
INTRODUCTION
Acute myeloid leukemia (AML) is characterized by the accumulation of somatically acquired genetic changes in hematopoietic progenitor cells, including gene mutations, copy number alterations and chromosomal translocation [1, 2]. Epigenetic aberrations, including methylation and genomic miRNA deregulation, also play an important role in AML classification and risk stratification. Traditionally, chromosome karyotype has been accepted to be the most important prognostic parameter in AML. However, nearly half of AML cases have a normal karyotype at diagnosis and are categorized as the intermediate-risk (IR) although they have significant clinical heterogeneity [3]. It is believed, therefore, that genetic mutational analysis may help these patients. Advances in identification of prognostic genetic alterations have facilitated greater detailed risk stratification [4, 5]. Previous studies have focused on several selected genes, for example, FLT3-ITD, NPM1 and CEBPA mutations have been incorporated in the European LeukemiaNet (ELN) risk classification system [6].A recent study by Hou and colleagues [7] reported that IR-AML patients could be re-stratified into three distinct prognostic groups according to the mutation status of FLT3-ITD, NPM1, CEBPA, IDH2, WT1, ASXL1, RUNX1 and DNMT3A.
Because tumor cells harbor hundreds of mutated genes and multiple mutations that often occur concomitantly, mutational screening for a panel of genes is biologically meaningful. Traditional sequencing platforms require large amounts of DNA to assess only one gene at a time, and it is labor-intensive and time-consuming, as well as logistically difficult for data integration in real time. Recently, next generation sequencing (NGS) has been shown to have great advantage and potential [8, 9] because of its massively parallel sequencing ability and high throughput multiplexing capacity, making simultaneously parallel and targeted sequencing of all genes of interest feasible, suggesting its value in routine clinical practice [10, 11]. One study recently conducted by the Cancer Genome Atlas Research Network analyzed the genomes of 200 adult AML patients by NGS, and mutations were classified into 9 categories. Nearly all AML samples had at least one mutation of these categories, and a complex interplay of genetic alterations was identified, partly explaining the genetic aberrations in AML. Thus, this approach will significantly impact future disease classification systems [12], but currently no such research is specifically directed toward IR-AML.
In this study, we applied a NGS platform to screen for mutations in 410 genes relevant to hematological malignancy and showed the value of this approach. Furthermore, we performed single and comprehensive mutational analyses associated with clinical features and propose new risk stratification model for IR-AML and its distinct molecular subgroups that will benefit from personalized therapy.
RESULTS
Clinical characteristics of the patients and panel genes
Clinical characteristics of the 95 patients are summarized in Table 1. Among these patients, fifty-five were males and forty were females. The median age was 45 years, ranging from 12 to 88 years and there were ninety-two adults and three children (≤15years). The median follow-up was 22.3 months (interquartile range, 7.5–38 m). Among our 95 IR-AML patients, excluding 10 patients who did not receive any chemotherapy or were only treated with low-dose cytosine arabinoside because of old age and/or poor performance status, sixty-five patients received conventional induction chemotherapy with one of the anthracyclines (idarubicin or doxorubicin) or mitoxantrone for 3 days and cytarabine for 7 days, another twenty patients received DCAG (decitabine 10 mg/m2 d1-5, aclarubicin 20mg d1,3,5, cytarabine 10mg/m2 q12h d1-5, G-CSF 300μg/day) regimen. After achieving complete remission, twenty-five patients received consolidation chemotherapy with conventional dose of cytarabine and anthracycline or mitoxantrone or with middle-/high-dose cytarabine and forty patients received hematopoietic stem cell transplantation for consolidation chemotherapy. Among them, 75 patients receiving more than 4 cycles of chemotherapy were used for survival analysis.
Table 1: Clinical and pathologic characteristics of 95 IR-AML patients
Characteristic |
Median(range) or Number |
|---|---|
Age at study entry (years) |
45 (12–88) |
Sex, no. (%) |
|
Male |
55/95 |
Female |
40/95 |
White-cell count at diagnosis |
|
WBC-G/L |
21.33 (0.87–405.13) |
Bone marrow blast at diagnosis, % |
64.4 (20–94.4) |
AML FAB subtype-no. (%) |
|
AML with minimal maturation: M0 |
0 |
AML without maturation: M1 |
4/95 |
AML with maturation: M2 |
23/95 |
Acute myelomonocytic leukemia: M4 |
28/95 |
Acute monoblastic or monocytic leukemia: M5 |
31/95 |
Acute erythroid leukemia: M6 |
5/95 |
Unclassified |
4/95 |
Immunophenotype |
|
CD13 |
73/95 |
CD33 |
86/95 |
CD34 |
68/95 |
CD117 |
77/95 |
MPO |
77/95 |
Cytogenetics |
|
Abnormal karyotype |
20/95 |
Normal karyotype |
75/95 |
Induction therapy |
|
IA/DA/MA |
65/95 |
DCAG |
20/95 |
Others |
10/95 |
Response evaluation |
|
Achieving CR |
65/95 |
Non-remission |
22/95 |
Unevaluated |
8/95 |
Consolidation therapy after CR |
|
Chemotherapy |
25/65 |
HSCT |
40/65 |
Abbreviation: WBC, white blood cell count. FAB, French American British. CR, complete remission. IA, idarubicin+ cytarabine. DA, daunorubicin+ cytarabine. MA, mitoxantrone+cytarabine. DCAG, decitabine+aclarubicin+cytarabine+ G-CSF. HSCT, hematopoietic stem cell transplantation.
The sequencing panel targets ~1.3 Mb of genomic content, consisting of the entire coding sequence of 410 genes relevant to the pathogenesis of hematologic malignancies. For this panel, 233 genes were originated from the COSMIC cancer database which contained somatic mutations in human blood cancer, and 23 genes originated from the Leukemia Gene Database (LeGenD). In addition, 154 genes were collected based on recent scientific and clinical literature (Supplementary Table S1).
Sensitivity of mutation detection
To create a 0–100% concentration gradient of tumor mutations in the Kasumi-1 cell line, Kasumi-1 cells were diluted with various ratios of K562 cells. Sequencing results from two different depths indicated at least 90% of the homozygous mutations in the cell line were correctly detected in a 10% solution of K562 cells and that 80% of the heterozygous mutations were identified in the 20% K562 cell solution. In contrast, false-positives were detected at 0 and 100% K562 cell solutions. Using the cell line alone, false-positives were less than 1% (Figure 1). In all IR-AML samples, tumor cells comprised more than 20% of the sample. Thus, detecting mutations is efficient when covering sequences above 200x. We counted allelic frequencies of unique SNPs in Kasumi-1 cells at each dilution and there was a concordance with each dilution which reflected the tumor concentration (Supplementary Figure S1).
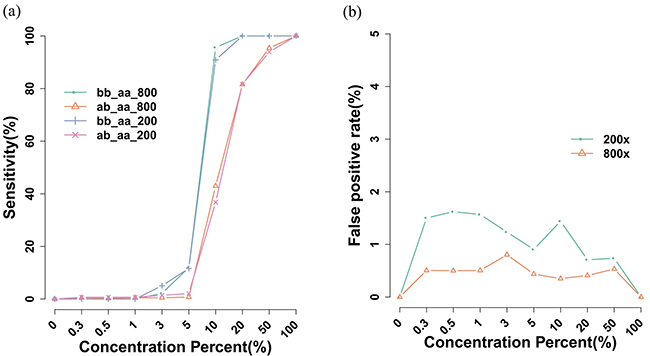
Figure 1: Sensitivity and false-positive mutation detection rates. a. In Kasumi-1 and K562 cell lines, mutation sensitivity can be calculated and more than 80% of unique mutations within Kasumi-1 can be detected. b. The false-positive rate revealed that more than 98.5% mutations were consistent with Kasumi-1 cell line data.
Frequencies of genetic alterations
In total, 8,766 single-nucleotide variants (SNVs) and 839 small insertion and deletions were detected in 95 IR-AML patients for the target region in the absence of matched control samples. We focused on the mutations that change with respect to exon amino acid coding regions, consensus splice-site regions, and RNA genes. A series of steps were used to remove germline and harmless mutations (Supplementary Methods). After filtering, an average of 3.6 mutations per sample were remained and 101 of 410 genes analyzed were identified (Supplementary Table S2). Each mutation position and gene type of some genes are shown in Supplementary Figure S2. Due to the limited amount of normal control samples, we did not confirm that all identified mutations were somatic mutations, so some may be rare SNPs. CEBPA and NPM1 were the two most commonly identified mutations (28.4% and 21.1% respectively), followed by DNMT3A (16.8%), FLT3-ITD (14.7%), NRAS (12.6%), IDH2 (11.6%) and WT1 (10.5%) mutations. Mutations of some genes like DNMT3A, IDH2, NRAS, TET2 and WT1 were shown in Supplementary Table S3 and these results were all validated by Sanger sequencing.
Conceptually, genetic alterations identified in AML have been grouped into class I mutations that promote growth and survival by activating intracellular signals, and class II mutations that block differentiation, impair subsequent apoptosis, and/or confer an advantage in self-renewal by altered transcription factors [13]. Presently, other family gene mutations have been classified according to their provisional gene function, such as epigenetic modifiers, tumor suppressors, the cohesin complex, and spliceosome genes [9, 12, 14]. In this study, class II mutations (NPM1, CEBPA, RUNX1, GATA2, ETV6, etc) were the mostly frequently identified (48/95, 50.53%), followed by class I mutations (FLT3, KIT, NRAS, KRAS, PTPN11, etc; 36/95, 37.89%), epigenetic modification mutations (DNMT3A, TET2, IDH1/2, ASXL1, EZH2, DOT1L, etc; 32/95, 33.68%), and tumor suppressor mutations (WT1, PHF6 and TP53) (14/95, 14.74%). Also, spliceosome genes (U2AF1, SRSF2 and SF3B1/2), cohesion complex genes (STAG2, RAD21, SMC1A and SMC3) and NOTCH family mutations were identified in 12 (12.63%), 10 (10.53%) and 3 (3.16%) patients, respectively (Figure 2).
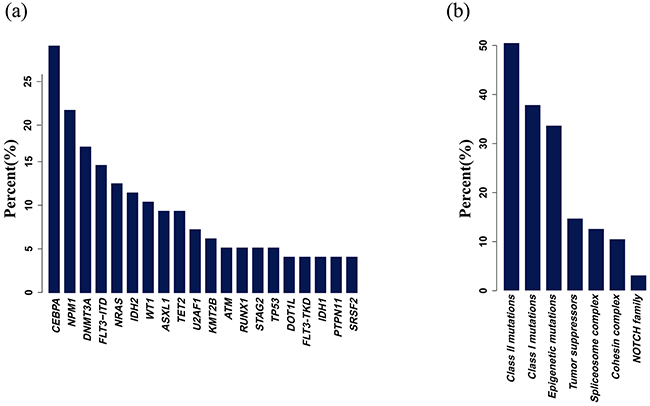
Figure 2: Mutation frequencies. a. Mutation frequencies that occurred in more than four samples. Mutations were identified in 101 of 410 genes analyzed. Only seven genes (CEBPA, NPM1, DNMT3A, FLT3-ITD, NRAS, IDH2 and WT1) were mutated in more than 10% of patients. b. Mutation frequencies according to functional classification. Mutations in class I and II and epigenetic modifiers were frequently identified.
Interaction of genetic alterations
Mutation profiles and several patterns of overlapping mutations were identified in our study. (Figure 3, Supplementary Figure S3). Significantly overlapped mutations were observed among FLT3-ITD, NPM1 and DNMT3A mutations; NRAS and WT1 mutations. In contrast, mutually exclusive mutations were observed between TET2 and IDH1/2 mutations; WT1 and TET2 mutations; NPM1 and CEBPA mutations; and between STAG2 and SMC3 mutations. Overall, we observed relationships of strong co-occurrences between class I and II and epigenetic modifying gene mutations, which also often coexisted with other family gene mutations such as the cohesion complex, NOTCH family and spliceosome mutations [8]. In contrast, mutations of the same gene classification were always mutually exclusive.

Figure 3: Mutation profile according to clinical features. Mutation profile according to clinical features (FAB category, clinical efficacy and karyotype). Significant mutations identified in AML patients are shown. Some mutations co-occurred or were exclusive and some were associated with specific clinical features. Yellow boxes indicate single mutations and pink boxes indicate double mutations.
Clinical features of single mutational analysis
Several associations between mutations and clinical features were observed. DNMT3A mutations occurred more frequently in older patients (>50 years-of-age, p<0.05) and NPM1 mutations were more frequently identified in patients older than 40 years-of-age (p<0.05). In addition, several mutations were identified to be associated with peripheral white leukocyte and bone marrow blast counts at diagnosis. FLT3-ITD was associated with high peripheral leukocyte counts and high numbers of bone marrow blast cells (p<0.05). In contrast, IDH2 mutations were associated with a lower leukocyte count (p<0.05) and some mutations were found to be associated with specific immunophenotype. Compared with the non-mutation group, NPM1 mutations were associated with lower CD34- and HLA-DR-positive rates (p<0.05) but with higher CD33-positive rates (p<0.05). DNMT3A mutations were associated with fewer CD34-, CD33- and CD117-positive rates (p<0.05). CEBPA mutations were associated with greater CD34- and CD7-positive rates (p<0.05) and IDH2 mutations were associated with fewer HLA-DR-positive rate (p<0.05) (Supplementary Table S4).
We also analyzed the association between mutations and CR. Using Pearson’s χ2 test, CEBPA mutations were identified as a favorable factor for achieving CR, whereas ASXL1, FLT3-ITD and DNMT3A mutations were unfavorable factors (Table 2). Multivariable logistic regression analysis showed that only FLT3-ITD and ASXL1 mutations were identified as unfavorable factors for achieving CR. We then analyzed the prognostic value of single-gene mutations. Using univariate analysis, FLT3-ITD, ASXL1 and DNMT3A mutations were identified as unfavorable prognostic factors for overall survival (OS) and disease-free survival (DFS), and CEBPA mutations were favorable factors for OS and DFS, while some other genes were not identified as prognosis factors (Figure 4, Figure 5, Table 3, Supplementary Figure S4). Multivariate COX regression analysis with stepwise selection showed that FLT3-ITD (HR: 3.271, 95% CI: 1.541-6.944, P=0.002) mutations were independent factors of poor prognosis, and CEBPA (HR: 0.407, 95% CI: 0.168-0.985, P=0.046) mutations were independent favorable factors.
Table 2: Gene mutations affecting CR
Mutations |
CR rate (%) |
P-value |
|
|---|---|---|---|
Pearson’s χ2 test |
Positive |
Negative |
|
ASXL1 |
4/9(44.4%) |
61/78(78.2%) |
0.042 |
DNMT3A |
8/15(53.3%) |
57/72(79.2%) |
0.036 |
FLT3-ITD |
7/14(50.0%) |
58/73(79.5%) |
0.020 |
CEBPA |
25/25(100%) |
40/62(64.5%) |
0.001 |
Mutations |
OR (95% CI) |
P-value |
|
ASXL1 |
0.148(0.034–0.649) |
0.011 |
|
FLT3-ITD |
0.185(0.053–0.644) |
0.008 |
|
Abbreviations: CR, complete remission; OR, odds ratio; CI, confidence interval. By Pearson’s χ2 test, CEBPA mutations were identified as favorable factors for achieving CR, while ASXL1, DNMT3A, FLT3-ITD mutations were as unfavorable factors. Multivariable logistic regression analysis showed that only FLT3-ITD and ASXL1 mutations were identified as unfavorable factors for achieving CR.
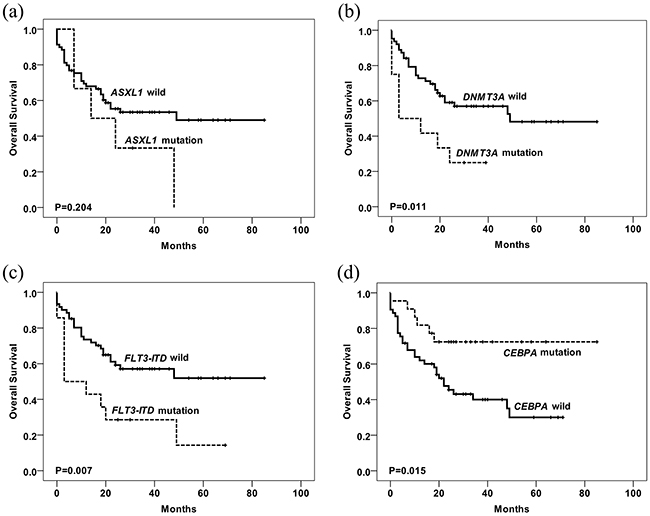
Figure 4: Kaplan-Meier curves of OS according to the mutations are shown. Overall survival stratified by mutational status. P value was estimated by the log-rank test. The mutated number of ASXL1 (a), DNMT3A (b), FLT3-ITD (c) and CEBPA (d) for survival analysis was 6, 12, 14 and 22, respectively. Patients with ASXL1, DNMT3A or FLT3-ITD mutations have worse survival than wild type groups, while patients with CEBPA mutations have better OS than those without mutations.
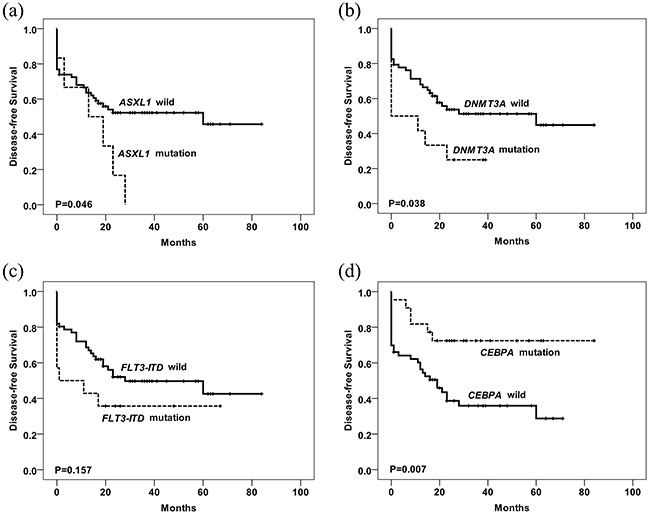
Figure 5: Kaplan-Meier curves of DFS according to the mutations are shown. Disease-free survival stratified by mutational status. P value was estimated by the log-rank test. The mutated number of ASXL1 (a), DNMT3A (b), FLT3-ITD (c) and CEBPA (d) for survival analysis was 6, 12, 14 and 22, respectively. Patients with ASXL1, DNMT3A or FLT3-ITD mutations have worse survival than wild type group, while patients with CEBPA mutations have better DFS than those without mutations.
Table 3: Univariate and Multivariate Analysis for DFS and OS
Univariate analysis |
Multivariate analysis |
|||
|---|---|---|---|---|
P-value |
Log rank χ2 test |
P-value |
HR (95% CI) |
|
OS |
||||
Age>50y |
0.001 |
11.574 |
>0.1 |
|
ASXL1 |
0.204 |
1.617 |
>0.1 |
|
DNMT3A |
0.011 |
6.399 |
>0.1 |
|
FLT3-ITD |
0.007 |
7.396 |
0.002 |
3.271 (1.541–6.944) |
CEBPA |
0.015 |
5.939 |
0.046 |
0.407 (0.168–0.985) |
HSCT |
<0.001 |
27.315 |
<0.001 |
0.151 (0.068–0.337) |
WBC count>30G/L |
0.066 |
3.368 |
>0.1 |
|
DFS |
||||
Age>50y |
0.001 |
11.613 |
>0.1 |
|
ASXL1 |
0.046 |
3.965 |
>0.1 |
|
DNMT3A |
0.038 |
4.293 |
>0.1 |
|
FLT3-ITD |
0.157 |
1.999 |
>0.1 |
|
CEBPA |
0.007 |
7.245 |
0.022 |
0.360 (0.150–0.865) |
HSCT |
<0.001 |
28.360 |
<0.001 |
0.191 (0.090–0.406) |
WBC count>30G/L |
0.404 |
0.697 |
>0.1 |
|
Abbreviations: P of the analysis is the p value of the Log rank test. HR is the value of the hazard ratio. 95% CI is the 95% confident interval of the hazard ratio.WBC, white blood cell count. HSCT, hematopoietic stem cell transplantation.
Clinical features of comprehensive analysis of multiple mutations
In our study, the most notably significant co-occurrence was identified among FLT3-ITD, NPM1 and DNMT3A mutations, which occurred in five patients (Table 4). NPM1mut/FLT3-ITDmut/ DNMT3Amut patients were older (median age, 61 years), mostly women (4/5, 80%) and had a heavy disease burden with greater leukocyte counts (p<0.05, median, 132.39×109/L) than NPM1wt/FLT3-ITDmut/DNMT3Amut, NPM1wt/FLT3-ITDneg/DNMT3Amut or NMP1mut/FLT3-ITDneg/DNMT3Amut groups (Supplementary Table S5). Morphologically, NPM1mut/FLT3-ITDmut/ DNMT3Amut AML was closely associated with myelomonocytic blast morphology, corresponding to M4 and M5 categories in the French-American-British (FAB) classification [15]. Among these five cases, three (60%) were M4 and two (40%) were M5. In terms of immunophenotype, all cases were positive for CD33 and CD13 and negative for surface and cytoplasmic CD3 expression. Myeloperoxidase expression was measured in 3/5 (60%) cases and blasts were positive for CD34 or CD56 in 2/5 (40%) cases. In accordance with myelomonocytic morphology, all cases expressed monocyte-associated CD64 antigen, of which two cases expressed additional monocyte-associated CD14 antigen. Additionally, no case was co-expressed with the B-cell marker CD19. Regarding prognosis, all five patients were non-remission after induction chemotherapy, and all died within six months (average survival time<3 m; range: 0.1-3.5 m).
Table 4: Clinical features of 5 patients with concurrent FLT3-ITD, NPM1 and DNMT3A mutations
Clinical features |
No. of Samples |
||||
|---|---|---|---|---|---|
D-2081 |
D-2959 |
D-2978 |
D-3128 |
D-3009 |
|
Age (y) |
73 |
61 |
43 |
56 |
66 |
Sex |
male |
female |
female |
female |
female |
Diagnosis |
M4 |
M5 |
M4 |
M4 |
M5 |
WBC at diagnosis (×109/L) |
129.21 |
170 |
332.77 |
132.39 |
70 |
BM percentage (%) |
80.40 |
68.40 |
88.40 |
48 |
85 |
Immunophenotype |
|||||
CD33 |
+ |
+ |
+ |
+ |
+ |
CD13 |
+ |
+ |
+ |
+ |
+ |
MPO |
- |
- |
+ |
+ |
+ |
CD3 |
- |
- |
- |
- |
- |
CD34 |
- |
- |
+ |
+ |
- |
CD56 |
+ |
+ |
- |
- |
- |
CD64 |
- |
+ |
+ |
+ |
+ |
CD14 |
- |
+ |
+ |
- |
- |
CD19 |
- |
- |
- |
- |
- |
DNMT3Amut type |
R882H |
R882C |
R882P |
R882H |
A910V |
NPM1 insertion type |
TCTG |
TCTG |
TCTG |
TCTG |
TGCA |
Response evaluation |
NR |
NR |
NR |
NR |
NR |
Survival time |
3.4 m |
3 m |
2 d |
1 w |
3.4 m |
In addition, another five patients had NPM1mut/IDH 1/2mut without FLT3-ITD (See Table 5). In these patients, blasts were positive for CD33 and myeloperoxidase and negative for surface and cytoplasmic CD3 expression. CD34 and CD56 expression occurred in one patient. All five patients had low leukocyte counts (range: 1.35-49.4×109/L). The four patients available for analysis all achieved CR.
Table 5: Clinical features of 5 patients with concurrent NPM1 and IDH1/2 without FLT3-ITD
Clinical features |
No. of Samples |
||||
|---|---|---|---|---|---|
D-2842 |
D-2848 |
D-2857 |
D-2862 |
D-2954 |
|
Age (y) |
53 |
36 |
50 |
47 |
54 |
Sex |
female |
female |
male |
female |
male |
Diagnosis |
M5 |
M2 |
M4 |
M4 |
AML |
Diagnosis time |
2011.8.15 |
2012.10.8 |
2012.7.4 |
2013.1.17 |
2010.9.20 |
WBC at diagnosis(×109/L) |
18.01 |
4.39 |
14.07 |
1.35 |
49.4 |
BM percentage (%) |
84.2 |
64.4 |
53.6 |
92.8 |
84 |
Immunophenotype |
|||||
CD33 |
+ |
+ |
+ |
+ |
+ |
CD13 |
+ |
+ |
+ |
- |
+ |
MPO |
+ |
+ |
+ |
+ |
+ |
CD3 |
- |
- |
- |
- |
- |
CD34 |
+ |
- |
- |
- |
+ |
CD56 |
+ |
- |
+ |
- |
- |
Response evaluation |
CR |
CR |
CR |
CR |
Unevaluated |
For overall prognosis, we evaluated a recently reported risk stratification system based on genetic status proposed by Patel’s group [4]. According to their system, our 95 IR-AML patients were clearly stratified into 3 distinct prognostic subgroups: a favorable group that accounts for 5.3%, an intermediate group, 70.5%, and a new emerging 24.2% unfavorable group. We then analyzed combinations of different mutations with respect to OS and these data agreed with previous reports (Supplementary Figure S5).
DISCUSSION
AML is a genetically heterogeneous disease resulting from complex interactions among different oncogenic pathways in leukemogenesis, so integrated mutational profiling via mutational analysis is highly valuable for thorough evaluation [6, 12]. In addition, one gene often has multiple mutation events and traditional sequencing platforms are challenging. NGS technology provides advantages of parallel sequencing and high throughput multiplexing ability, facilitating routine and simultaneous parallel and targeted sequencing of all genes. In our study, cell dilution data indicated that our current pipeline can detect genomic aberrations when at least 10% tumor content is present, and it is sufficient for any de novo non-M3 AML patients. The tumor allelic frequency can reflect the concentration of tumor correctly, even as dilute as 0.3%.
Using a second detection method, for each cell line dilution, we observed that at 10%, 100% homozygous and 93% heterozygous mutations were correctly detected in 800x coverage data. With more coverage, detection sensitivity can be much lower than 10%. The lowest detection sensitivity we achieved was 1.5% with a 50% detection rate (Supplementary Figure S6). Other methods may offer detection as low as 0.02% but detection rates are not reported [16]. Thus, our approaches might have potential for detecting minimal residual disease.
In this study, we comprehensively analyzed mutations of 410 genes using a NGS platform and identified 9,605 mutations in 95 IR-AML samples. Consistent with previous reports [4, 8, 17, 18]], only seven genes (CEBPA, NPM1, DNMT3A, FLT3-ITD, NRAS, IDH2 and WT1) were mutated in more than 10% of study patients but CEBPA mutations were more frequent [19]. This finding may be due to patient selection as all patients belong to the intermediate risk group and might be related with racial factors. Likewise, the CEBPA mutation rate in CN-AML that reported in Taiwan was also higher, that is, about 35% [20]. Moreover, we analyzed another 26 newly diagnosed AML who own bone marrow and paired control saliva samples to verify the reliability of our method. The result showed that the mutation frequency of genes by two methods (with or without matched control samples) was almost consistent (Supplementary Figure S7), especially for those clinically relevant and important genes with recognized mutation hotspots [21–25], suggesting that the variants of our 95 IR-AML cases represent somatic mutations. In our study, DNMT3A was identified as being frequently overlapped with FLT3-ITD and NPM1 mutations. In contrast, TET2 and IDH1/2 mutations, WT1 and TET2 mutations, NPM1 and CEBPA mutations, and cohesion complex genes (STAG2 and SMC3) were found to be mutually exclusive, and this is consistent with biological evidence that these mutations are functionally involved in a shared mechanism of hematopoietic transformation [26, 27]. Such cooperative and exclusive mutational patterns may indicate a definite role in the pathogenesis of AML, but additional studies are required for further validation.
We observed correlation between mutations and immunophenotype. For example, NPM1mut patients had higher CD33-positive rates and fewer CD34- and HLA-DR-positive rates, whereas CEBPA mutations were associated with high expression of CD34 and CD7. This is the first report to document this observation [28, 29]. Consistent with other studies [30], all four patients with NPM1mut/FLT3-ITDmut/DNMT3Amut were positive for CD33 and CD13, and negative for surface and cytoplasmic CD3 expression. Additionally, another study [31] indicated that patients with NPM1mut /FLT3wt were always CD34- and CD56-; while those with NPM1wt /FLT3-ITDmut were CD34+ and TdT+. However, we did not observe this in our study. Our results showed some specific correlation between mutation and phenotype, but deducing the phenotypic features from specific somatic mutations is challenging, and further studies are needed to verify how different mutation combinations could lead to distinct clinical and phenotypic profile.
The achievement of CR after induction therapy has been accepted as a precondition for long-term survival. If no CR is achieved after induction therapy, the possible mortality of AML can reach 75% within one year [32]. The CR rate varies across different molecular alteration groups. In our study, CEBPA mutations were found to be favorable factors for achieving CR, whereas ASXL1, DNMT3A and FLT3-ITD mutations were unfavorable factors, data consistent with previous reports [19, 33, 34]. Combining all mutation events, Loghavi’s group [7, 30, 35] confirmed that NPM1mut/FLT3mut/DNMT3Amut patients seem to have worse clinical outcomes than those with FLT3mut/DNMT3Amut and those with NPM1mut/DNMT3Amut. In this study, four patients with NPM1mut/FLT3-ITDmut/DNMT3Amut were all non-remission after induction chemotherapy and all died within six months.
The prognostic impact of each mutation and/or co-occurring mutations needs individual and independent clarification. Studies indicate that some mutations have definite prognostic significance and FLT3-ITD, NPM1, CEBPA mutations had been incorporated into the ELN stratification system [2, 36–39]. In this study, FLT3-ITD, ASXL1 and DNMT3A mutations were identified to be associated with adverse OS or DFS, while CEBPA mutations were associated with favorable prognosis. Although we couldn’t ensure their treatment uniform owing to the retrospective study, the treatment between each mutation and wild group for survival analysis was relatively balanced. Due to the limited number of our cases, we didn’t further group these patients according to whether receiving HSCT, but the proportion of transplant patients in two groups analyzed for survival analysis was relatively equivalent (Supplementary Table S6). Moreover, we conducted Multivariate COX regression analysis to adjust the impact of HSCT, FLT3-ITD and CEBPA mutations were identified as independent prognostic factors.
As multiple mutations often coexist in a single patient and more novel mutations were discovered, comprehensive analysis of combined multiple gene mutations is required. However, due to our limited number of cases, we could not scientifically propose a new prognostic model by integrating multiple mutations including those novels. According to the prognostic model recently proposed by Patel’s group [4], the intermediate-risk population was reduced by about one-third in our study, thereby guiding different clinical treatment choices for future prospective study. Ultimately, AML treatment may be personalized based on risk stratification. Studies indicated that allo-HSCT can lead to better clinical outcomes for patients with unfavorable-risk cytogenetics in the first CR [40–42]. For patients with adverse-risk genotypes (other than NPM1mut/FLT3-ITDneg or mutated CEBPA), allo-HSCT in the first CR was also suggested [40, 43–45]. Similarly, in this study, patients with unfavorable molecular genotypes seem to have superior OS when they received allo-HSCT compared to those who did not. For those with intermediate-risk cytogenetics, studies showed that auto-HSCT may represent an alternative therapeutic approach, as some had similar clinical outcomes with allo-HSCT. Still, all of these studies lacked further molecular stratification to the patients [45, 46]. In our study, five patients with intermediate molecular genotypes who received auto-HSCT had similar OS but less transplantation-related morbidity and mortality than those who received allo-HSCT. Prospective studies with more patients are required to verify such observations.
In conclusion, simultaneous screening of multiple genes with high sensitivity and specificity make target sequencing a valuable next-generation sequencer for massive mutation screening of AML samples. Comprehensive genomic analysis based on NGS technology has identified additional novel mutations and several cooperative and exclusive mutation patterns. Elucidation of underlying important relationships among mutated genes and novel pathways provides a theoretical basis for comprehensive understanding of AML biology and molecular pathogenesis. Finally, the relationship between mutations and clinical features such as leukocyte count at diagnosis, immunophenotype and effect assessments can be explored. Integrating data from these multiple sources can increase predictive efficacy for outcomes with traditional treatment to avoid over- or under- treatment. Most importantly, integrated mutational profiling will improve clinical AML management by providing a framework for IR-AML risk stratification and insight into potential therapeutic targets. A proportion of IR-AML defined by cytogenetics alone can be reclassified into favorable or unfavorable risk group according to molecular genotype. More detailed genetic information and large-scale prospective studies are required in the future to evaluate prognosis, predict treatment responses and supervise clinical decisions in AML.
MATERIALS AND METHODS
Patients and methods
From September 2008 and March 2014, 95 newly diagnosed de novo non-M3 AML patients with IR cytogenetics and healthy adults were recruited for mutational analysis. Patients with antecedent hematological diseases or therapy-related AML were excluded. The diagnosis and classification of AML were based on the FAB Cooperative Group Criteria [47]. Then, 32 peripheral blood samples from the healthy adults were collected as target background control templates. Another 26 bone marrow and matched control saliva samples from newly diagnosed AML were collected to test the reliability of our method to identify somatic mutations. Genomic DNA was extracted from patient bone marrow samples using the standard methods [48]. Metaphase chromosomes were banded by trypsin-Giemsa technique and karyotyped according to the International System for Human Cytogenetic Nomenclature. Flow cytometric analysis was performed on a BD Calibur Flow Cytometer or FC500 Flow Cytometer, and data were analyzed by using Cell Quest or CXP software.
Target sequencing and variant analysis
All 410 known or putative genes for leukemia were processed for mutations in all IR-AML, 26 newly diagnosed AML and 32 healthy samples by target sequencing. NimbelGen SeqCap EZ Choice was used according to the manufacturer’s protocol with modification. Multiplexed libraries were sequenced using 100-bp paired-end runs on an Illumina HiSeq 2500.
Reads were aligned, using Burrows-Wheeler alignment (BWA) tool, to human genomic reference sequences (HG19, NCBI built 37) [49]. To identify of SNPs and INDELs, GATK with recommended parameters was performed [50], Pindel (0.2.4) was performed to identify the FLT3 internal tandem duplications (ITD) [51]. All mutations were annotated by ANNOVAR software using some resources (details were in Supplementary Method) [52]. For 26 newly diagnosed AML samples, two methods were used to identify somatic mutations. The method described was applied in the absence of matched saliva samples, and VarScan with the default parameters was used in the present of matched saliva samples. Somatic mutations identified from the two methods were compared. A subset of somatic mutations detected by target sequencing was independently confirmed by Sanger sequencing.
Cell line dilution
To determine the effect of varying tumor content on the ability of target-Seq to detect SNP, cells from an AML1-ETO fusion-positive Kasumi-1 cell line was diluted with cells from an BCR-ABL fusion-positive K562 cell line to decrease Kasumi-1 cell content as follows 100, 50, 20, 10, 5, 3, 1, 0.5, 0.3 and 0% (Supplementary Table S7). Cells were resuspended in aliquots of 1×106/ml. A total of 1μg DNA from each of these dilutions was used for library construction.
Statistical analysis
A Mann-Whitney U-test was used to compare the differences in continuous variables. Analysis of frequencies was performed using Fisher’s exact test or Pearson’s χ2 test for larger tables. The logistic regression model was used to identify risk factors for achieving CR. Survival analysis was performed by the Kaplan-Meier method and a Cox proportional hazards model was used to assess the prognostic significance of the clinical variables. OS was defined as the time from diagnosis to death from any cause or last follow-up. DFS was defined as the time from the day achieving CR to the date of relapse, death, or last follow-up. Patients undergoing HSCT were not censored at the time of transplantation. A two-sided p-value< 0.05 was considered statistically significant. All statistical analyses were performed with the SPSS 19.0.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81370635 and 81470010), National Public Health Grant Research Foundation (No.201202017) and the capital of the public health project (Z111107067311070), Z-park Industrial Technology Alliance 2013 Foundation.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Sanders MA and Valk PJ. The evolving molecular genetic landscape in acute myeloid leukaemia. Current opinion in hematology. 2013; 20:79–85.
2. Schlenk RF and Dohner H. Genomic applications in the clinic: use in treatment paradigm of acute myeloid leukemia. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2013; 2013:324–330.
3. Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ, Burnett AK and National Cancer Research Institute Adult Leukaemia Working G. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010; 116:354–365.
4. Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, Socci ND, Heguy A, Cherry A, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. The New England journal of medicine. 2012; 366:1079–1089.
5. Ofran Y and Rowe JM. Genetic profiling in acute myeloid leukaemia--where are we and what is its role in patient management. British journal of haematology. 2013; 160:303–320.
6. Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management. American journal of hematology. 2013; 88:318–327.
7. Hou HA, Lin CC, Chou WC, Liu CY, Chen CY, Tang JL, Lai YJ, Tseng MH, Huang CF, Chiang YC, Lee FY, Kuo YY, Lee MC, Liu MC, Liu CW, Lin LI, et al. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia. 2014; 28:50–58.
8. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, McMichael JF, Wallis JW, Lu C, Shen D, Harris CC, Dooling DJ, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012; 481:506–510.
9. Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012; 150:264–278.
10. Metzker ML. Sequencing technologies - the next generation. Nature reviews Genetics. 2010; 11:31–46.
11. Cagnetta A, Adamia S, Acharya C, Patrone F, Miglino M, Nencioni A, Gobbi M and Cea M. Role of genotype-based approach in the clinical management of adult acute myeloid leukemia with normal cytogenetics. Leukemia research. 2014; 38:649–659.
12. Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine. 2013; 368:2059–2074.
13. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellstrom-Lindberg E, Tefferi A and Bloomfield CD. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009; 114:937–951.
14. Abdel-Wahab O and Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood. 2013; 121:3563–3572.
15. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR and Sultan C. Proposals for the classification of the myelodysplastic syndromes. British journal of haematology. 1982; 51:189–199.
16. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo BW, Jr., Alizadeh AA and Diehn M. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nature medicine. 2014; 20:548–554.
17. Naoe T and Kiyoi H. Gene mutations of acute myeloid leukemia in the genome era. International journal of hematology. 2013; 97:165–174.
18. Grossmann V, Schnittger S, Kohlmann A, Eder C, Roller A, Dicker F, Schmid C, Wendtner CM, Staib P, Serve H, Kreuzer KA, Kern W, Haferlach T and Haferlach C. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood. 2012; 120:2963–2972.
19. Taskesen E, Bullinger L, Corbacioglu A, Sanders MA, Erpelinck CA, Wouters BJ, van der Poel-van de Luytgaarde SC, Damm F, Krauter J, Ganser A, Schlenk RF, Lowenberg B, Delwel R, Dohner H, Valk PJ and Dohner K. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood. 2011; 117:2469–2475.
20. Lin LI, Chen CY, Lin DT, Tsay W, Tang JL, Yeh YC, Shen HL, Su FH, Yao M, Huang SY and Tien HF. Characterization of CEBPA mutations in acute myeloid leukemia: most patients with CEBPA mutations have biallelic mutations and show a distinct immunophenotype of the leukemic cells. Clinical cancer research. 2005; 11:1372–1379.
21. Janke H, Pastore F, Schumacher D, Herold T, Hopfner KP, Schneider S, Berdel WE, Buchner T, Woermann BJ, Subklewe M, Bohlander SK, Hiddemann W, Spiekermann K and Polzer H. Activating FLT3 mutants show distinct gain-of-function phenotypes in vitro and a characteristic signaling pathway profile associated with prognosis in acute myeloid leukemia. PloS one. 2014; 9:e89560.
22. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. The New England journal of medicine. 2005; 352:254–266.
23. Ahmad F, Mohota R, Sanap S, Mandava S and Das BR. Molecular evaluation of DNMT3A and IDH1/2 gene mutation: frequency, distribution pattern and associations with additional molecular markers in normal karyotype Indian acute myeloid leukemia patients. Asian Pacific journal of cancer prevention. 2014; 15:1247–1253.
24. Chou WC, Huang HH, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, Chang YC, Lee FY, Liu MC, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010; 116:4086–4094.
25. Tyner JW, Erickson H, Deininger MW, Willis SG, Eide CA, Levine RL, Heinrich MC, Gattermann N, Gilliland DG, Druker BJ and Loriaux MM. High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009; 113:1749–1755.
26. Shih AH, Abdel-Wahab O, Patel JP and Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nature reviews Cancer. 2012; 12:599–612.
27. Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, Ishikawa S, Sato-Otsubo A, Nagae G, Nishimoto A, Haferlach C, Nowak D, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nature genetics. 2013; 45:1232–1237.
28. Liu YR, Zhu HH, Ruan GR, Qin YZ, Shi HX, Lai YY, Chang Y, Wang YZ, Lu D, Hao L, Li JL, Li LD, Jiang B and Huang XJ. NPM1-mutated acute myeloid leukemia of monocytic or myeloid origin exhibit distinct immunophenotypes. Leukemia research. 2013; 37:737–741.
29. Iriyama N, Asou N, Miyazaki Y, Yamaguchi S, Sato S, Sakura T, Maeda T, Handa H, Takahashi M, Ohtake S, Hatta Y, Sakamaki H, Honda S, Taki T, Taniwaki M, Miyawaki S, et al. Normal karyotype acute myeloid leukemia with the CD7+ CD15+ CD34+ HLA-DR + immunophenotype is a clinically distinct entity with a favorable outcome. Annals of hematology. 2014; 93:957–963.
30. Loghavi S, Zuo Z, Ravandi F, Kantarjian HM, Bueso-Ramos C, Zhang L, Singh RR, Patel KP, Medeiros LJ, Stingo F, Routbort M, Cortes J, Luthra R and Khoury JD. Clinical features of De Novo acute myeloid leukemia with concurrent DNMT3A, FLT3 and NPM1 mutations. Journal of hematology & oncology. 2014; 7:74.
31. Dalal BI, Mansoor S, Manna M, Pi S, Sauro GD and Hogge DE. Detection of CD34, TdT, CD56, CD2, CD4, and CD14 by flow cytometry is associated with NPM1 and FLT3 mutation status in cytogenetically normal acute myeloid leukemia. Clinical lymphoma, myeloma & leukemia. 2012; 12:274–279.
32. Schlenk RF, Benner A, Hartmann F, del Valle F, Weber C, Pralle H, Fischer JT, Gunzer U, Pezzutto A, Weber W, Grimminger W, Preiss J, Hensel M, Frohling S, Dohner K, Haas R, et al. Risk-adapted postremission therapy in acute myeloid leukemia: results of the German multicenter AML HD93 treatment trial. Leukemia. 2003; 17:1521–1528.
33. Mrozek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, Metzeler KH, Schwind S, Wu YZ, Kohlschmidt J, Pettenati MJ, Heerema NA, Block AW, Patil SR, Baer MR, Kolitz JE, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. Journal of clinical oncology. 2012; 30:4515–4523.
34. Gaidzik VI, Schlenk RF, Paschka P, Stolzle A, Spath D, Kuendgen A, von Lilienfeld-Toal M, Brugger W, Derigs HG, Kremers S, Greil R, Raghavachar A, Ringhoffer M, Salih HR, Wattad M, Kirchen HG, et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: results of the AML Study Group (AMLSG). Blood. 2013; 121:4769–4777.
35. Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, Miyazaki Y, Sakura T, Ozawa Y, Usui N, Kanamori H, Kiguchi T, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014; 28:1586–1595.
36. Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrozek K, Radmacher MD, Kohlschmidt J, Nicolet D, Whitman SP, Wu YZ, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. Journal of clinical oncology. 2012; 30:742–750.
37. Gaidzik VI, Paschka P, Spath D, Habdank M, Kohne CH, Germing U, von Lilienfeld-Toal M, Held G, Horst HA, Haase D, Bentz M, Gotze K, Dohner H, Schlenk RF, Bullinger L and Dohner K. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. Journal of clinical oncology. 2012; 30:1350–1357.
38. Ohgami RS, Ma L, Merker JD, Gotlib JR, Schrijver I, Zehnder JL and Arber DA. Next-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutations. Mod Pathol. 2015; 28:706–714.
39. Schnittger S, Eder C, Jeromin S, Alpermann T, Fasan A, Grossmann V, Kohlmann A, Illig T, Klopp N, Wichmann HE, Kreuzer KA, Schmid C, Staib P, Peceny R, Schmitz N, Kern W, et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. 2013; 27:82–91.
40. Koreth J, Schlenk R, Kopecky KJ, Honda S, Sierra J, Djulbegovic BJ, Wadleigh M, DeAngelo DJ, Stone RM, Sakamaki H, Appelbaum FR, Dohner H, Antin JH, Soiffer RJ and Cutler C. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. Jama. 2009; 301:2349–2361.
41. Yanada M, Matsuo K, Emi N and Naoe T. Efficacy of allogeneic hematopoietic stem cell transplantation depends on cytogenetic risk for acute myeloid leukemia in first disease remission: a metaanalysis. Cancer. 2005; 103:1652–1658.
42. Schetelig J, Schaich M, Schafer-Eckart K, Hanel M, Aulitzky WE, Einsele H, Schmitz N, Rosler W, Stelljes M, Baldus CD, Ho AD, Neubauer A, Serve H, Mayer J, Berdel WE, Mohr B, et al. Hematopoietic cell transplantation in patients with intermediate and high-risk AML: results from the randomized Study Alliance Leukemia (SAL) AML 2003 trial. Leukemia. 2015; 29:1060–1068.
43. Kanate AS, Pasquini MC, Hari PN and Hamadani M. Allogeneic hematopoietic cell transplant for acute myeloid leukemia: Current state in 2013 and future directions. World journal of stem cells. 2014; 6:69–81.
44. Liu YC, Hsiao HH, Lin PM, Yang WC, Chang CS, Liu TC, Hsu JF, Yang MY and Lin SF. Prognostic implication of molecular aberrations in cytogenetically normal acute myeloid leukemia patients receiving allogeneic hematopoietic stem cell transplantation. Genetics and molecular research. 2013; 12:5414–5423.
45. Ma Y, Wu Y, Shen Z, Zhang X, Zeng D and Kong P. Is allogeneic transplantation really the best treatment for FLT3/ITD-positive acute myeloid leukemia? A systematic review. Clinical transplantation. 2015; 29:149–160.
46. Guieze R, Cornillet-Lefebvre P, Lioure B, Blanchet O, Pigneux A, Recher C, Bonmati C, Fegueux N, Bulabois CE, Bouscary D, Vey N, Delain M, Turlure P, Himberlin C, Harousseau JL, Dreyfus F, et al. Role of autologous hematopoietic stem cell transplantation according to the NPM1/FLT3-ITD molecular status for cytogenetically normal AML patients: a GOELAMS study. American journal of hematology. 2012; 87:1052–1056.
47. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR and Sultan C. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Annals of internal medicine. 1985; 103:620–625.
48. Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S, Asou N, Kuriyama K, Jinnai I, Shimazaki C, Akiyama H, Saito K, Oh H, Motoji T, Omoto E, Saito H, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999; 93:3074–3080.
49. Kimura K and Koike A. Ultrafast SNP analysis using the Burrows-Wheeler transform of short-read data. Bioinformatics. 2015; 31:1577–1583.
50. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M and DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010; 20:1297–1303.
51. Luthra R, Patel KP, Reddy NG, Haghshenas V, Routbort MJ, Harmon MA, Barkoh BA, Kanagal-Shamanna R, Ravandi F, Cortes JE, Kantarjian HM, Medeiros LJ and Singh RR. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. 2014; 99:465–473.
52. Wang K, Li M and Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010; 38:e164.