
Introduction
The Hedgehog (HH)/GLI pathway plays a central role in the control of vertebrate development and tissue homeostasis of adult mammalian organisms, while its uncontrolled activation or inefficient termination has been implicated in a number of human malignancies including cancers of the skin, brain, hematopoietic system, lung, ovary and of the gastrointestinal tract [1]. Targeting oncogenic HH/GLI signaling in cancer and cancer stem cells has therefore emerged as promising therapeutic strategy for many malignant diseases with high medical need [2, 3].
Precise control of canonical HH/GLI signaling is an intricate process involving numerous regulatory processes. Briefly, in the absence of HH ligand, the twelve-transmembrane protein Patched (PTCH) represses HH signaling by preventing the G-protein coupled receptor-like protein Smoothened (SMO) to translocate to the primary cilium, an antenna-like compartment central to the activation of canonical HH/GLI signaling. Binding of HH to its receptor PTCH revokes the repressive effect of PTCH thereby allowing SMO to enter the primary cilium and initiate signaling. In the cilium, SMO activates the GLI zinc finger transcription factors GLI2/3 by preventing the formation of GLI repressor forms and by releasing GLIs from their negative regulator Suppressor of Fused (SUFU). Translocation of GLI2/3 activator forms to the nucleus induces HH/GLI target gene expression, including the potent transcriptional activator and oncogene GLI1 (for detailed reviews see [4-7]).
The etiologic role of HH signaling in cancer has triggered numerous efforts to develop HH pathway antagonists targeting the essential HH effector SMO [8]. In 2012, the FDA approved vismodegib, a small molecule inhibitor of SMO, for the treatment of non-melanoma skin cancer. Vismodegib provides a remarkable therapeutic benefit to patients with advanced or metastatic basal cell carcinoma (BCC) [3], a malignancy caused by loss of PTCH or mutational activation of SMO [9]. Despite several successful trials with BCC patients and case studies with medulloblastoma (MB) patients, the therapeutic efficacy of SMO targeting is challenged by acquired and de novo drug resistance [10-12]. Furthermore, clinical trials with SMO inhibitors so far have failed to prove a clear therapeutic benefit for patients with non-BCC malignancies including colorectal, ovarian and pancreatic cancer [13, 14].
De novo resistance to SMO targeting can – at least in part – be explained by the uncoupling of GLI activation from canonical SMO-dependent HH signaling. Various molecular cues and genetic alterations responsible for SMO-independent GLI activation in cancer cells have been identified. Oncogenic receptor tyrosine kinases, RAS/MAP kinase, PI3K/AKT/S6K, DYRK1A, PKC and histone deacetylases can enhance the transcriptional activity of GLI in human cancer cells [15-21]. Likewise, genetic loss of SUFU results in constitutive GLI activation independent of SMO signaling [22]. In pancreatic cancer, TGFβ/SMAD signaling is able to induce expression of GLI activator forms [23] and in Ewing Sarcoma the EWS-FLI1 oncogene directly stimulates GLI1 expression [24].
GLI proteins, particularly GLI1, act as potent oncogenic drivers by promoting a variety of malignant traits including proliferation, survival, invasion and metastasis (reviewed in [7]). GLI1 also represents a critical determinant of tumor-initiating cancer stem cells in several entities such as glioblastoma, colorectal cancer and pancreatic cancer [16, 25-27]. These oncogenic properties together with the capacity of GLI1 to integrate and relay common SMO-independent cancer-promoting cues such as receptor-tyrosine kinase pathways, PI3K and MAP kinase signaling render GLI1 an attractive molecular target for cancer therapy. However, unlike kinase inhibition, direct targeting of transcription factors is generally considered challenging. Some recent studies demonstrated successful inhibition, though with yet unclear clinical relevance and specificity [28-32].
We therefore turned our focus to kinases as well established therapeutic targets to identify druggable effectors involved in promoting both canonical and SMO-independent GLI activation in cancer. Candidate kinases include members of the Dual-Specificity Tyrosine-Phosphorylation-Regulated Kinase (DYRK) family, which have been shown to positively and negatively modify HH signaling and to have oncogenic functions in solid cancers known to be associated with HH/GLI signaling including pancreatic cancer [33]. The DYRK family comprises two subfamilies with a total of five members [34]. Of note, the class I DYRK family member DYRK1A is able to enhance GLI1 activity, while the closely related yet functionally distinct class I member DYRK1B has been shown to increase HH ligand expression and prevent autocrine HH pathway activation [15, 35]. By contrast, the class II family member DYRK2 negatively affects HH/GLI signaling by triggering the destabilization and degradation of GLI2/3 transcription factors (Figure 1A) [36]. Whether DYRK family members can serve as therapeutic targets in HH/GLI-associated cancer entities has not yet been addressed.
In this study we analyzed the role of class I DYRK members and identified DYRK1B as critical player in both SMO-inhibitor sensitive and resistant settings. Furthermore, we introduce a novel small molecule DYRK1B inhibitor with potent in vitro and in vivo activity targeting GLI dependent cancer cells. We propose that small molecule inhibition of DYRK1B represents a novel and promising approach to target HH/GLI-associated cancers including malignancies with acquired or de novo resistance to SMO inhibitors.
Results
Chemical inhibition of class I DYRK members impairs HH/GLI pathway activation
Members of the DYRK family can modulate GLI activity in opposite directions. While DYRK2 promotes GLI degradation [36], overexpression of the class I family member DYRK1A is able to promote the transcriptional activity of GLI1 (Figure 1A) [15]. Whether small-molecule modulation of DYRK is able to inhibit HH/GLI signaling in cancer cells has not yet been addressed.
To study whether inhibition of DYRK1 kinases affects oncogenic HH/GLI signaling, we first measured the effect of the known class I DYRK inhibitor harmine [37] on HH/GLI pathway activity. As assay system we employed HH-responsive, SMO-inhibitor sensitive human medulloblastoma cells (DAOY) (Figure 1B) [38] and measured changes in the expression of the known HH target genes GLI1 and PTCH as quantitative read-out for pathway activity.
Smoothened Agonist (SAG) treatment of DAOY cells induced mRNA expression of the HH targets GLI1 and PTCH, which was effectively repressed by the SMO inhibitor vismodegib (vismo) and notably, also by the DYRK1 inhibitor harmine in a concentration dependent manner with an IC50(harmine) of 10.9 µM (Figure 1C, 1D). Similar to vismodegib, harmine prevented the accumulation of GLI1 protein in response to SAG treatment (Figure 1E), together suggesting that class I DYRK family members may serve as targets for HH/GLI signal inhibition by non-SMO antagonists.
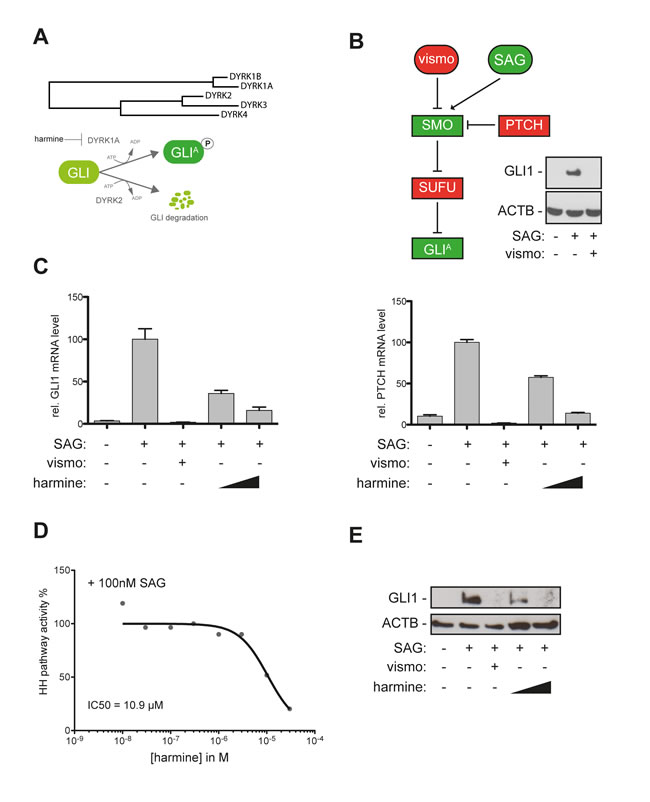
Figure 1: The DYRK1 inhibitor harmine blocks canonical HH/GLI signaling. A. Evolutionary distance of DYRK family members and mode of action of distinct DYRK members on GLI activation (DYRK1A) and GLI degradation (DYRK2). B. DAOY human medulloblastoma cells harbor a responsive canonical HH/GLI signaling system. Treatment with the SMO agonist SAG (100nM) results in activation of GLI1 expression that is quantitatively abolished in the presence of the clinically approved SMO inhibitor vismodegib (vismo) (0.5 µM). Treatment with recombinant sonic HH protein yielded comparable results (data not shown). C. qPCR analysis showing repression of GLI1 mRNA (left) and PTCH mRNA expression (right panel) in SAG-stimulated DAOY cells in response to vismodegib (0.5 µM) or harmine treatment (10 µM and 20 µM). D. Analysis of concentration-dependent inhibition of HH pathway activity and IC50 calculation of 10.9 µM for the natural DYRK inhibitor harmine. E. Efficient inhibition of GLI1 protein expression in SAG-stimulated DAOY cells either treated with vismodegib (0.5 µM) or harmine (10 µM and 20 µM).
Identification of DYRK1B as critical effector in canonical HH/GLI signaling
The beta-carboline alkaloid harmine is a potent inhibitor of both class I family members DYRK1A and DYRK1B and of monoamine oxidase-A [37, 39]. To validate the involvement of DYRK1 kinases in HH pathway regulation and to address whether DYRK1A, DYRK1B or both kinases account for the repressive effect of harmine on HH/GLI pathway activity, we performed genetic RNA-interference mediated perturbation experiments in cells with activated canonical HH/GLI signaling. In SAG-stimulated human medulloblastoma cells, stable, lentiviral shRNA inhibition of DYRK1A moderately reduced GLI1 and PTCH mRNA expression by 55 and 40 percent, respectively (Figure 2A, 2B). By contrast, knockdown of DYRK1B efficiently abolished HH target gene expression below levels of unstimulated cells. We confirmed the contribution of DYRK1B to HH target gene activation with a second, less functional shRNA against DYRK1B (shD1B#2) resulting in reduction of GLI1 and PTCH mRNA expression by more than 50 and 60 percent, respectively, compared to SAG-stimulated cells transduced with scrambled control shRNA (Figure 2A, 2B). Analysis of GLI1 protein expression confirmed mRNA expression data. Inhibition of DYRK1A moderately reduced GLI1 protein levels in SAG-treated DAOY cells, while depletion of DYRK1B efficiently eliminated GLI1 protein expression (Figure 2C). Reduction of DYRK1B expression using shD1B#2 with moderate or shD1B#3 with little functionality repressed GLI1 protein expression proportional to the shRNA functionality (Figure 2C). RNAi-mediated inhibition of DYRK1B did not impair the formation of the primary cilium, an antenna-like compartment essential for HH/GLI signal transduction [4], suggesting that HH/GLI inhibition by DYRK1B targeting is not due to impaired ciliogenesis (Figure S1). The requirement of Dyrk1b for Hh/Gli signaling was also obvious in murine Ptch-deficient BCC cells [40]. Harmine treatment of murine BCC cells reduced Gli1 expression in a concentration-dependent manner (Figure 2D and Figure S2), and like in human medulloblastoma cells, RNAi mediated perturbation of Dyrk1b efficiently inhibited Gli1 protein expression, while depletion of Dyrk1a did not. Taken together, these data identify DYRK1B as druggable target for the inhibition of oncogenic HH/GLI signaling.
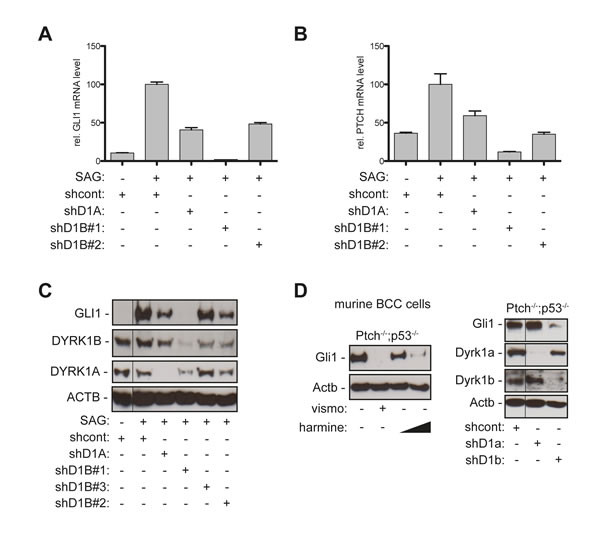
Figure 2: Genetic perturbation of DYRK1B interferes with canonical HH/GLI pathway activation. A.-B. qPCR analysis of GLI1 (A) or PTCH mRNA expression (B) in SAG-treated DAOY cells stably transduced with scrambled control shRNA (shcont), shRNA against DYRK1A (shD1A), or two shRNAs against DYRK1B (shD1B#1, shD1B#2). C. Western blot analysis of GLI1 protein expression in SAG-stimulated DAOY cells expressing the respective lentiviral shRNA constructs and a third shRNA against DYRK1B (shD1B#3). D. Western blot analysis of Ptch-deficient murine BCC cell lines showing abrogation of Gli1 expression by harmine treatment (left panel) (10 µM and 20 µM) and by shRNA against Dyrk1b (shD1b) but not by shRNA against Dyrk1a (shD1a) (right panel). Fine black lines indicate cropping of intermediate lanes from the same Western blots. ACTB/Actb: human/mouse beta actin loading control.
Targeting DYRK1B inhibits HH/GLI signaling in SMO-inhibitor resistant cells
Overcoming de novo and acquired resistance to SMO inhibitors is a major challenge in the treatment of HH-associated cancers, underlining the high medical need for HH inhibitors acting independent of SMO. To map DYRK1B within the HH/GLI signaling cascade, we performed epistasis experiments by using PTCH and SUFU-deficient cells displaying SMO-dependent and SMO-independent pathway activation, respectively. In addition, we analyzed the role of DYRK1B in the regulation of GLI1 expression in pancreatic cancer and Ewing sarcoma cells, where TGFβ/RAS and the EWS-FLI1 oncogene, respectively, control GLI1 expression independent of canonical SMO activity (Figure 3A) [23, 24].
As a first approach we triggered SMO-independent GLI activation in human medulloblastoma cells (DAOY) by RNAi mediated knockdown of SUFU, a critical negative GLI regulator acting downstream of SMO (Figure 3A) [41]. As shown in Figure 3B, SUFU-knockdown in DAOY cells led to activation of GLI1 expression that was resistant to vismodegib treatment. In contrast to SMO inhibition, RNAi-mediated depletion of DYRK1B but not of DYRK1A largely prevented GLI1 expression and moderately reduced GLI2 expression while leaving levels and processing of GLI3 unchanged (Figure 3C).
We confirmed the role of Dyrk1b in the regulation of Gli1 using mouse embryonic fibroblasts (MEF) deficient in either Ptch or Sufu. In line with Dyrk1b acting downstream of Smo, Dyrk1b inhibition by harmine treatment reduced Gli1 expression in Ptch-/- and Sufu-/- MEF (Figure 3D), while vismodegib abolished Gli1 expression only in Ptch-/- but not in Sufu-/- cells (data not shown).
In light of these findings we hypothesized that targeting DYRK1B may represent a novel strategy to inhibit oncogenic GLI1 activity in cancer cells with non-canonical, SMO-independent GLI1 activation. As proof of concept we tested this hypothesis in pancreatic cancer and Ewing sarcoma cell models. Pancreatic cancer cells have been shown to express GLI1 in response to TGFβ and RAS signaling independent of SMO activation [23, 42]. To test whether DYRK1B contributes to the control of GLI1 expression in pancreatic cancer cells, we analyzed PANC-1 cells for GLI1 expression in response to DYRK1 inhibition. PANC-1 cells display high levels of DYRK1B and express detectable levels of GLI1 protein. GLI1 protein levels remained unaffected upon vismodegib treatment, indicating SMO-independent GLI1 regulation and de novo resistance to SMO targeting (Figure 3E, left panel). Of note, RNAi targeting of DYRK1B but not of DYRK1A resulted in loss of GLI1 protein expression and in reduced mRNA levels of the GLI1 target BCL2 [43] (Figure 3E right panel and 3F).
Ewing sarcoma cells harboring the EWS-FLI1 oncogenic fusion gene have been shown to express GLI1 [44] in response to direct binding of the EWS-FLI1 oncoprotein to the GLI1 promoter. Consequently, EWS cells (here A673 cells) display SMO-independent GLI1 expression that is resistant to vismodegib treatment [24] (Figure 3G, left panel). In agreement with the previous data in SUFU-deficient and pancreatic cancer cells, inhibition of DYRK1B but not of DYRK1A reduced GLI1 protein levels (Figure 3G, right panel). Together, these data identify DYRK1B as possible therapeutic target to overcome SMO-inhibitor resistance in GLI1-dependent cancer cells.
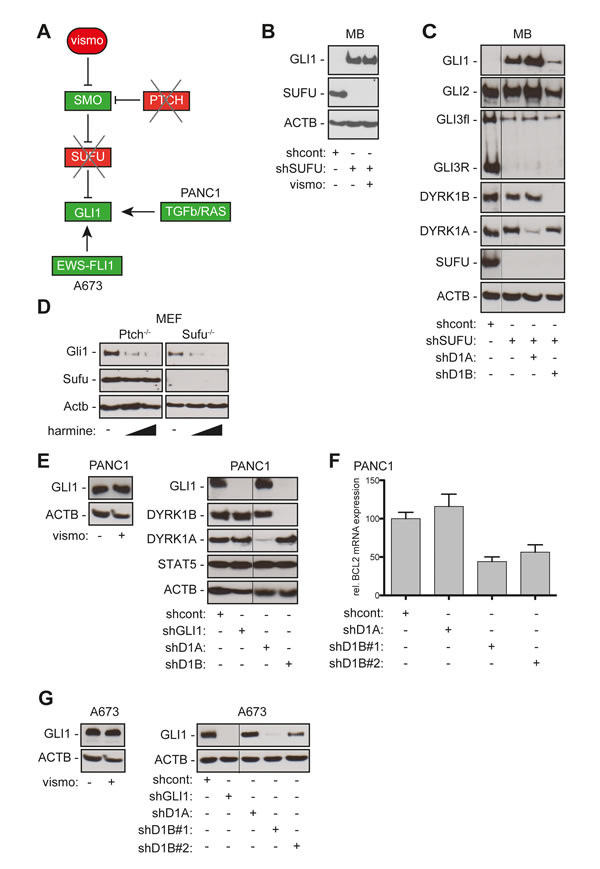
Figure 3: DYRK1B targeting inhibits SMO-dependent and SMO-independent activation of GLI. A. Illustration of HH/GLI signaling and SMO-targeting in PTCH- or SUFU-deficient cells. In PANC-1 pancreatic cancer cells, TGFb and RAS control GLI1 expression independent of SMO. In Ewing sarcoma cells (A673) the EWS-FLI1 oncoprotein directly activates GLI1 expression. B. shRNA-mediated depletion of SUFU renders DAOY cells resistant to SMO inhibition by vismodegib (vismo). Western blot analysis showing that stable expression of shRNA against SUFU (shSUFU) results in activation of GLI1 expression. Note that GLI1 expression in SUFU depleted cells is resistant to SMO inhibition by vismodegib. shcont: scrambled control shRNA. C. Western blot showing GLI1, GLI2 and GLI3 expression in wild-type and SUFU-deficient DAOY cells in response to DYRK1A/B knock-down. Note that RNAi against DYRK1B (shD1B) but not against DYRK1A (shD1A) strongly reduces GLI1 and moderately reduces GLI2 expression in SUFU-depleted (shSUFU) DAOY cells. GLI3 expression and processing are unaffected by DYRK1A and DYRK1B targeting. D. Harmine treatment (10 µM and 20 µM) inhibits Gli1 protein expression in both Ptch-deficient and Sufu-deficient mouse embryonic fibroblasts (MEF). E. Human pancreatic adenocarcinoma cells PANC-1 express detectable levels of GLI1 protein in response to TGFb/SMAD and RAS signaling [23]. GLI1 expression in PANC-1 cells is independent of SMO activity since vismodegib (vismo) treatment does not affect GLI1 protein levels (left panel). RNAi against GLI1 (shGLI1) and against DYRK1B (shD1B) but not against DYRK1A (shD1A) efficiently represses GLI1 protein expression. DYRK1B targeting does not affect non-HH/GLI effectors such as STAT5 (or STAT3 and CTNNB, data not shown). F. qPCR analysis of PANC-1 cells showing that inhibition of DYRK1B (shD1B#1, shD1B#2) but not of DYRK1A (shD1A) reduces expression of the GLI target BCL2. G. GLI1 expression in the Ewing sarcoma cell line A673 harboring the EWS-FLI1 oncogene is resistant to SMO inhibition by vismodegib (vismo) treatment (left panel). While shRNA against DYRK1A (shD1A) does not affect GLI1 expression in A673 cells, knock down of DYRK1B with two distinct shRNAs (shD1B#1, shD1B#2) decreases GLI1 expression (right panel). shGLI1 knockdown demonstrates specificity of the anti-GLI1 antibody used for detection of GLI1 in PANC-1 and A673 cells. ACTB/Actb: human/mouse beta actin loading control. Fine black lines indicate cropping of intermediate lanes from the same Western blots.
A novel DYRK1 inhibitor as potent antagonist of HH/GLI signaling
Having identified DYRK1B as novel drug target for the inhibition of oncogenic HH/GLI signaling, we set out to screen for small molecule inhibitors with potent activity against DYRK1B and pharmacological properties suitable for in vivo administration and therapy.
To identify inhibitors of HH signaling we screened in house kinase inhibitors for activity in Shh-light2 reporter gene assay. This led to the identification of a novel DYRK1 inhibitor referred to as DYRKi (Figure 4A) that inhibited reporter gene activity with an IC50 of 3.7 µM (Figure 4B) without affecting viability and inhibited the target kinase DYRK1B with an IC50 of 90 nM in extracellular in vitro ATP competition assays using recombinant protein (data not shown). The compound was shown to be nontoxic, and demonstrated favorable selectivity against other kinases in the DiscoveRX KINOMEScan (selectivity factor S (35)=0.078) (Figure S3 A-D). DYRKi demonstrated in vivo a dose proportional exposure of up to 100 mg/kg. Administration of 100 mg/kg resulted in a plasma concentration of ~10 µM over 8 h, and a terminal t1/2 of ~3 h, demonstrating suitable pharmacokinetics for once daily dosage (Figure S3 A, B).
Importantly and in line with our DYRK1B RNAi data, DYRKi treatment of SAG-treated SUFU-positive (WT MB +SAG) and untreated SUFU-depleted (∆Sufu MB) human medulloblastoma cells prevented GLI1 mRNA expression at comparable IC50 concentrations of 1.16 µM and 1.04 µM, respectively (Figure 4C). By contrast, vismodegib treatment prevented GLI1 expression only in SUFU-positive cells but not in SUFU-depleted SMO-inhibitor resistant cells (Figure 4D). DYRKi-treated cells were viable and fully responsive to non-HH stimuli such as EGF, indicating that DYRKi-mediated HH-pathway inhibition is not due to unspecific cytotoxic effects of the compound (Figure S4). Furthermore, DYRKi treatment did not significantly change DYRK1B protein levels in DAOY, PANC-1 or A673 cells (Figure S5). Analysis of GLI1 protein expression in SUFU-positive and SUFU-deficient human medulloblastoma cells (Figure 4E, 4F) and Sufu knockout mouse embryonic fibroblasts (Figure 4G) further corroborated the potent activity of DYRKi to inhibit GLI activity in SMO-inhibitor sensitive and resistant cells. Notable, addition of the clinically approved proteasome inhibitor bortezomib largely reversed the negative effect of DYRK1 inhibition on GLI1 and GLI2 by DYRKi (Figure 4H), suggesting that DYRK1B plays a critical role in preventing the GLI activator forms GLI1 and GLI2 from proteasome-mediated degradation, consistent with the documented role of DYRK1B in protein stabilization [34]. Since we were unable to show direct phosphorylation of GLI1 by DYRK1B (data not shown), the detailed mechanisms of GLI1 (and GLI2) protein stabilization by DYRK1B remain unclear.
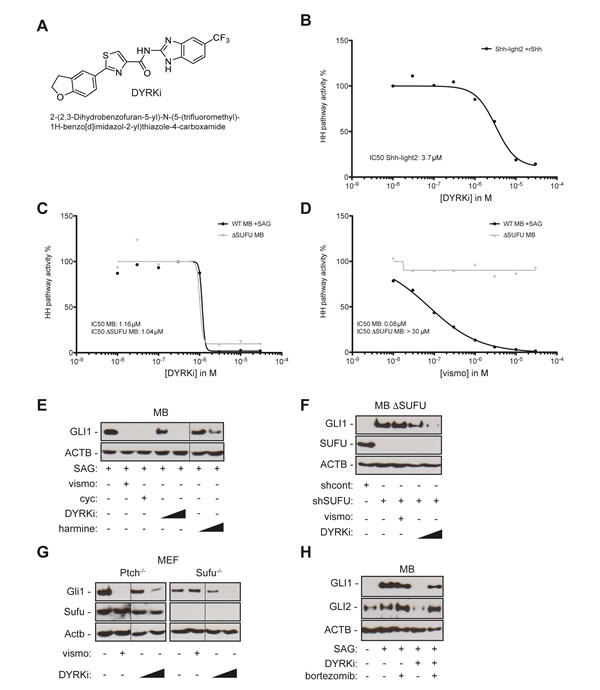
Figure 4: A novel DYRK1 inhibitor efficiently repressing SMO-dependent and SMO-independent GLI1 expression. A. Chemical structure of DYRKi, a novel DYRK1 inhibitor. B. Concentration-dependent inhibition of Hh/Gli signaling in murine Gli luciferase reporter cells by DYRKi resulting in an IC50 of 3.7 µM. C. DYRKi efficiently blocks HH pathway activity in both SAG-stimulated wild-type human medulloblastoma cells (DAOY, WT MB +SAG) and SMO-inhibitor resistant, SUFU depleted medulloblastoma cells (ΔSUFU MB). D. shRNA mediated depletion of SUFU expression renders SAG-stimulated human medulloblastoma cells resistant to SMO inhibition by vismodegib (vismo). Data in C and D were calculated as a function of GLI1 mRNA expression in the respective samples. GLI1 mRNA expression was determined by qPCR. GLI1 mRNA levels of SAG-treated/solvent controls (wild-type DAOY) or solvent-only treated SUFU depleted DAOY cells were set to 100 percent. E. GLI1 protein expression in SAG-stimulated DAOY medulloblastoma cells treated with SMO-antagonists vismodegib (vismo, 0.5 µM), cyclopamine (cyc, 5 µM) or with DYRK1 inhibitors DYRKi (1 µM and 5 µM) or harmine (10 µM and 20 µM). F. Inhibition of GLI1 protein expression in SUFU depleted DAOY medulloblastoma (shSUFU) cells by DYRKi treatment (1 µM and 5 µM). Note that vismodegib (vismo, 0.5 µM) fails to reduce GLI1 expression. shcont: scrambled control shRNA. G. DYRKi treatment (1 µM and 5 µM) represses Gli1 protein expression in both Ptch-deficient and Sufu-deficient mouse embryonic fibroblasts isolated from Ptch- and Sufu knockout mice, respectively. Vismodegib (vismo) inhibits Gli1 expression in Ptch-deficient cells only. H.Treatment with the proteasome inhibitor bortezomib largely reverses the suppressive effect of DYRKi on GLI1 and GLI2 protein expression, supporting a model of post-translational regulation of GLI1 and GLI2 stability by DYRK1B. Fine black lines indicate cropping of intermediate lanes from the same Western blots. ACTB/Actb: human/mouse beta actin loading control.
DYRK1B targeting impairs oncogenic growth of GLI-dependent pancreatic cancer cells
In previous work, others and we have provided evidence for a critical tumorigenic role of GLI1 in pancreatic cancer cells, including reduced formation of tumor-initiating spheroids in vitro and impaired in vivo tumor growth [45-47]. Having shown that GLI1 expression in PANC-1 cells depends on DYRK1B, we therefore addressed whether inhibition of DYRK1B is able to phenocopy the anti-tumorigenic effect of GLI1 inhibition in pancreatic cancer cells. We first monitored the effect of DYRK1B inhibition on tumor-initiating spheroid formation of GLI1-dependent yet SMO-inhibitor resistant PANC-1 cells in 3D cultures [46]. As shown in Figure 5A and 5B, treatment with the SMO inhibitor vismodegib did not affect clonogenic growth, while harmine and DYRKi treatment significantly reduced large spheroid formation. Pre-treatment of pancreatic cancer cells with DYRKi or vismodegib prior to seeding into 3D cultures for clonogenic growth assays yielded similar results. Again, DYRKi reduced the formation of tumor-initiating spheres while vismodegib did not (Figure S6). In agreement with the critical role of DYRK1B in GLI1 expression, RNA-interference against DYRK1B but not against DYRK1A inhibited clonogenic growth of GLI1-dependent pancreatic cancer cells. Note that neither chemical nor genetic inhibition of DYRK1B significantly reduced viability of cells in planar cultures and equal numbers of viable cells were used in all assays.
To test the in vivo relevance of these data, we performed xenograft assays with two distinct GLI1-dependent pancreatic cancer cell lines (PANC-1 and L3.6pl) [46]. In line with in vitro data, stable knockdown of DYRK1B effectively abolished the engraftment and in vivo tumor growth of PANC-1 and L3.6pl pancreatic cancer cells (Figure 5C, 5D). In addition, oral administration of DYRKi significantly reduced in vivo tumor growth of pancreatic cancer cells (Figure 5E).
Taken together, these results demonstrate that inhibition of DYRK1B efficiently represses GLI1 expression and reduces the malignant properties of GLI1 dependent cancer cells including pancreatic cancer cells with resistance to SMO inhibitors.
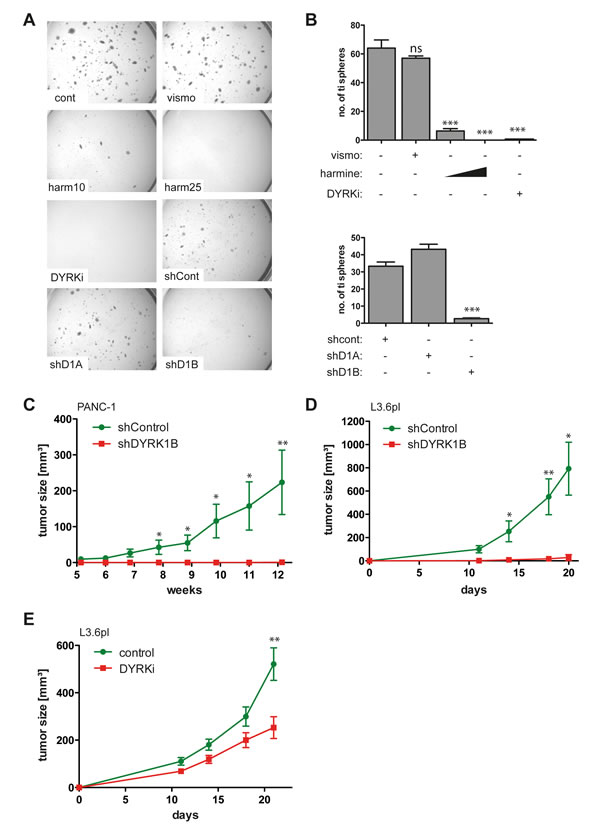
Figure 5: DYRK1B targeting inhibits the malignant properties of GLI1-dependent human pancreatic cancer cells. A.-B. Growth of GLI1 expressing PANC-1 spheres in 3-D cultures. Sphere formation is resistant to SMO inhibition by vismodegib (vismo) and shRNA against DYRK1A. By contrast, harmine (10 µM and 25 µM), DYRKi (5 µM) and shRNA against DYRK1B (shD1B) efficiently prevent the formation of tumor-initiating spheres. Sphere formation shown in A) was quantitatively analyzed and the number of tumor-initiating (ti) spheres plotted in B). For all experiments, identical numbers of live cells were seeded into 3D matrix cultures. We noted that inhibition of DYRK1B does not simply induce cell death but prevents the formation of large spheres formed by highly clonogenic, putative tumor-initiating cells [16]. C.-D. Xenograft analysis of in vivo tumor growth of PANC-1 (n=7) in C. and highly metastatic GLI1-dependent L3.6pl pancreatic cancer cell lines (n=6) in D. shControl: cells lentivirally transduced with scramble control shRNA, shDYRK1B: cancer cells stably expressing shRNA against DYRK1B. E. Oral administration of DYRKi (100 mg/kg/d) significantly reduces in vivo tumor growth of GLI1-dependent pancreatic cancer cells (L3.6pl) (n=20). Control mice (n=8) received solvent only (control). * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001;
Discussion
The widespread aberrant activation of HH/GLI in human cancers and its causal role in tumor initiation and growth explain the attractiveness and rationale of targeting HH/GLI signaling in cancer. Most efforts to identify selective HH pathway inhibitors have so far concentrated on targeting the essential pathway effector SMO. In 2012, the FDA approved the first-in class oral SMO inhibitor vismodegib for the treatment of advanced and metastatic basal cell carcinoma with striking therapeutic efficacy, though severe side effects and the rapid development of acquired SMO inhibitor resistance pose significant limitations to the clinical application of drugs targeting SMO [10, 11, 48]. Furthermore, clinical trials on colorectal, ovarian and pancreatic cancer failed to demonstrate therapeutic efficacy of SMO inhibition [13], which may at least in part be due to SMO-independent activation of oncogenic GLI activity by for instance TGFβ, RAS, PI3K/AKT/S6K or genetic deletion of the GLI repressor SUFU (reviewed in [7, 49]). Together, these potential shortfalls of SMO inhibitors call for the identification of targets regulating oncogenic GLI downstream of SMO.
In the present study, we identified DYRK1B as cell-autonomous positive regulator of GLI activity. We have shown that genetic and chemical perturbation of DYRK1B represses the expression of the GLI1 oncogene in a variety of settings, including human brain and pancreatic cancer and murine basal cell carcinoma cells. Importantly, DYRK1B targeting abolished GLI1 expression in SMO-inhibitor sensitive and SMO-inhibitor resistant cells including SUFU deficient medulloblastoma, GLI1-dependent pancreatic cancer [16, 23] and Ewing sarcoma cells expressing GLI1 in response to the EWS-FLI1 oncoprotein [24, 44]. This places DYRK1B function downstream of SMO/SUFU to promote GLI1 oncogene expression.
Mechanistically, we propose that D1B enhances the stability of GLI activator forms [50], as inhibition of the proteasome machinery neutralizes the negative regulatory effect of DYRK1B targeting on GLI1/2 expression. Whether stabilization of GLI1 and GLI2 involves direct phosphorylation by DYRK1B or depends on alternative indirect mechanisms is unclear at present and requires future in-depth analysis of post-translational GLI modifications.
In light of the disappointing outcomes of several clinical trials with SMO inhibitors, targeting oncogenic GLI transcription factors downstream of SMO has emerged as promising alternative therapeutic strategy. Other non-SMO Hedgehog pathway inhibitors including kinase inhibitors or epigenetic modifiers [19, 31, 51], inhibitors of GLI DNA binding and/or post-transcriptional GLI activation [28, 30, 52-54], or GLI antagonists with yet unidentified targets have recently been reported [29], supporting the promising therapeutic potential of targeting oncogenic GLI transcription factors in settings where SMO-inhibition is inefficient. However, except for arsenic trioxide and pyrvinium [51, 52, 54], lack of suitable pharmacological properties, limited potency or toxicity is a concern for many of the GLI antagonists identified. The identification of DYRKi as a novel DYRK1 inhibitor with suitable pharmacokinetics and anti-GLI1 activity provides proof-of-concept for oncogenic HH/GLI inhibition by DYRK1B targeting in malignancies with de novo or acquired SMO inhibitor resistance.
Previous studies have already highlighted the complex regulatory role of DYRK family members in HH/GLI signaling. For instance, DYRK2 can directly phosphorylate GLI2 thereby promoting its degradation, while overexpression of DYRK1A can enhance GLI transcriptional activity. DYRK1B, the closest homologue of DYRK1A, can act downstream of RAS to prevent autocrine and promote paracrine HH signaling in RAS mutant cancer cells [35]. The same study has also shown a repressive effect of oncogenic RAS and its downstream effector DYRK1B on GLI expression, contrasting our findings and those of other independent studies that support a GLI-activating role of DYRK1B and RAS, respectively [23, 42, 55]. Whether this discrepancy is due to different experimental conditions (e.g. serum, confluency, cell line passages, transient versus stable long-term knockdown approaches) is unclear.
Our findings that DYRK1B rather than DYRK1A acts as critical positive regulator of HH/GLI add another example of HH/GLI regulation by DYRK kinases with possible therapeutic relevance and also highlight the highly complex and context-dependent control of HH/GLI activity [7].
Using SMO-inhibitor resistant pancreatic cancer cells we also confirm a previous report showing that DYRK1B targeting in pancreatic cancer cells has pronounced therapeutic efficacy in vitro and in vivo, though the molecular mechanisms including the link to HH/GLI signaling remained unidentified in this study [56]. In the present study we show that interfering with DYRK1B dramatically impairs SMO-independent GLI1 expression in RAS mutant pancreatic cancer cells and phenocopies the anti-tumorigenic effect of GLI1 targeting [16]. Since GLI activator function is critical for tumor-initiating cells [16, 25, 25, 57] and also involved in the development of Ras-driven murine pancreatic cancer within the epithelial tumor compartment [23, 47, 55], we speculate that chemical DYRK1B inhibition may be able to repress the growth of pancreatic cancer due to negative regulation of SMO-independent GLI1 in tumor and tumor-initiating cells, consistent with the abrogation of tumor engraftment in response to genetic DYRK1B inhibition. Intriguingly, up to 10 percent of pancreatic cancers harbor genomic amplifications of DYRK1B and display DYRK1B overexpression in the epithelial compartment of the tumors [33]. Targeting DYRK1B may therefore overcome the inefficient therapeutic response of pancreatic cancer patients to SMO-inhibitors that target canonical paracrine signaling in the tumor environment rather than non-canonical GLI activity in the epithelial tumor compartment [58-61]. It will be important to address in future studies whether DYRK1B overexpression and amplification correlate with increased epithelial GLI1 protein levels and therapeutic response to DYRK1B inhibition in patient samples.
In summary, the identification of DYRK1B as drug target for the inhibition of oncogenic HH/GLI signaling in SMO-inhibitor resistant cancers and the introduction of DYRKi as novel DYRK1B inhibitor with suitable pharmacologic properties provide a basis for future efforts to translate these findings to clinical testing.
Materials and Methods
Cell culture and inhibitors
DAOY cells (American Type Culture Collection, ATCC, Manassas, VA, USA) were grown as described previously [38]. BSZ2 cells were grown as described in [40] and PANC-1 (ATCC) cells as described in [46].
Ptch-/- and Sufu-/- embryonic fibroblasts were grown in DMEM supplemented with 10% fetal bovine serum (FBS), antibiotics and for Sufu-/- cells also with L-glutamine. During assays and treatments of confluent cells, FBS was reduced to 0.5%. For 3-dimensional (3D) cultures, 5 x 103 cells were seeded in 12-well plates as described previously [62]. 3D spheroid cultures were grown for 4-6 weeks at 37°C in a humidified atmosphere containing 5% CO2. Colony formation was documented on a stereomicroscope with Cell^D Image capture system (Olympus, Tokyo, Japan) and quantified using Colony Counter Software (Microtec Nition, Chiba, Japan). Smoothened agonist SAG (Axxora, Farmingdale NY, USA), harmine (Thermo Fisher Scientific, Waltham, MA, USA), vismodegib, cyclopamine (LC Laboratories, Woburn, MA, USA), GANT61 (Merck Chemicals Ltd., Darmstadt, Germany) and DYRKi were dissolved in dimethylsulfoxide (DMSO, Sigma, St. Louis, MO, USA).
RNA interference and lentiviral transduction
Stable RNAi knockdown experiments were performed by lentiviral shRNA transductions as described in [16]. The following shRNA constructs selected from the Mission TRC shRNA library (Sigma) were used: shRNA DYRK1A (TRCN0000000526), shRNA DYRK1B#1 (TRCN0000002139), shRNA DYRK1B#2 (TRCN0000355722), shRNA DYRK1B#3 (TRCN0000355721), shSUFU (TRCN0000019466) and scrambled control shRNA (SHC002) (Sigma). The shGLI1 construct has been described in [16]. The functionality of shRNAs was validated by Western blot analysis using antibodies listed below.
RNA isolation, qPCR and Western blot analysis
Total RNA was isolated using TRI-reagent (Molecular Research Center Inc., Cincinnati, OH, USA) followed by LiCl purification. Precipitated and purified RNA was used for cDNA synthesis using Superscript II reverse transcriptase (Life Technologies, Thermo Fisher Scientific) according to the manufacturer´s instructions. qPCR was done on a Rotorgene Q (Qiagen, Venlo, Netherlands) using GoTaq qPCR Mastermix reagent (Promega, Fitchburg, WI, USA). HH target genes were identified with primers as described in [16].
For Western blot analysis, proteins were visualized with horseradish peroxidase-conjugated secondary antibodies in combination with enhanced chemiluminescence detection system (GE Health Care, Chalfont St Giles, United Kingdom). The following antibodies were used: anti-GLI1 (V812; Cell Signaling Technology, Danvers, MA, USA), anti-GLI2 (H-300, Santa Cruz Biotechnology, Dallas, TX, USA), anti-GLI3 (R&D Systems, Minneapolis, MN, USA), anti-DYRK1A, anti-DYRK1B (Cell Signaling Technology), anti-SUFU (C-15, Santa Cruz Biotechnology), anti-STAT3 (BD Biosciences, San Jose, CA, USA), anti-STAT5 (3H7, Cell Signaling Technology), anti-Beta Catenin (Cell Signaling Technology), anti-ACTB (Santa Cruz Biotechnology)
HH/GLI reporter assays
2.5 x 104 Gli Reporter - NIH3T3 cells (AMS Biotechnology Ltd., Abingdon, United Kingdom) were seeded per well into a white 96 well plate. After overnight incubation, compounds were added for 1h prior to HH pathway stimulation with 1 µg/ml murine SHH (R&D Systems). After incubation for 24h the cells were investigated for viability using CellTiter-FluorTM Kit (Promega) and for reporter gene activity using ONE-Glo™ Luciferase Assay System (Promega).
Identification and synthesis of the DYRK1 inhibitor DYRKi
We applied the KINOMEscan™ screening platform to quantify interactions between test compounds and more than 450 human kinases and disease relevant mutant variants. The assay was performed at DiscoveRX (San Diego, CA, USA) according to the protocol description available at http://www.discoverx.com/targets/kinase-target-biology. The Image shown in Figure S3 was generated using TREEspot™ Software Tool and reprinted with permission from KINOMEscan®, a division of DiscoveRX Corporation (San Diego, CA, USA).
DYRK1B-Kinase Assay was performed at Reaction Biology Corporation (Reaction Biology Corp., Malvern, PA, USA) according to following protocol. The substrate DYRKtide was prepared in fresh Base Reaction Buffer (20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/ml BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO) and required cofactors were added. Recombinant DYRK1B was added to the substrate solution and gently mixed. Compound dilution series in DMSO were added to the reaction, followed 20 min later by addition of a mixture of ATP and 33P ATP (specific activity 0.01 µCi/µl final) to a final concentration of 10 µM. Reactions were carried out at 25°C for 120 min, followed by spotting the reactions onto P81 ion exchange filter paper. Unbound phosphate was removed by extensive washing of the filters in 0.75% phosphoric acid. After subtraction of background derived from control reactions containing inactive enzyme, kinase activity data were expressed as the percent remaining kinase activity in test samples compared to vehicle (DMSO) reactions. IC50 values and curve fits were obtained using Prism (Graph Pad Software, La Jolla, CA, USA).
Synthesis of DYRKi was done as follows: to a solution of 2-(2,3-dihydro-1-benzofuran-5-yl)-1,3-thiazole-4-carboxylic acid (1.00 g. 4.04 mmol) in 20 ml N,N-dimethylformamide, 5-(trifluoromethyl)-1H-benzo[d]imidazol-2-amine (895 mg, 4.45 mmol), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (1.53 g, 4.04 mmol), 4-dimethylaminopyridine (49 mg, 0.40 mmol) and N,N-diisopropylethylamine (1.76 ml, 10.11 mmol) were added. The reaction mixture was stirred for 72 h at room temperature, poured into ice water and the formed precipitate dried and purified by flash column chromatography (DCM/MeOH 95:5 to 0:100). The crude product was suspended in Et2O, filtered and dried. The product was obtained as a white solid (1.07 g, 2.49 mmol, 62 % yield). mp: 232.5; 1H NMR (400 MHz, DMSO-d6) d 12.51 (bs, 1 H), 11.81 (bs, 1 H), 8.50 (s, 1 H), 8.04 (bs, 1 H), 7.85 (dd, J=8.33 Hz, J=1.91 Hz, 1 H), 7.77 (bs, 1 H), 7.62 (d, J=8.34 Hz, 1 H), 7.40 (dd, J=8.46 Hz, J=1.44 Hz, 1 H), 4.57 (t, J=8.75 Hz, 2 H), 3.16-3.30 (m, 2 H); 13C NMR (75 MHz, DMSO-d6) d 168.0, 162.1, 159.9, 148.4, 148.2, 128.6, 127.4, 126.9, 125.8, 125.1, 123.7, 123.3, 122.1, 121.7, 118.0, 118.0, 117.9, 109.2, 71.7, 28.5; analysis (calcd., found for C20H13F3N4O2S): C (55.81, 55.56), H (3.04, 3.17), F (13.24, 13.1), N (13.02, 12.98), S (7.45, 7.18); LC/MS [M+H]+: 431.0.
Xenograft experiments
For in vivo tumor growth studies 1 x 106 PANC-1 or 1 x 105 L3.6pl pancreatic cancer cells in 25% Matrigel (BD Biosciences) were injected subcutaneously into the lower flanks of Foxn1nu nude mice (Charles River Laboratories, Wilmington, MA, USA). For in vivo treatment, DYRKi was dissolved in sun flower oil and administered at 100mg/kg/d by oral gavage. Tumor volume was measured with a caliper and calculated according to the formula [4/3 x π x (length/2) x (width/2) x (height/2)].
Acknowledgments
We are grateful to Drs. Stephan Teglund and Rune Toftgard for Sufu and Ptch knockout cells and to Mag. Andrea Loipetzberger, Dr. Aniela Sommer and Dr. Ilse Foissner for technical assistance with confocal microscopy.
Conflicts of interest
MZ, SM, SH, JL, DV are or were employees of 4SC group (4SC Discovery GmbH and 4SC AG), 4SC Discovery GmbH is developing novel DYRK1B inhibitors for cancer therapy. 4SC AG and 4SC Discovery GmbH contributed to the costs for testing DYRK inhibitors. No other authors declared any conflict of interest.
Grant support
This work was financially supported by the priority program “Biosciences and Health” of the Paris-Lodron University of Salzburg, the Austrian Science Fund projects P20652 and P25629 and the Marie Curie Initial Training Network HEALING. Novel DYRK inhibitors were provided by 4SC Discovery GmbH.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Teglund S and Toftgard R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochimica et biophysica acta. 2010; 1805:181-208.
2. Merchant AA and Matsui W. Targeting Hedgehog--a cancer stem cell pathway. Clinical cancer research. 2010; 16:3130-3140.
3. Basset-Seguin N, Sharpe HJ and de Sauvage FJ. Efficacy of Hedgehog Pathway Inhibitors in Basal Cell Carcinoma. Molecular cancer therapeutics. 2015.
4. Eggenschwiler JT and Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007; 23:345-373.
5. Hui CC and Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011; 27:513-537.
6. Ingham PW, Nakano Y and Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet. 2011; 12:393-406.
7. Aberger F and Ruiz i Altaba A. Context-dependent signal integration by the GLI code: the oncogenic load, pathways, modifiers and implications for cancer therapy. Semin Cell Dev Biol. 2014; 33:93-104.
8. van den Heuvel M and Ingham PW. smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature. 1996; 382:547-551.
9. Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008; 8:743-754.
10. Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang AL, Oro AE and Tang JY. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer cell. 2015; 27:342-353.
11. Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, Tsui V, Durham AB, Dlugosz AA, Haverty PM, Bourgon R, Tang JY, Sarin KY, Dirix L, Fisher DC, Rudin CM, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer cell. 2015; 27:327-341.
12. Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, Scales SJ, Gould SE, Yauch RL and de Sauvage FJ. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011; 71:435-444.
13. Ng JM and Curran T. The Hedgehog’s tale: developing strategies for targeting cancer. Nature reviews Cancer. 2011; 11:493-501.
14. Jimeno A, Weiss GJ, Miller WH, Jr., Gettinger S, Eigl BJ, Chang AL, Dunbar J, Devens S, Faia K, Skliris G, Kutok J, Lewis KD, Tibes R, Sharfman WH, Ross RW and Rudin CM. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clinical cancer research. 2013; 19:2766-2774.
15. Mao J, Maye P, Kogerman P, Tejedor FJ, Toftgard R, Xie W, Wu G and Wu D. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J Biol Chem. 2002; 277:35156-35161.
16. Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, Zoidl K, Schnidar H, Hache H, Bauer HC, Solca F, Hauser-Kronberger C, Ermilov AN, Verhaegen ME, Bichakjian CK, Dlugosz AA, Nietfeld W, et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol Med. 2012; 4:218-233.
17. Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, Beermann F and Ruiz IAA. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:5895-5900.
18. Riobo NA, Lu K, Ai X, Haines GM and Emerson CP, Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:4505-4510.
19. Atwood SX, Li M, Lee A, Tang JY and Oro AE. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature. 2013; 494:484-488.
20. Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, Li CW, Hsu JL, Miller SA, Wang X, Lee DF, Hsu JM, Huo L, Labaff AM, Liu D, Huang TH, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer cell. 2012; 21:374-387.
21. Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, Pietrosanti L, De Smaele E, Ferretti E, Miele E, Pelloni M, De Simone G, Pedone EM, Gallinari P, Giorgi A, Steinkuhler C, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010; 12:132-142.
22. Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, Scherer SW, Dura WT, Wainwright B, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002; 31:306-310.
23. Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME and Hanahan D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009; 23:24-36.
24. Beauchamp E, Bulut G, Abaan O, Chen K, Merchant A, Matsui W, Endo Y, Rubin JS, Toretsky J and Uren A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem. 2009; 284:9074-9082.
25. Clement V, Sanchez P, de Tribolet N, Radovanovic I and Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007; 17:165-172.
26. Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P and Ruiz i Altaba A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009; 1:338-351.
27. Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, Karikari C, Alvarez H, Iacobuzio-Donahue C, Jimeno A, Gabrielson KL, Matsui W and Maitra A. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007; 67:2187-2196.
28. Lauth M, Bergstrom A, Shimokawa T and Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:8455-8460.
29. Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K, Sun M, Rack PG, Sinha S, Wu JJ, Solow-Cordero DE, Jiang J, Rowitch DH and Chen JK. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:14132-14137.
30. Infante P, Mori M, Alfonsi R, Ghirga F, Aiello F, Toscano S, Ingallina C, Siler M, Cucchi D, Po A, Miele E, D’Amico D, Canettieri G, De Smaele E, Ferretti E, Screpanti I, et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. Embo J. 2015; 34:200-217.
31. Tang Y, Gholamin S, Schubert S, Willardson MI, Lee A, Bandopadhayay P, Bergthold G, Masoud S, Nguyen B, Vue N, Balansay B, Yu F, Oh S, Woo P, Chen S, Ponnuswami A, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014; 20:732-740.
32. Kreso A, van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T, Cao L, Baiazitov R, Du W, Sydorenko N, Moon YC, Gibson L, Wang Y, Leung C, Iscove NN, Arrowsmith CH, et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. 2014; 20:29-36.
33. Friedman E. Mirk/Dyrk1B in cancer. Journal of cellular biochemistry. 2007; 102:274-279.
34. Becker W. Emerging role of DYRK family protein kinases as regulators of protein stability in cell cycle control. Cell Cycle. 2012; 11:3389-3394.
35. Lauth M, Bergstrom A, Shimokawa T, Tostar U, Jin Q, Fendrich V, Guerra C, Barbacid M and Toftgard R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nature structural & molecular biology. 2010; 17:718-725.
36. Varjosalo M, Bjorklund M, Cheng F, Syvanen H, Kivioja T, Kilpinen S, Sun Z, Kallioniemi O, Stunnenberg HG, He WW, Ojala P and Taipale J. Application of active and kinase-deficient kinome collection for identification of kinases regulating hedgehog signaling. Cell. 2008; 133:537-548.
37. Gockler N, Jofre G, Papadopoulos C, Soppa U, Tejedor FJ and Becker W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. The FEBS journal. 2009; 276:6324-6337.
38. Gotschel F, Berg D, Gruber W, Bender C, Eberl M, Friedel M, Sonntag J, Rungeler E, Hache H, Wierling C, Nietfeld W, Lehrach H, Frischauf A, Schwartz-Albiez R, Aberger F and Korf U. Synergism between Hedgehog-GLI and EGFR signaling in Hedgehog-responsive human medulloblastoma cells induces downregulation of canonical Hedgehog-target genes and stabilized expression of GLI1. PLoS One. 2013; 8:e65403.
39. Kim H, Sablin SO and Ramsay RR. Inhibition of monoamine oxidase A by beta-carboline derivatives. Archives of biochemistry and biophysics. 1997; 337:137-142.
40. So PL, Langston AW, Daniallinia N, Hebert JL, Fujimoto MA, Khaimskiy Y, Aszterbaum M and Epstein EH, Jr. Long-term establishment, characterization and manipulation of cell lines from mouse basal cell carcinoma tumors. Experimental dermatology. 2006; 15:742-750.
41. Kogerman P, Grimm T, Kogerman L, Krause D, Unden AB, Sandstedt B, Toftgard R and Zaphiropoulos PG. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999; 1:312-319.
42. Ji Z, Mei FC, Xie J and Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem. 2007; 282:14048-14055.
43. Bigelow RL, Chari NS, Unden AB, Spurgers KB, Lee S, Roop DR, Toftgard R and McDonnell TJ. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J Biol Chem. 2004; 279:1197-1205.
44. Zwerner JP, Joo J, Warner KL, Christensen L, Hu-Lieskovan S, Triche TJ and May WA. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene. 2008; 27:3282-3291.
45. Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME and Hanahan D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009; 23:24-36.
46. Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, Zoidl K, Schnidar H, Hache H, Bauer H-C, Solca F, Hauser-Kronberger C, Ermilov AN, Verhaegen ME, Bichakjian CK, Dlugosz AA, Nietfeld W, et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. Embo Molecular Medicine. 2012; 4:218-233.
47. Rajurkar M, De Jesus-Monge WE, Driscoll DR, Appleman VA, Huang H, Cotton JL, Klimstra DS, Zhu LJ, Simin K, Xu L, McMahon AP, Lewis BC and Mao J. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc Natl Acad Sci U S A. 2012; 109:E1038-1047.
48. Tang JY, Mackay-Wiggan JM, Aszterbaum M, Yauch RL, Lindgren J, Chang K, Coppola C, Chanana AM, Marji J, Bickers DR and Epstein EH, Jr. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. The New England journal of medicine. 2012; 366:2180-2188.
49. Ruiz i Altaba A, Mas C and Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007; 17:438-447.
50. Huntzicker EG, Estay IS, Zhen H, Lokteva LA, Jackson PK and Oro AE. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006; 20:276-281.
51. Li B, Fei DL, Flaveny CA, Dahmane N, Baubet V, Wang Z, Bai F, Pei XH, Rodriguez-Blanco J, Hang B, Orton D, Han L, Wang B, Capobianco AJ, Lee E and Robbins DJ. Pyrvinium attenuates hedgehog signaling downstream of smoothened. Cancer Res. 2014; 74:4811-4821.
52. Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, Peaceman D, Ozdemirli M, Rodriguez O, Macdonald TJ, Albanese C, Toretsky JA and Uren A. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. The Journal of clinical investigation. 2011; 121:148-160.
53. Kim J, Lee JJ, Kim J, Gardner D and Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:13432-13437.
54. Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, Kim J, Chen B, King EM, Borodovsky A, Riggins GJ, Epstein EH, Jr., Beachy PA and Rudin CM. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer cell. 2013; 23:23-34.
55. Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA and Hebrok M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006; 20:3161-3173.
56. Deng X and Friedman E. Mirk kinase inhibition blocks the in vivo growth of pancreatic cancer cells. Genes cancer. 2014; 5:337-347. doi: 10.18632/genesandcancer.29.
57. Santini R, Vinci MC, Pandolfi S, Penachioni JY, Montagnani V, Olivito B, Gattai R, Pimpinelli N, Gerlini G, Borgognoni L and Stecca B. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells. 2012; 30:1808-1818.
58. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, Nannini-Pepe M, Kotkow K, Marsters JC, Rubin LL and de Sauvage FJ. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008; 455:406-410.
59. Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S, Nagle JM, Deshpande V, Boucher Y, Kato T, Chen JK, Willmann JK, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:E3091-3100.
60. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer cell. 2014; 25:735-747.
61. Hwang RF, Moore TT, Hattersley MM, Scarpitti M, Yang B, Devereaux E, Ramachandran V, Arumugam T, Ji B, Logsdon CD, Brown JL and Godin R. Inhibition of the hedgehog pathway targets the tumor-associated stroma in pancreatic cancer. Mol Cancer Res. 2012; 10:1147-1157.
62. Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M and Aberger F. Epidermal Growth Factor Receptor Signaling Synergizes with Hedgehog/GLI in Oncogenic Transformation via Activation of the MEK/ERK/JUN Pathway. Cancer Res. 2009; 69:1284-1292.