Cancer Institute NSW scholarship. KBM and MO’R are funded by CR-UK. The Bavarian study (BAV) was supported by ELAN Funds of the University of Erlangen-Nuremberg. HSK would like to thank Ira Schwaab for her tireless work on sample preparation. The Belgian study (BEL) was funded by Nationaal Kankerplan and we would like to thank Gilian Peuteman, Thomas Van Brussel and Dominiek Smeets for technical assistance. The Japanese study (JPN) was funded by a Grant-in-Aid for the Third Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare. The International Collaborative Ovarian Neoplasm study (ICON)7 trial team would like to thank the Medical Research Council (MRC) Clinical Trial Unit (CTU) at the University of London (UCL), the ICON7 Translational Research Sub-group, and the University of Leeds for their work on the coordination of samples and data from the ICON7 trial. The LAX study (Women’s Cancer Program) was supported by the American Cancer Society Early Detection Professorship (120950-SIOP-06-258-06-COUN) and Entertainment Industry Foundation. Funding for MALOVA (MAL) was provided by research grant RO1 CA 61107 from the National Cancer Institute, Bethesda, MD; research grant 94 222 52 from the Danish Cancer Society, Copenhagen, Denmark; and the Mermaid I project. The Mayo Clinic study (MAYO) was supported by R01 CA122443, P50 CA136393. The Oregon study (ORE) was funded by the Sherie Hildreth Ovarian Cancer Research Fund and the OHSU Foundation. We would like to thank all members of Scottish Gynaecological Clinical Trials group and the SCOTROC1 investigators. SCOTROC1 (SRO) was funded by Cancer Research UK, and the SCOTROC biological studies were supported by Cancer Research UK (grant C536/A6689). RSH receives support from NIH/NIGMS grant K08GM089941, NIH/NCI grant R21 CA139278, NIH/NIGMS grant UO1GM61393, University of Chicago Cancer Center Support Grant (#P30 CA14599) and Breast Cancer SPORE Career Development Award.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61: 69-90.
2. Markman M. Pharmaceutical management of ovarian cancer : current status. Drugs. 2008; 68: 771-89.
3. Marchetti C, Pisano C, Facchini G, Bruni GS, Magazzino FP, Losito S, Pignata S. First-line treatment of advanced ovarian cancer: current research and perspectives. Expert Rev Anticancer Ther. 2010; 10: 47-60.
4. Johnatty SE, Beesley J, Paul J, Fereday S, Spurdle AB, Webb PM, Byth K, Marsh S, McLeod H, Group AS, Harnett PR, Brown R, DeFazio A, et al. ABCB1 (MDR 1) polymorphisms and progression-free survival among women with ovarian cancer following paclitaxel/carboplatin chemotherapy. Clin Cancer Res. 2008; 14: 5594-601.
5. Tian C, Ambrosone CB, Darcy KM, Krivak TC, Armstrong DK, Bookman MA, Davis W, Zhao H, Moysich K, Gallion H, DeLoia JA. Common variants in ABCB1, ABCC2 and ABCG2 genes and clinical outcomes among women with advanced stage ovarian cancer treated with platinum and taxane-based chemotherapy: a Gynecologic Oncology Group study. Gynecol Oncol. 2012; 124: 575-81.
6. Johnatty SE, Beesley J, Gao B, Chen X, Lu Y, Law MH, Henderson MJ, Russell AJ, Hedditch EL, Emmanuel C, Fereday S, Webb PM, Australian Ovarian Cancer Study G, et al. ABCB1 (MDR1) polymorphisms and ovarian cancer progression and survival: a comprehensive analysis from the Ovarian Cancer Association Consortium and The Cancer Genome Atlas. Gynecol Oncol. 2013; 131: 8-14.
7. Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012; 90: 7-24.
8. Stadler ZK, Thom P, Robson ME, Weitzel JN, Kauff ND, Hurley KE, Devlin V, Gold B, Klein RJ, Offit K. Genome-wide association studies of cancer. J Clin Oncol. 2010; 28: 4255-67.
9. Azzato EM, Pharoah PD, Harrington P, Easton DF, Greenberg D, Caporaso NE, Chanock SJ, Hoover RN, Thomas G, Hunter DJ, Kraft P. A genome-wide association study of prognosis in breast cancer. Cancer Epidemiol Biomarkers Prev. 2010; 19: 1140-3.
10. Kiyotani K, Mushiroda T, Tsunoda T, Morizono T, Hosono N, Kubo M, Tanigawara Y, Imamura CK, Flockhart DA, Aki F, Hirata K, Takatsuka Y, Okazaki M, et al. A genome-wide association study identifies locus at 10q22 associated with clinical outcomes of adjuvant tamoxifen therapy for breast cancer patients in Japanese. Hum Mol Genet. 2012; 21: 1665-72.
11. Rafiq S, Tapper W, Collins A, Khan S, Politopoulos I, Gerty S, Blomqvist C, Couch FJ, Nevanlinna H, Liu J, Eccles D. Identification of inherited genetic variations influencing prognosis in early-onset breast cancer. Cancer Res. 2013; 73: 1883-91.
12. Shu XO, Long J, Lu W, Li C, Chen WY, Delahanty R, Cheng J, Cai H, Zheng Y, Shi J, Gu K, Wang WJ, Kraft P, et al. Novel genetic markers of breast cancer survival identified by a genome-wide association study. Cancer Res. 2012; 72: 1182-9.
13. Tan XL, Moyer AM, Fridley BL, Schaid DJ, Niu N, Batzler AJ, Jenkins GD, Abo RP, Li L, Cunningham JM, Sun Z, Yang P, Wang L. Genetic variation predicting cisplatin cytotoxicity associated with overall survival in lung cancer patients receiving platinum-based chemotherapy. Clin Cancer Res. 2011; 17: 5801-11.
14. Wu C, Xu B, Yuan P, Miao X, Liu Y, Guan Y, Yu D, Xu J, Zhang T, Shen H, Wu T, Lin D. Genome-wide interrogation identifies YAP1 variants associated with survival of small-cell lung cancer patients. Cancer Res. 2010; 70: 9721-9.
15. Wu X, Ye Y, Rosell R, Amos CI, Stewart DJ, Hildebrandt MA, Roth JA, Minna JD, Gu J, Lin J, Buch SC, Nukui T, Ramirez Serrano JL, et al. Genome-wide association study of survival in non-small cell lung cancer patients receiving platinum-based chemotherapy. J Natl Cancer Inst. 2011; 103: 817-25.
16. Yang JJ, Cheng C, Yang W, Pei D, Cao X, Fan Y, Pounds SB, Neale G, Trevino LR, French D, Campana D, Downing JR, Evans WE, et al. Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA. 2009; 301: 393-403.
17. Daly AK. Genome-wide association studies in pharmacogenomics. Nat Rev Genet. 2010; 11: 241-6.
18. Evans WE, McLeod HL. Pharmacogenomics—drug disposition, drug targets, and side effects. N Engl J Med. 2003; 348: 538-49.
19. Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med. 2011; 364: 1144-53.
20. Watters JW, Kraja A, Meucci MA, Province MA, McLeod HL. Genome-wide discovery of loci influencing chemotherapy cytotoxicity. Proc Natl Acad Sci U S A. 2004; 101: 11809-14.
21. Dolan ME, Newbold KG, Nagasubramanian R, Wu X, Ratain MJ, Cook EH, Jr., Badner JA. Heritability and linkage analysis of sensitivity to cisplatin-induced cytotoxicity. Cancer Res. 2004; 64: 4353-6.
22. Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer. 2012; 19: 197-208.
23. Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010; 34: 816-34.
24. Grabe N. AliBaba2: context specific identification of transcription factor binding sites. In Silico Biol. 2002; 2: S1-15.
25. Deshane J, Kim J, Bolisetty S, Hock TD, Hill-Kapturczak N, Agarwal A. Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J Biol Chem. 2010; 285: 16476-86.
26. Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ, Helleday T, Legube G, La Thangue NB, Porter AC, Humphrey TC. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014; 7: 2006-18.
27. Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013; 4: 2126.
28. Osterberg L, Levan K, Partheen K, Delle U, Olsson B, Sundfeldt K, Horvath G. Potential predictive markers of chemotherapy resistance in stage III ovarian serous carcinomas. BMC Cancer. 2009; 9: 368.
29. De Rijck J, Bartholomeeusen K, Ceulemans H, Debyser Z, Gijsbers R. High-resolution profiling of the LEDGF/p75 chromatin interaction in the ENCODE region. Nucleic Acids Res. 2010; 38: 6135-47.
30. Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012; 8: e1002717.
31. Daugaard M, Kirkegaard-Sorensen T, Ostenfeld MS, Aaboe M, Hoyer-Hansen M, Orntoft TF, Rohde M, Jaattela M. Lens epithelium-derived growth factor is an Hsp70-2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007; 67: 2559-67.
32. Matsui H, Lin LR, Singh DP, Shinohara T, Reddy VN. Lens epithelium-derived growth factor: increased survival and decreased DNA breakage of human RPE cells induced by oxidative stress. Invest Ophthalmol Vis Sci. 2001; 42: 2935-41.
33. Huang RS, Johnatty SE, Gamazon ER, Im HK, Ziliak D, Duan S, Zhang W, Kistner EO, Chen P, Beesley J, Mi S, O’Donnell PH, Fraiman YS, et al. Platinum sensitivity-related germline polymorphism discovered via a cell-based approach and analysis of its association with outcome in ovarian cancer patients. Clin Cancer Res. 2011; 17: 5490-500.
34. Singh DP, Fatma N, Kimura A, Chylack LT, Jr., Shinohara T. LEDGF binds to heat shock and stress-related element to activate the expression of stress-related genes. Biochem Biophys Res Commun. 2001; 283: 943-55.
35. Daugaard M, Baude A, Fugger K, Povlsen LK, Beck H, Sorensen CS, Petersen NH, Sorensen PH, Lukas C, Bartek J, Lukas J, Rohde M, Jaattela M. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat Struct Mol Biol. 2012; 19: 803-10.
36. Cavalluzzo C, Christ F, Voet A, Sharma A, Singh BK, Zhang KY, Lescrinier E, De Maeyer M, Debyser Z, Van der Eycken E. Identification of small peptides inhibiting the integrase-LEDGF/p75 interaction through targeting the cellular co-factor. J Pept Sci. 2013; 19: 651-8.
37. Vets S, Kimpel J, Volk A, De Rijck J, Schrijvers R, Verbinnen B, Maes W, Von Laer D, Debyser Z, Gijsbers R. Lens epithelium-derived growth factor/p75 qualifies as a target for HIV gene therapy in the NSG mouse model. Mol Ther. 2012; 20: 908-17.
38. Satoh J, Kawana N, Yamamoto Y. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio. 2013; 7: 139-52.
39. Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010; 466: 707-13.
40. Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, Voight BF, Bonnycastle LL, Jackson AU, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009; 41: 56-65.
41. Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010; 123: 725-31.
42. Bhargavan B, Chhunchha B, Fatma N, Kubo E, Kumar A, Singh DP. Epigenetic repression of LEDGF during UVB exposure by recruitment of SUV39H1 and HDAC1 to the Sp1-responsive elements within LEDGF promoter CpG island. Epigenetics. 2013; 8: 268-80.
43. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81: 559-75.
44. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006; 38: 904-9.
45. McEvoy BP, Montgomery GW, McRae AF, Ripatti S, Perola M, Spector TD, Cherkas L, Ahmadi KR, Boomsma D, Willemsen G, Hottenga JJ, Pedersen NL, Magnusson PK, et al. Geographical structure and differential natural selection among North European populations. Genome Res. 2009; 19: 804-14.
46. Lu Y, Chen X, Beesley J, Johnatty SE, Defazio A, Australian Ovarian Cancer Study Study G, Lambrechts S, Lambrechts D, Despierre E, Vergotes I, Chang-Claude J, Hein R, Nickels S, et al. Genome-wide association study for ovarian cancer susceptibility using pooled DNA. Twin Res Hum Genet. 2012; 15: 615-23.
47. Meyer KB, Maia AT, O’Reilly M, Teschendorff AE, Chin SF, Caldas C, Ponder BA. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008; 6: e108.
48. Lee JS, Kim Y, Kim IS, Kim B, Choi HJ, Lee JM, Shin HJ, Kim JH, Kim JY, Seo SB, Lee H, Binda O, Gozani O, et al. Negative regulation of hypoxic responses via induced Reptin methylation. Mol Cell. 2010; 39: 71-85.
49. Ghoussaini M, et al. Evidence that breast cancer risk at the 2q35 locus is mediated through IGFBP5 regulation. Nat Commun. 2014; 4:4999.
50. Richard DJ, Bolderson E, Cubeddu L, Wadsworth RI, Savage K, Sharma GG, Nicolette ML, Tsvetanov S, McIlwraith MJ, Pandita RK, Takeda S, Hay RT, Gautier J, et al. Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature. 2008; 453: 677-81.