
INTRODUCTION
Human hepatocellular carcinoma (HCC) is the second leading cause of cancer-related death, causing an estimated over half a million deaths annually [1, 2]. Metastasis is the leading cause of most cancer-related deaths [3], especially in HCC [4]. Adhesion-junction proteins, which are intercellular junctions abundant in normal epithelia and reduced in cancers [5], were found to contribute to carcinogenesis and metastasis of many types of cancer [6]. Adhesion-junctions are formed by transmembrane E-cadherin, β-catenin, and α-catenin [5]. α-catenin integrates adhesion-junctions with the actin cytoskeleton and could promote intercellular adhesion [5]. α-catenin is a putative tumor suppressor in myeloid leukemia [7], glioblastoma [8], skin cancer [9, 10] and breast cancer [11, 12]. Among the three α-catenin coding genes, CTNNA1 participates the carcinogenesis of multiple types of cancers [12, 13], and mutates in HCC [14]. CTNNA2 mutates in gastric cancer [15] and laryngeal carcinomas [16]. CTNNA3 is a tumor suppressor and frequently mutates in laryngeal carcinomas [16] and low-expressed in urothelial carcinoma of the bladder [17]. The role of CTNNA2 and CTNNA3 in HCC haven’t been found yet.
miRNAs are small, non-coding RNAs that negatively regulate the expression of target genes by mRNA degradation or translational repression [18]. miRNAs function as important regulators in cancer microenvironment [19, 20]. Many miRNAs, like miR-3127 [21], miR-494 [22], miR-42509 [23], participate the carcinogenesis of HCC by inhibiting their target genes. Therefore, miRNAs are also included in our study.
In this study, we analyzed the expression of the three α-catenin coding genes in HCC using microarray data of HCC samples and normal liver controls with bioinformatics methods and identified that CTNNA3 was down-regulated in HCC. CCK8 and Transwell assays revealed that CTNNA3 inhibited proliferation, migration and invasion of HCC cells. The silence of CTNNA3 resulted in increased proliferating cell nuclear antigen (PCNA), decreased cell cycle inhibitor p21Cip1/Waf1 and Akt signal activation, as well as the increased matrix metallopeptidase MMP-9. miR-425 inhibited CTNNA3 in HCC. miR-425 directly bound to the 3′untranslated region of CTNNA3 and inhibited CTNNA3 to promote the proliferation, migration and invasion of HCC cells.
RESULTS
CTNNA3 was down-regulated in HCC
The comparison of gene expression between HCC and normal healthy controls indicated that CTNNA3 was down-regulated (P-value < 0.05 and LgFC < –1) in 78 of 81 human HCC samples. The other two α-catenin coding genes, CTNNA1 and CTNNA2, were not deregulated in the analyzed HCC dataset. Therefore, CTNNA3 was selected for further investigation.
CTNNA3 inhibited HCC cell proliferation
We then explored the potential impact of CTNNA3 on HCC cell proliferation in HepG2, MHCC97H and HCCLM3 cell lines. HepG2, MHCC97H and HCCLM3 cells were transfected with CTNNA3 overexpression vector or siRNA or inactive controls (Figure 1). CCK8 assay indicated that the cell proliferations were enhanced in all of the CTNNA3-siRNA-transfected HCC cell lines compared with inactive-control-transfected HCC cell lines (Figure 2A). Conversely, CTNNA3 overexpression vector inhibited the cell proliferations of the HepG2, MHCC97H and HCCLM3 cells (Figure 2A).
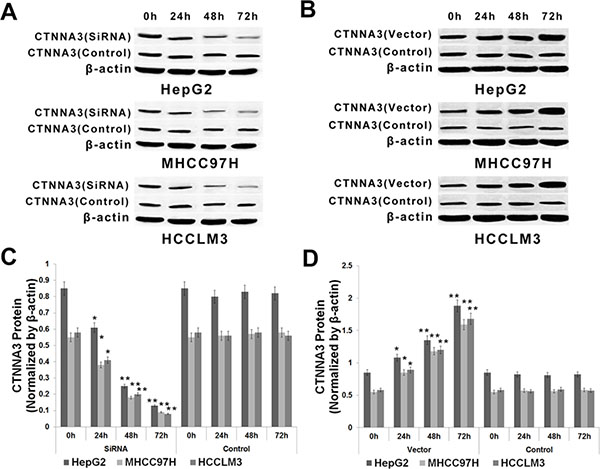
Figure 1: Expression of CTNNA3 in HCC cells transfected with CTNNA3 expression vector, siRNA or inactive controls. (A, C) Protein of CTNNA3 decreased with time after transfection with CTNNA3 siRNA in HepG2, MHCC97H and HCCLM3 cells. (B, D) Protein of CTNNA3 increased with time after transfection with CTNNA3 overexpression vector in HepG2, MHCC97H and HCCLM3 cells; *p < 0.05, **p < 0.01, and ***p < 0.001.
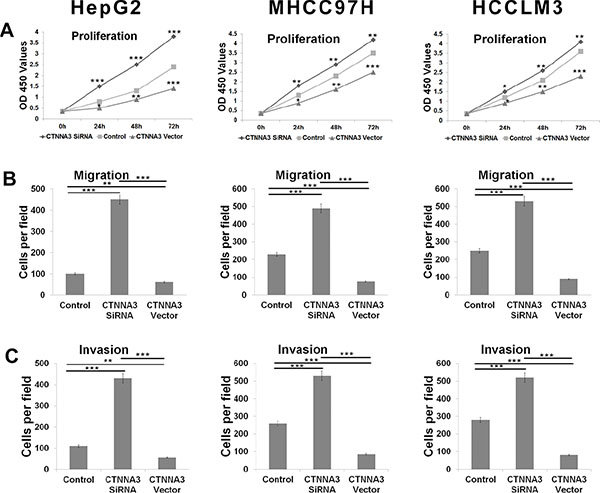
Figure 2: CTNNA3 regulates HCC cell proliferation, migration and invasion. (A) Growth of HCC cells was shown after transfection with CTNNA3 siRNA or overexpression vector or inactive control. The growth index as assessed at 0, 24, 48 and 72 h. (B) Transwell analysis of HCC cells migration after treatment with CTNNA3 siRNA or overexpression vector or inactive control. (C) Transwell analysis of HCC cells invasion after treatment with CTNNA3 siRNA or overexpression vector or inactive control; *p < 0.05, **p < 0.01, and ***p < 0.001.
CTNNA3 inhibited HCC cell cycle progression
As CTNNA3 inhibtied HCC cell proliferation, cell cycle analysis was performed to examine how CTNNA3 affectes the cell cycle. Flow cytometric analysis showed that the percentage of CTNNA3 overexpression cells at G1 phase increased comparing to control cells. This phenomenon was associated with a concomitant decrease of cells at the S phases of the cell cycle (Figure 3B). Moreover, the percentage of CTNNA3 knockdown cells at G1 phase decreased comparing to control cells. And it was associated with a concomitant increase of cells at the S phases of the cell cycle (Figure 3C).
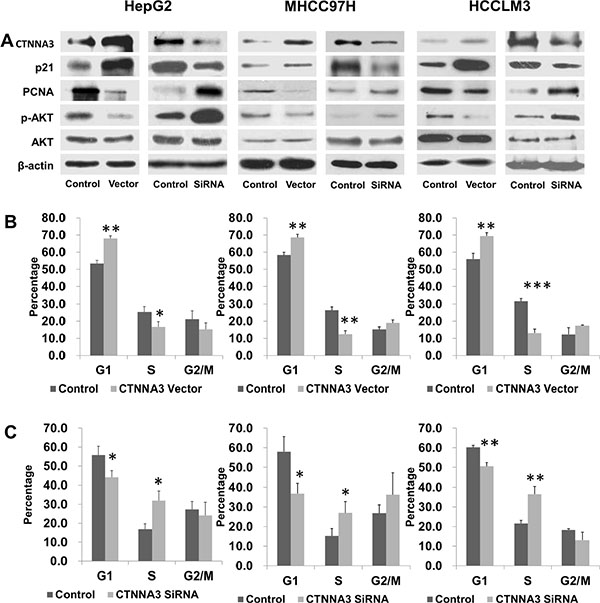
Figure 3: CTNNA3 regulates HCC cell cycle progression. (A) Changed expression of CTNNA3 significant changed the levels of phosphorylated Akt, p21Cip1/Waf1 and PCNA (B) Cell cycle analysis of CTNNA3 overexpression cells and control cells. (C) Cell cycle analysis of CTNNA3 knockdown cells and control cells. *p < 0.05, **p < 0.01, and ***p < 0.001.
In order to investigate the mechanisms underlying the above changes in cell cycle progression, several cell cycle-related proteins were compared between CTNNA3 overexpression cells, CTNNA3 knockdown cells and control cells using western blot. Expression changes of CTNNA3 did not cause significant deregulation of Cyclin A1, Cyclin A2, Cyclin D1, Cyclin D3 or Cyclin E2 (data not shown), but resulted in significant changes of the cell cycle inhibitor p21Cip1/Waf1 and the proliferation marker PCNA (Figure 3A). CTNNA3 knockdown cells showed decreased protein levels of p21Cip1/Waf1 and increased protein levels of PCNA (Figure 3A). Conversely, CTNNA3 overexpression cells showed increased protein levels of p21Cip1/Waf1 and decreased protein levels of PCNA (Figure 3A). These results supported the inhibition effect of CTNNA3 on HCC cell cycle progression.
Since Akt signaling can lead to down-regulation of the CDK inhibitor p21Cip1/Waf1 [24], we examined the effect of CTNNA3 on inhibiting Akt signaling by measuring the protein level of phosphorylated and unphosphorylated Akt. Western blot analysis showed that phosphorylated Akt was decreased in CTNNA3 overexpression cells (Figure 3A), revealing that CTNNA3 resulted in inhibited Akt signaling, which in turn leads to up-regulation of p21Cip1/Waf1 and concomitant down-regulation of PCNA.
CTNNA3 inhibited HCC cell migration and invasion
We explored the potential impact of CTNNA3 on HCC cell migration and invasion in HepG2, MHCC97H and HCCLM3 cell lines. Cells were transfected with CTNNA3 overexpression vector or siRNA or inactive controls. Transwell assay indicated that the cell migration and invasion were promoted in all of the CTNNA3-siRNA-transfected HCC cell lines compared with inactive-control-transfected HCC cell lines (Figure 2B and 2C). Conversely, CTNNA3 overexpression vector could inhibit the migration and invasion of the HepG2, MHCC97H and HCCLM3 cells (Figure 2B and 2C).
It has been reported that Akt promotes cancer cell invasion by increasing matrix metallopeptidase (MMP-9) [25]. This leads to the hypothesis that MMP-9 may also participate the molecular mechanisms underlying CTNNA3 related inhibition of cell migration and invasion. Therefore, we investigated expression of MMP-9 in CTNNA3 knockdown cells, CTNNA3 overexpression cells and control cells with and without 12-O-tetradecanoylphorbol- 13-Acetate (TPA) treatment, which is an activator of protein kinase C that can promote cancer cell invasion via MMP-9 induction [26]. The results indicated that CTNNA3 inhibited MMP-9 in TPA-treated cells (Figure 4). These results indicated that MMP-9 may be part of the molecular mechanisms that underlying the cell invasion inhibition by CTNNA3 in HCC.
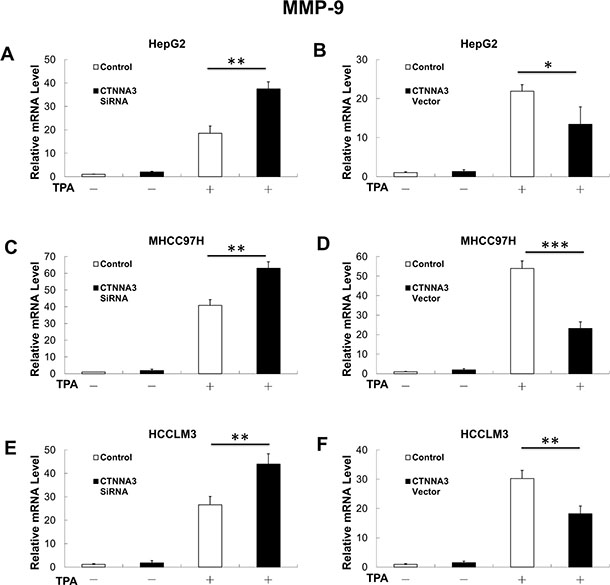
Figure 4: MMP-9 mRNA expression is negatively related with in CTNNA3 HCC cells. (A) TPA-induced MMP-9 mRNA expression was increased in CTNNA3 knockdown HepG2 cells. (B) TPA-induced MMP-9 mRNA expression was decreased in CTNNA3 overexpression HepG2 cells. (C) TPA-induced MMP-9 mRNA expression was increased in CTNNA3 knockdown MHCC97H cells; (D) TPA-induced MMP-9 mRNA expression was decreased in CTNNA3 overexpression MHCC97H cells. (E) TPA-induced MMP-9 mRNA expression was increased in CTNNA3 knockdown HCCLM3 cells. (F) TPA-induced MMP-9 mRNA expression was decreased in CTNNA3 overexpression HCCLM3 cells. *p < 0.05, **p < 0.01, and ***p < 0.001.
MiR-425 targeted and inhibited CTNNA3 in HCC cells
As shown above, the down-regulation of CTNNA3 may contribute to the carcinogenesis of HCC. Here come the question: How CTNNA3 is down-regulated in human HCC? miRNAs are inhibitors of gene expression [18]. It lead to the hypothesis that CTNNA3 might be inhibited by miRNA. Therefore, bioinformatics analysis was performed to predict the potential regulation interactions between miRNAs and CTNNA3. As predicted by miRanda [27] and TargetScan [28], 23 miRNAs have high complementarity to the 3′UTR of CTNNA3. Among them, only miR-425 was found to be up-regulated (P-value < 0.05 & LgFC > 0.5) in the 13 HCC samples. Therefore, miR-425 was selected for further validation using wet experiments. The results indicated that miR-425 reduced the protein levels of CTNNA3 in HCC cells (Figure 5A and 5C), while miR-425 inhibitors increased the protein levels of CTNNA3 in HCC cells (Figure 5B and 5D). Then the effect of miR-425 on the translation of CTNNA3 mRNA into protein was assessed by a luciferase reporter assay (Figure 5E and 5F). miR-425 reduced the luciferase activity of the reporter gene with the wild type CTNNA3 3′UTR construct, but not with the mutant construct (Figure 5E). Meanwhile the miR-425 inhibitor enhanced the luciferase activity of the reporter gene with the wild type CTNNA3 3′UTR construct, but not with the mutant construct (Figure 5F). These evidences indicated that miR-425 inhibited CTNNA3 by directly targeting its 3′UTR region.
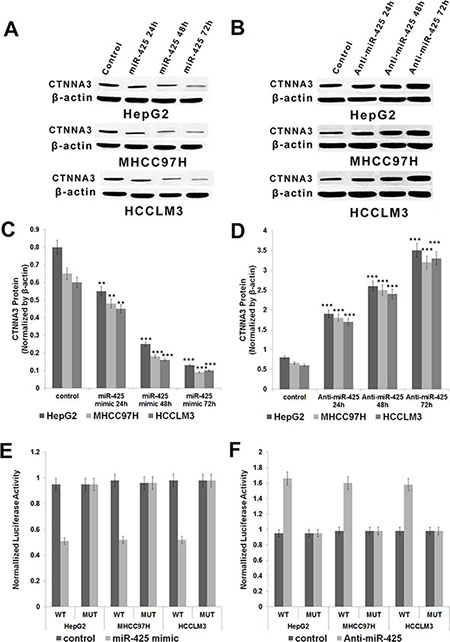
Figure 5: miR-425 targets and negatively regulates CTNNA3 in HCC cells. (A. C) Protein of CTNNA3 decreased with time after transfection with miR-425 mimics in HepG2, MHCC97H and HCCLM3 cells. (B, D) Protein of CTNNA3 increased with time after transfection with anti-miR-425 in HepG2, MHCC97H and HCCLM3 cells; (E) The analysis of the relative luciferase activities of CTNNA3-WT, CTNNA3-MUT in HepG2, MHCC97H and HCCLM3 cells after transfection with miR-425 mimics. (F) The analysis of the relative luciferase activities of CTNNA3-WT, CTNNA3-MUT in HepG2, MHCC97H and HCCLM3 cells after transfection with anti-miR-425; *p < 0.05, **p < 0.01, and ***p < 0.001.
miR-425 promoted HCC cell proliferation, migration and invasion by inhibiting CTNNA3
We explored the potential impacts of miR-425 on cell proliferation, migration and invasion in the HepG2, MHCC97H and HCCLM3 cell lines. HCC cells were transfected with miR-425 mimics or inhibitors or inactive control cel-mir-67, respectively. The results of CCK-8 proliferation assay revealed that the cell proliferation was promoted in all of the miR-425-mimics-transfected cell lines comparing to the inactive control cel-mir-67-transfected cell lines (Figure 6A). Meanwhile, miR-425 inhibitor decreased the proliferation of all the three HCC cell lines (Figure 6A). Flow cytometric analysis showed that miR-425 mimics promoted the transformation from G1 phase to S phases of the cell cycle (Figure 7B). At molecular level, the miR-425 mimics inhibited CTNNA3, and in turn resulted in increased PCNA, decreased p21Cip1/Waf1 and activated Akt (Figure 7A). These results revealed that the miR-425 promoted cell proliferation by inhibiting CTNNA3.
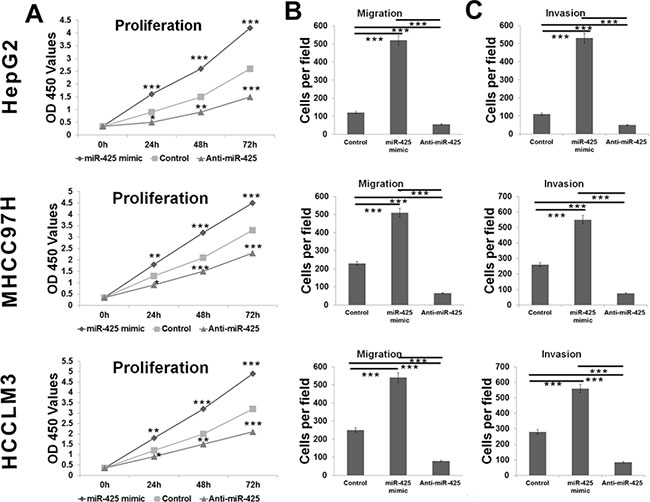
Figure 6: miR-425 promotes HCC cell proliferation, migration and invasion. (A) Growth of HCC cells was shown after transfection with miR-425 mimics or inhibitor or inactive control. The growth index as assessed at 0, 24, 48 and 72 h. (B) Transwell analysis of HCC cells migration after treatment with miR-425 mimics, inhibitors or inactive control. (C) Transwell analysis of HCC cells invasion after treatment with miR-425 mimics, inhibitors or inactive control. *p < 0.05, **p < 0.01, and ***p < 0.001.
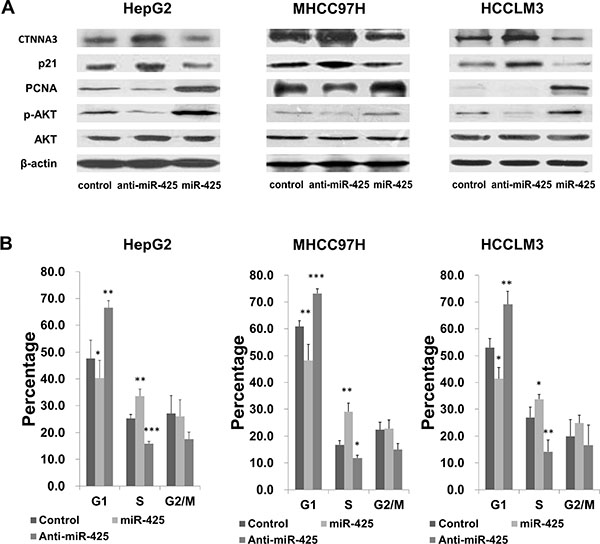
Figure 7: miR-425 regulates HCC cell cycle progression by inhibiting CTNNA3. (A) miR-425 significant changed the levels of CTNNA3, phosphorylated Akt, p21Cip1/Waf1 and PCNA. (B) Cell cycle analysis of HCC cells transfected with miR-425 mimics or inhibitor or inactive control. *p < 0.05, **p < 0.01, and ***p < 0.001.
Migration and invasion assay indicated that miR-425 mimics promoted the migration and invasion of HCC cells, whereas miR-425 inhibitors reduced migration and invasion of HCC cells (Figure 6B and 6C). At molecular level, the miR-425 mimics increased the mRNA levels of MMP-9 in TPA-treated cells, while miR-425 inhibitors decreased the mRNA levels of MMP-9 (Figure 8).
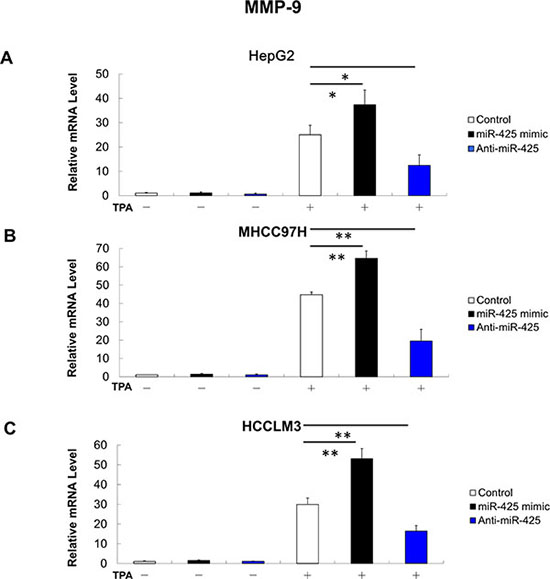
Figure 8: MMP-9 mRNA expression is positively related with miR-425 in HCC cells. (A) MMP-9 mRNA expression in HepG2 cells with or without TPA treatment after transfection of miR-425 mimics or inhibitor or inactive control. (B) MMP-9 mRNA expression in MHCC97H cells with or without TPA treatment after transfection of miR-425 mimics or inhibitor or inactive control. (C) MMP-9 mRNA expression in HCCLM3 cells with or without TPA treatment after transfection of miR-425 mimics or inhibitor or inactive control. *p < 0.05, **p < 0.01, and ***p < 0.001.
CTNNA3 suppresses the tumor growth of the HCC cell xenograft in nude mice
To explore the relationship between miR-425/CTNNA3 axis and tumorigenesis in vivo, the xenograft model of HCC cells in nude mice was adopted. We used lentiviral vectors for the stable expression of CTNNA3, CTNNA3 siRNA (Sh CTNNA3), miR-425 and miR-425 inhibitor (anti-miR-425) in MHCC97H cells. MHCC97H cells that stably expressed CTNNA3, ShCTNNA3, miR-425, anti-miR-425, or their controls were injected subcutaneously into each flank of nude mice. The tumor weights and volumes were monitored. Then the growth curves of the tumors were plotted accordingly. We found that CTNNA3 overexpression decreased tumor growth and miR-425 promoted tumor growth in vivo (Figure 9A–9C). We further performed IHC staining for Ki67 and PCNA in the tumors. Compared with the negative control, CTNNA3 overexpression suppressed proliferative activity, as indicated by the percentage of cells positive for Ki67 and PCNA staining. In contrast, miR-425 overexpression increased the percentages of Ki67- and PCNA-positive cells (Figure 10). These in vivo observations confirmed the key role of miR-425/CTNNA3 axis in the control of HCC cell growth and may serve as potential therapeutic targets for HCC treatment.
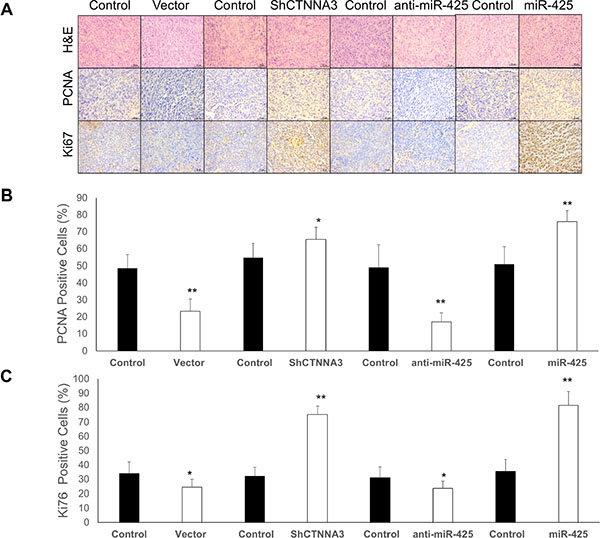
Figure 9: CTNNA3 suppresses the tumor growth of the HCC cell xenograft in nude mice. (A) Representative image of tumors in nude mice after the injection of MHCC97H cells stably expressing CTNNA3, ShCTNNA3, miR-425, anti-miR-425 and their controls (n = 8, 4 is shown). (B) Quantification of tumor weights of xenograft tumors in mice. (C) Growth curves of xenograft tumors in mice. *p < 0.05, **p < 0.01, and ***p < 0.001.
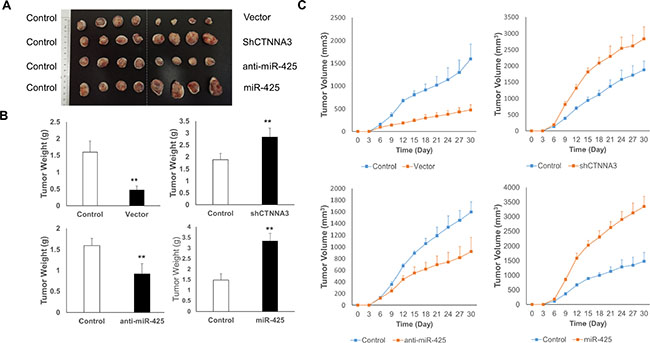
Figure 10: IHC staining for Ki67 and PCNA in the xenograft tumors in nude mice. (A) Representative images of tumor samples that were stained with hematoxylin and eosin (H & E), Ki67, PCNA by IHC. (B) The percentages of PCNA-positive cells. (C) The percentages of Ki67-positive cells. *p < 0.05, **p < 0.01, and ***p < 0.001.
DISCUSSION
HCC is the second leading cause of cancer-related death, causing an estimated over half a million deaths annually [1, 2]. Theα-catenin coding genes, CTNNA1, CTNNA2 and CTNNA3, participate carcinogenesis of multiple cancers, such as laryngeal carcinomas [16], oropharyngeal squamous cell carcinomas [29] and breast cancer [12]. In this study, we analyzed gene microarray data of HCC samples and normal liver controls using bioinformatics methods with a special focus on the three α-catenin coding genes. The results indicated that CTNNA3 was down-regulated in HCC (LgFC < –1).
In order to explore the function of CTNNA3 in HCC, we silenced the expression of CTNNA3 in HCC cell lines using siRNAs (Figure 1A and 1C). An important finding of this study is that CTNNA3 inhibited the proliferation marker PCNA, implying that CTNNA3 is an important proliferation inhibitor of HCC. It was supported by the fact that silence of CTNNA3 in HCC cells results in increased cell proliferation (Figure 2A). Moreover, CTNNA3 inhibited the cell cycle in G1-S and increased p21Cip1/Waf1 (Figure 3). Several studies showed that p21Cip1/Waf1 inhibits HCC cell proliferation, and p21Cip1/Waf1 could be inhibited by the Akt signaling pathway [24]. These findings are consistent with our results (Figure 3). These observations indicated that CTNNA3 suppresses HCC cell proliferation at least in part through inhibiting the Akt signaling and in turn, increasing p21Cip1/Waf1.
Invasion and metastasis are characteristic features of HCC and major poor prognostic factor in HCC patients. Degradation of the extracellular matrix and the basement membrane is necessary for invasion and metastasis. CTNNA3 encodes an adhesion-junction protein. It explains why CTNNA3 inhibits the migration and invasion of the HCC cells. Moreover, it’s interesting to find that CTNNA3 inhibits HCC invasion not only by itself. MMP-9, which plays an important role in the proteolytic destruction of the extracellular matrix and basement membranes, was found to be inhibited by CTNNA3 (Figure 4). MMP-9 would be increased by Akt signaling pathways [30]. In this study, we showed that CTNNA3 inhibits the Akt signaling in HCC cells. Therefore, CTNNA3 may inhibit cell invasiveness by inhibiting MMP-9 via Akt signaling pathway.
Then we explored the potential mechanism of impaired CTNNA3 expression in HCC. The miRNAs, which inhibit gene expression and function as important regulators in cancer microenvironment [20, 31], were selected as the research focus. Bioinformatics analysis indicated the potential regulatory interaction between miR-425 and CTNNA3. Luciferase assay and western blot confirmed that miR-425 directly targets the 3′UTR region of CTNNA3 and inhibit its expression (Figure 5). The inhibition of CTNNA3 by miR-425 in turn resulted in increased PCNA, decreased p21Cip1/Waf1 and Akt activation (Figure 7A), as well as the increased MMP-9 (Figure 8). CCK-8 and transwell assay showed that miR-425 promoted cell proliferation, migration and invasion of HCC cell lines (Figure 6). Moreover, miR-425 promotes the tumor growth of the HCC cell xenograft in nude mice, while CTNNA3 suppresses the growth. These results revealed that miR-425 promotes cell proliferation, migration and invasion by inhibiting CTNNA3 in HCC.
In general, our study highlights the crucial role of CTNNA3 in human HCC by inhibiting cell proliferation and invasion. And CTNNA3 is inhibited by miR-425. The miR-425/CTNNA3 axis provide insights into the mechanisms underlying human HCC, and may be involved in potential therapeutics of HCC.
MATERIALS AND METHODS
Gene and miRNA profile data collection
Gene and miRNA microarray data of HCC and normal control samples were collected from GEO database (http://www.ncbi.nlm.nih.gov/gds, GSE62232, GSE67882, and GSE40744). After quality control with limma package on R platform, 81 patients with HCC and 10 healthy controls were selected for gene expression analysis, 13 HCC patients and 15 healthy controls were selected for miRNA expression analysis.
Identification of differentially expressed genes and miRNAs
The differentially expressed genes and miRNAs in patients with HCC were identified with Limma package on R platform using download gene and miRNA microarray data as mentioned above. The cutline for differentially expressed gene and miRNA is P value < 0.05 (T-test).
miRNA target genes prediction
Human miRNA target genes prediction were performed using miRNA sequences downloaded from the Rfam website (http://www.sanger.ac.uk/Software/Rfam) on January 2015. A uniform system for microRNA annotation was used to retrieve 3′UTR sequences data for human genes [32] RepeatMasker [33] was used to mask repetitive elements in these sequences. The miRanda [27] and TargetScan [28] methods were used to predict the target genes of miRNAs. The predicted target genes that supported by both of the methods were selected for further analysis.
HCC cell lines and cell culture
The human HCC cell lines HepG2, MHCC97H and HCCLM3 was obtained from the Chinese Center for Type Cultures Collections (CCTCC, Wuhan, China). The HepG2, MHCC97H and HCCLM3 cell lines were cultured in RPMI 1640 media (Life Technologies, Beijing, China) and supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Beijing, China). All the Cells were maintained at 37°C in a humidified atmosphere with 5% CO2.
Cell transfection
Full-length CTNNA3 coding sequences were amplified and cloned into a pcDNA™ 3.1 expression vector (Life Technologies, Beijing, China), following manufacturer’s protocol. HepG2, MHCC97H and HCCLM3 cell lines were seeded in 24-well plates at 3 × 105 cells/wells and incubated overnight. Transfection of the CTNNA3 pcDNA™ 3.1 vectors, inactive control mock pcDNA™ 3.1 vectors, CTNNA3 siRNAs, inactive control scrambled siRNAs, miR-425 miRNA mimics, anti-miR-425, inactive control cel-mir-67 (Life Technologies, Beijing, China), or pMIR-Report vectors were taken using Lipofectamine 2000 transfection reagent (Life Technologies, Beijing, China) with 1 μg/ml DNA plasmid or 300 nmol of miRNA, respectively. Total proteins of HepG2, MHCC97H and HCCLM3 cells were isolated at 24 h, 48 h and 72 h after transfection.
Cell migration and invasion assay
Cell invasion and migration were detected using a transwell chamber assay (Corning, Beijing, China) with or without Matrigel (Life Technologies, Beijing, China). For the determination of HepG2, MHCC97H and HCCLM3 cell invasion, transwell chamber was placed into a 24-well plate, then coated with 30 μl Matrigel, and incubated at 37°C for 40 minutes. In transwell assay with or without Matrigel, HepG2, MHCC97H and HCCLM3 cells were trypsinized and seeded in chambers at the density of 6 × 104 cells/well at 48 h after the transfection. These cells were cultured in RPMI 1640 medium with 2% serum. And 600 μl of 10% FBS-1640 was added to the lower chamber. After 24 h, migrated HepG2, MHCC97H and HCCLM3 cells were fixed in the 100% methanol for 30 minutes. Those non-migrated HepG2, MHCC97H and HCCLM3 cells were removed by cotton swabs. After that cells on the bottom surface of the membrane were fixed and stained with the 0.1% crystal violet for 30 minutes. Images of HepG2, MHCC97H and HCCLM3 cells were taken using a phase-contrast microscope.
Cell proliferation assay
Cell proliferation was detected using a Cell Counting Kit-8 assay (Dojindo, Kumamoto, Japan). HepG2, MHCC97H and HCCLM3 cells were plated in 24-well plates at 3 × 105 cells/well. Then HCC cells were incubated at 37°C in 10% CCK-8 diluted with normal culture medium for color conversion. Proliferation rate was measured at 24 h, 48 h and 72 h after transfection.
Cell cycle analysis
6 × 105 cells were synchronized by serum starvation for 24 h. Then the cells were induced to re-enter the cell cycle by changing with 10% fetal bovine serum for 24 h. floating and adherent cells were harvested and fixed using 75% ethanol overnight at 4°C. After that, cells were incubated with RNase A for 30 min at 37°C. Then cells were stained with propidium iodide. Cell cycle was determined by flow cytometry.
Luciferase assay
HepG2, MHCC97H and HCCLM3 cells were seeded in 24-well plates at 3 × 105 cells/well and then incubated for 24 hours. Then the HepG2, MHCC97H and HCCLM3 cells were co-transfected with 0.6 μg of pGL3-CTNNA1–3′UTR or pGL3-CTNNA1–3′UTR Mut plasmid, or 0.06 ng of the phRL-SV40 control vector (Promega, Beijing, China), and 100 nM miR-425 mimics or miR-425 inhibitors or control RNA using Lipofectamine 2000 (Invitrogen, Beijing, China). The renilla and firefly luciferase activities were measured by a dual luciferase assay (Promega, Beijing, China) at 24 h after transfections.
Western blot
Isolated proteins from HepG2, MHCC97H and HCCLM3 cells at 24 h, 48 h and 72 h after transfections were separated by 12% SDS-PAGE gel and transferred onto nitrocellulose membranes (Bio-Rad, Beijing, China). Membranes were blocked with 5% non-fat milk and incubated with anti-CTNNA3 antibody (Abcam, Beijing, China) or anti-β-actin antibody (Abcam, Beijing, China). After extensive washes, the secondary antibody (Abcam, Beijing, China) was added to the system. Finally, Immunoreactive protein bands were determined using the Enhanced Chemiluminescence (ECL) system.
Real-time reverse transcription quantitative PCR
Total RNA was extracted with Trizol reagent (Invitrogen, Beijing, China) according to the instructions. The cDNA was synthesized from total RNA with MMLV reverse transcriptase (Invitrogen, Beijing, China) and random hexamers. Real-time PCRs were performed using ABI 7300 Sequence Detection System (Life, Beijing, China). Relative quantification was determined by normalization to the amount of GAPDH. Primers used for real-time PCR were designed by Primer Express 3.0 and synthesized in Invitrogen.
In vivo tumorigenicity assay
Tumor cells (5 × 106) were injected subcutaneously into each flank of 4-week-old male Balb/c athymic nude mice (n = 8 per group). Mice were monitored every three day and euthanized one month later. Then tumors were dissected and weighed. The tumor volume was calculated as V (volume, mm3) = 0.5 × L (length, mm) × W2 (width, mm2).
Immunohistochemistry (IHC)
IHC was performed on tissue microarray chips (Outdo, Shanghai, China). Single serial sections were made from xenograft tumor samples. The slides were probed with the following primary antibodies: mouse anti–human Ki67, and mouse anti–human PCNA (Abcam, Beijing, China). Then the slides were incubated with HRP-conjugated goat anti–mouse secondary antibodies. The proteins were visualized in situ with DAB chromogenic substrate.
Statistical analysis
Experiments were repeated at least three times to achieve confident results. Statistical analysis was performed using R. All the data were presented as means ± S.D. Differences between groups were determined with Student’s t-test. Statistical analysis was considered to be significant when P value < 0.05.
ACKNOWLEDGMENTS
The Project was supported by the National Natural Science Foundation of China (Grant No. 61372151).
CONFLICTS OF INTEREST
Authors have declared that no competing interest exists.
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer Journal international du cancer. 2015; 136:E359–386.
2. Flores A, Marrero JA. Emerging trends in hepatocellular carcinoma: focus on diagnosis and therapeutics. Clinical Medicine Insights Oncology. 2014; 8:71–76.
3. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011; 331:1559–1564.
4. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014; 63:844–855.
5. Kobielak A, Fuchs E. Alpha-catenin: at the junction of intercellular adhesion and actin dynamics. Nature reviews Molecular cell biology. 2004; 5:614–625.
6. Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003; 112:535–548.
7. Liu TX, Becker MW, Jelinek J, Wu WS, Deng M, Mikhalkevich N, Hsu K, Bloomfield CD, Stone RM, DeAngelo DJ, Galinsky IA, Issa JP, Clarke MF, et al. Chromosome 5q deletion and epigenetic suppression of the gene encoding alpha-catenin (CTNNA1) in myeloid cell transformation. Nature medicine. 2007; 13:78–83.
8. Ji H, Wang J, Fang B, Fang X, Lu Z. alpha-Catenin inhibits glioma cell migration, invasion, and proliferation by suppression of beta-catenin transactivation. Journal of neuro-oncology. 2011; 103:445–451.
9. Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell. 2011; 144:782–795.
10. Silvis MR, Kreger BT, Lien WH, Klezovitch O, Rudakova GM, Camargo FD, Lantz DM, Seykora JT, Vasioukhin V. alpha-catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Science signaling. 2011; 4:ra33.
11. Bajpai S, Feng Y, Krishnamurthy R, Longmore GD, Wirtz D. Loss of alpha-catenin decreases the strength of single E-cadherin bonds between human cancer cells. The Journal of biological chemistry. 2009; 284:18252–18259.
12. Piao HL, Yuan Y, Wang M, Sun Y, Liang H, Ma L. alpha-catenin acts as a tumour suppressor in E-cadherin-negative basal-like breast cancer by inhibiting NF-kappaB signalling. Nature cell biology. 2014; 16:245–254.
13. Majewski IJ, Kluijt I, Cats A, Scerri TS, de Jong D, Kluin RJ, Hansford S, Hogervorst FB, Bosma AJ, Hofland I, Winter M, Huntsman D, Jonkers J, et.al. An alpha-E-catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. The Journal of pathology. 2013; 229:621–629.
14. Janku F, Kaseb AO, Tsimberidou AM, Wolff RA, Kurzrock R. Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next generation sequencing. Oncotarget. 2014; 5:3012–3022. doi: 10.18632/oncotarget.1687.
15. Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, Chan KH, Chan AS, Tsui WY, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nature genetics. 2014; 46:573–582.
16. Fanjul-Fernandez M, Quesada V, Cabanillas R, Cadinanos J, Fontanil T, Obaya A, Ramsay AJ, Llorente JL, Astudillo A, Cal S, Lopez-Otin C. Cell-cell adhesion genes CTNNA2 and CTNNA3 are tumour suppressors frequently mutated in laryngeal carcinomas. Nature Communications. 2013; 4:2531.
17. Meehan M, Melvin A, Gallagher E, Smith J, McGoldrick A, Moss C, Goossens S, Harrison M, Kay E, Fitzpatrick J, Dervan P, Mc Cann A. Alpha-T-catenin (CTNNA3) displays tumour specific monoallelic expression in urothelial carcinoma of the bladder. Genes, chromosomes and cancer. 2007; 46:587–593.
18. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136:215–233.
19. Suzuki HI, Katsura A, Matsuyama H, Miyazono K. MicroRNA regulons in tumor microenvironment. Oncogene. 2015; 34:3085–94.
20. Kohlhapp FJ, Mitra AK, Lengyel E, Peter ME. MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene. 2015; 34:5857–68.
21. Jiang J, Zhang Y, Guo Y, Yu C, Chen M, Li Z, Tian S, Sun C. MicroRNA-3127 promotes cell proliferation and tumorigenicity in hepatocellular carcinoma by disrupting of PI3K/AKT negative regulation. Oncotarget. 2015; 6:6359–6372. doi: 10.18632/oncotarget.3438.
22. Chuang KH, Whitney-Miller CL, Chu CY, Zhou Z, Dokus MK, Schmit S, Barry CT. MiR-494 is A Master Epigenetic Regulator of Multiple Invasion-Suppressor MicroRNAs via Targeting TET1 in Invasive Human HCC Tumors. Hepatology. 2015; 62:466–80.
23. Liu T, Zhang X, Sha K, Liu X, Zhang L, Wang B. miR-709 up-regulated in hepatocellular carcinoma, promotes proliferation and invasion by targeting GPC5. Cell proliferation. 2015; 48:330–337.
24. Thaler S, Hahnel PS, Schad A, Dammann R, Schuler M. RASSF1A mediates p21Cip1/Waf1-dependent cell cycle arrest and senescence through modulation of the Raf-MEK-ERK pathway and inhibition of Akt. Cancer research. 2009; 69:1748–1757.
25. Gao Y, Guan Z, Chen J, Xie H, Yang Z, Fan J, Wang X, Li L. CXCL5/CXCR2 axis promotes bladder cancer cell migration and invasion by activating PI3K/AKT-induced upregulation of MMP2/MMP9. Int J Oncol. 2015; 47:690–700.
26. Kim JM, Noh EM, Kim MS, Hwang JK, Hwang HY, Ryu DG, Kim HJ, Yu HN, You YO, Kim JS, Youn HJ, Kwon KB, Jung SH, Lee YR. Decursin prevents TPA-induced invasion through suppression of PKCalpha/p38/NF-kappaB-dependent MMP-9 expression in MCF-7 human breast carcinoma cells. International journal of oncology. 2014; 44:1607–1613.
27. John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS biology. 2004; 2:e363.
28. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120:15–20.
29. Gao G, Kasperbauer JL, Tombers NM, Cornell MD, Smith DI. Prognostic significance of decreased expression of six large common fragile site genes in oropharyngeal squamous cell carcinomas. Translational oncology. 2014; 7:726–731.
30. Chen JS, Huang XH, Wang Q, Huang JQ, Zhang LJ, Chen XL, Lei J, Cheng ZX. Sonic hedgehog signaling pathway induces cell migration and invasion through focal adhesion kinase/AKT signaling-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9 in liver cancer. Carcinogenesis. 2013; 34:10–19.
31. Suzuki HI, Katsura A, Matsuyama H, Miyazono K. MicroRNA regulons in tumor microenvironment. Oncogene. 2015; 34:3085–3094.
32. Kasprzyk A, Keefe D, Smedley D, London D, Spooner W, Melsopp C, Hammond M, Rocca-Serra P, Cox T, Birney E. EnsMart: a generic system for fast and flexible access to biological data. Genome research. 2004; 14:160–169.
33. Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics / editoral board, Andreas D Baxevanis [et al]. 2009; Chapter 4:Unit 4 10.